

Abstract
The effect of noradrenaline on the volume-sensitive chloride current (ICl(swell)) was studied with conventional whole-cell recording techniques in freshly dispersed isolated smooth muscle cells of the rabbit portal vein. In the absence of receptor antagonists, noradrenaline produced an increase in the amplitude of ICl(swell) in some cells and a decrease in others. In the presence of the β-adrenoceptor antagonist propranolol, noradrenaline increased ICl(swell) and in the presence of the α1-adrenoceptor antagonist prazosin, noradrenaline reduced ICl(swell). The phospholipase C (PLC) inhibitor U73122 reduced the amplitude of ICl(swell) whereas the inactive analogue U73343 had no effect. The phorbol esters phorbol-12-myristate-13-acetate (PMA) and phorbol-12,13-dibutyrate (PDBu) increased the amplitude of ICl(swell) by approximately 60 and 100 %, respectively, in a voltage-independent fashion. Inhibitors of protein kinase C (PKC) chelerythrine and calphostin-C decreased the amplitude of ICl(swell) in a concentration-dependent but voltage-independent manner. Bath application of 8-Br-cAMP decreased ICl(swell) by about 60 % whereas the inhibitor of protein kinase A (PKA) KT5720 increased the amplitude of ICl(swell) by approximately 80–90 %. In the presence of propranolol, chelerythrine prevented the increase of ICl(swell) by noradrenaline; in the presence of prazosin, KT5720 blocked the inhibitory action of noradrenaline. The results show that in rabbit portal vein myocytes noradrenaline enhances ICl(swell) by acting on α1-adrenoceptors and reduces ICl(swell) by stimulating β-adrenoceptors. The data suggest that the potentiating and inhibitory effects of noradrenaline are mediated, respectively, by PKC and PKA.
A volume-sensitive chloride current (ICl(swell)) has been recorded in many cell types and several physiological roles have been proposed for this conductance including volume regulation and cell proliferation (e.g. see Okada, 1997). Recently ICl(swell) has also been identified in vascular smooth muscle cells (Yamazaki et al. 1998; Greenwood & Large, 1998) and it has been suggested that ICl(swell) may also be involved in controlling vascular contractility (Greenwood & Large, 1998; Nelson, 1998; Greenwood & Large, 1999; Graves et al. 2000). This proposed function is based on the fact that in smooth muscle the chloride equilibrium potential (ECl, about −20 to −30 mV) is much more positive than the resting membrane potential. Consequently, activation of ICl(swell) depolarises the membrane potential and increases the open probability of voltage-dependent calcium channels resulting in an influx of Ca2+.
The possible involvement of ICl(swell) in vascular contraction was based primarily on pharmacological studies in which it was found that inhibitors of ICl(swell) blocked the myogenic response in rat cerebral arteries (Nelson et al. 1997); Cl− efflux has also been recorded during the myogenic response (Doughty & Langton, 2001). In addition the contraction of rat coronary arteries produced by inhibition of nitric oxide (NO) synthesis was also blocked by antagonists of ICl(swell) (Graves et al. 2000). These observations suggested that ICl(swell) may contribute to vascular contraction in addition to its other proposed roles, such as cell proliferation. It is well established that agents that cause smooth muscle contraction also stimulate mitogenesis (e.g. see Tolloczko et al. 2000).
Previously we demonstrated that the vasodilator substance NO has a dual effect on ICl(swell) in rabbit portal vein myocytes. NO increased the amplitude of ICl(swell) by cGMP-dependent phosphorylation while NO decreased ICl(swell) in a cGMP-independent manner (Ellershaw et al. 2000). In the present work we have investigated the effects of the sympathetic neuro-effector transmitter noradrenaline on ICl(swell) in rabbit portal vein cells. The results show that noradrenaline acts on α-adrenoceptors to increase ICl(swell), an effect which is mimicked by phorbol esters and blocked by inhibitors of protein kinase C (PKC) and therefore may be mediated by PKC. In addition there is a β-adrenoceptor-mediated decrease of ICl(swell) which is blocked by an inhibitor of protein kinase A (PKA) and which is similar to the effect of 8-Br-cAMP implicating a role for PKA in the inhibitory effect of noradrenaline.
METHODS
Cell preparation
New Zealand White rabbits (2-3 kg) were killed by injection of a lethal dose of sodium pentobarbitone (120 mg kg−1 i.v.) into the ear vein in accordance with Home Office regulations. Portal veins were excised, cleaned of fat and connective tissue and the exposed muscle sheet was cut into strips which were then immersed in physiological salt solution (PSS) containing 100 μm CaCl2 at 37 °C. Single smooth muscle cells were prepared by first incubating the tissue with protease Type 14 (0.2 mg ml−1, Sigma, UK) for 5 min. The tissue was then washed in PSS, containing 100 μm Ca2+, and incubated for a further 10 min in collagenase Type 4 (1 mg ml−1, Sigma, UK). The digested tissue was then triturated using a wide bore Pasteur pipette in order to liberate single smooth muscle cells. Isolated cells were transferred to PSS containing 0.75 mm CaCl2, placed on cover slips for storage at 4 °C and were used within 6 h of isolation. All experiments were carried out at room temperature (21-24 °C).
Electrophysiological recording
Membrane currents were recorded with a List LM PCA amplifier using the whole-cell patch clamp technique. All voltage protocols were generated by the CED (Cambridge, UK) Voltage Clamp program and evoked currents were analysed using the corresponding CED analysis package, filtering at 3 kHz and sampling at a rate of 5 kHz. Further analysis and graphics were produced using Microcal Origin (Northampton, USA). Changes in junction potentials between the pipette and bath solutions were minimised by use of a KCl-agar bridge linking the main chamber to a side bath in which the reference electrode was located. The voltage-dependent characteristics of the hypotonicity-activated current were investigated by applying a voltage-ramp every 15 s. The ramp protocol consisted of stepping the voltage from the holding potential of −50 to −100 mV for 50 ms followed by continuously changing the voltage from −100 to +100 mV at a rate of 250 mV s−1 in isotonic PSS and hypotonic solutions. The leak-subtracted current voltage (I-V) relationship of the volume-sensitive chloride current was calculated by subtracting the control I-V curve in isotonic solution from that in hypotonic solution when the current had reached equilibrium.
Solutions
Normal PSS used for dissection contained (mm): NaCl 126, KCl 6, MgCl2 1.2, CaCl2 1.5, Hepes 10, glucose 11 and was adjusted to pH 7.2 with NaOH. To remove contaminating K+ currents experiments were performed using K+-free internal and external solutions. The normal K+-free extracellular solution was composed of (mm): NaCl 126, MgCl2 1.2, CaCl2 1.5, Hepes 10, glucose 11 and was adjusted to pH 7.2 with NaOH. Nicardipine (5 μm) was also included to inhibit voltage-dependent Ca2+ currents. In all experiments the K+-free pipette solution contained (mm): CsCl 126, MgCl2 1.2, Hepes 10, glucose 11, and the pH was adjusted to 7.2 with CsOH. In the present study the volume-sensitive chloride current was activated by substituting normal K+-free PSS, i.e. with 126 mm NaCl, with an external solution in which the NaCl concentration was reduced to 75 mm. This procedure evokes ICl(swell) which has no cationic contribution (Ellershaw et al. 2000) and is due to cell swelling and not a change in ionic strength (Greenwood & Large, 1998). To eliminate any contamination from Ca2+-activated Cl− currents 10 mm EGTA was added to the pipette solution in most experiments although in some experiments 1 mm EGTA was used for comparison. There was no difference in the results obtained with 1 and 10 mm EGTA. As noradrenaline evokes a non-selective cation current (Icat) in these cells (Helliwell & Large; 1996) we performed experiments to determine the influence of Icat in these experiments by including 1 mm CdCl2 in the bathing solution which totally blocks Icat (A. S. Aromolaran & W. A. Large, unpublished data). In other experiments we replaced external NaCl and pipette CsCl with N -methyl-d-glucamine (NMDG) Cl to block all non-selective cation conductances.
Chemicals
Calphostin-C, phorbol-12-myristate-13-acetate (PMA), phorbol-12,13-dibutyrate (PDBu), 8-Br-cAMP, U73122, U73443 and KT5720 were purchased from Calbiochem (La Jolla, CA, USA). Noradrenaline bitartrate, isoprenaline hydrochloride, prazosin hydrochloride, phenylephrine hydrochloride, chelerythrine chloride, cadmium chloride, N -methyl-d-glucamine (NMDG) and 4,4′-diisothiocyanato-stilbene-2,2′-disulfonic acid (DIDS) were purchased from Sigma (Poole, UK). Reagents were dissolved in dimethylsulphoxide which at the highest concentration used (0.1 %) had no effect on ICl(swell).
Statistics
All data are shown as the mean ±s.e.m. of n cells in at least three rabbits. Student's t test was used to compare mean values and statistical significance was set at P < 0.05.
RESULTS
Effect of noradrenaline on ICl(swell)
Previously it has been shown that application of hypotonic solution to rabbit portal vein myocytes produces cell swelling and the simultaneous activation of a volume-sensitive Cl− current, designated ICl(swell), which is sustained during the continued presence of the hypotonic solution (Greenwood & Large, 1998; Ellershaw et al. 2000). On application of 75 mm NaCl hypotonic solution, the maximum width of the cell increased from 12 ± 1 μm to 16 ± 2 μm (n = 16). First we investigated the effects of noradrenaline on evoked ICl(swell) in rabbit portal vein myocytes.
In the absence of pharmacological receptor antagonists, bath-applied noradrenaline (10 μm) produced a complex range of effects. In some cells (3 out of 8 cells) ICl(swell) was increased by noradrenaline (Fig. 1A) and this effect was not voltage dependent (Fig. 1B). The maximum potentiation by noradrenaline was 35–45 % and occurred within 5–10 min. In other cells (3 out of 8 cells) noradrenaline decreased ICl(swell) and interestingly in some cells this reduction was associated with oscillation of ICl(swell) (Fig. 1C). The maximum inhibition of ICl(swell) produced by 10 μm noradrenaline was about 50 % and was not voltage dependent (Fig. 1D). In two of eight cells 10 μm noradrenaline produced no net effect on membrane conductance. As it has been shown that both α- and β-adrenoceptors are present in the rabbit portal vein (Holman et al. 1968) it is possible that pathways elicited by activation of these receptors might modulate ICl(swell). Consequently further experiments were conducted in the presence of propranolol, an antagonist of β-adrenoceptors, or prazosin, an antagonist of α1-adrenoceptors.
Figure 1.
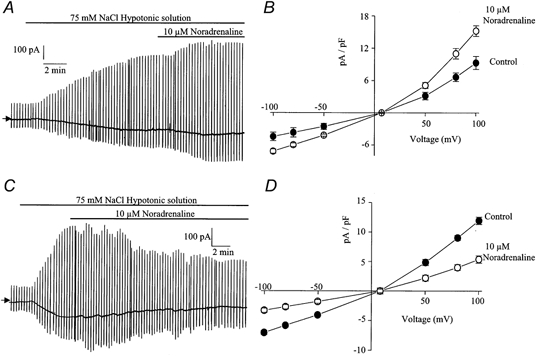
The effect of noradrenaline on ICl(swell)
A, representative trace illustrating the increase of ICl(swell) produced by 10 μm noradrenaline. In all traces vertical deflections represent the ramp protocol from a holding potential of −50 mV and in this and subsequent figures the arrow to the left of the trace indicates zero current level. B, leak-subtracted current-voltage relationship (see Methods) of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 10 μm noradrenaline (○). In this and subsequent figures mean current densities were calculated as pA pF−1. Each point is the mean ±s.e.m. of 5 cells. C, representative trace illustrating the inhibitory effect of 10 μm noradrenaline on ICl(swell) in another cell. Note the oscillation of ICl(swell) as it is inhibited. D, leak-subtracted current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 10 μm noradrenaline (○). Each point is the mean ±s.e.m. of 6 cells.
In the presence of 1 μm propranolol, application of noradrenaline to the bathing solution produced an increase in the amplitude of ICl(swell) which reached a plateau after approximately 5–10 min. An example of this effect is shown in Fig. 2A where it can be seen that the noradrenaline-induced increase was reversed on washout of the drug. The increase of ICl(swell) by noradrenaline was concentration dependent (Fig. 2B) and was reversible on removal from the bathing solution. The concentration producing 50 % of the maximum effect (EC50), measured from Fig. 2B, was approximately 5 μm. The potentiating effect of noradrenaline on ICl(swell) was not voltage dependent and the increase produced by 10 μm noradrenaline was 65 ± 9 % (n = 5) at −50 mV and 62 ± 9 % (n = 5) at +100 mV.
Figure 2.
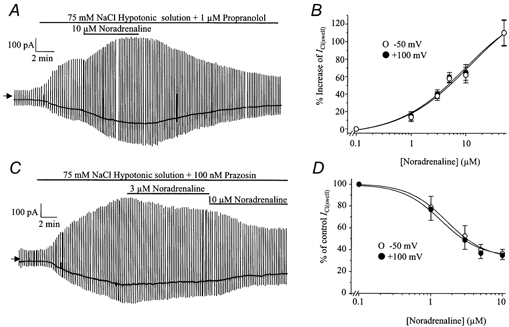
The effect of noradrenaline on ICl(swell) in the presence of propranolol or prazosin
A, representative trace illustrating the increase of ICl(swell) produced by 10 μm noradrenaline in the presence of 1 μm propranolol. B, concentration-effect curve of the noradrenaline-induced increase of ICl(swell) at −50 mV (○) and +100 mV(•). Each point is the mean ±s.e.m. of 5 cells. C, representative trace illustrating the inhibitory effect of 3 and 10 μm noradrenaline on ICl(swell) in the presence of prazosin (100 nm). D, concentration-effect curve of the noradrenaline-induced inhibition of ICl(swell) at −50 mV (○) and +100 mV (•). The vertical axis represents the percentage of the current before noradrenaline was added. Each point is the mean ±s.e.m. of 6 cells.
The potentiating effect of noradrenaline was not due to a change in cell size. Under hypotonic conditions maximum cell width was 15 ± 2 μm and following application of 10 μm noradrenaline, the maximum cell width was 15 ± 2 μm (n = 5). Similarly, in all further experiments agents which had an effect on the amplitude of ICl(swell) did not alter cell size.
In the presence of 100 nm prazosin, noradrenaline always inhibited ICl(swell) with a similar time course to the excitatory effect. An example of a typical trace is shown in Fig. 2C. This inhibitory effect was also concentration dependent (Fig. 2D) and the concentration producing 50 % of maximum inhibition (IC50) was approximately 1.5 μm. The inhibitory effect of noradrenaline was voltage-independent and the inhibition produced by 10 μm noradrenaline was 64 ± 4 % (n = 6) at −50 mV and 65 ± 4 % (n = 6) at +100 mV. The inhibition produced by noradrenaline was, at least partially, reversible on washout of this agent. The inhibitory effect of noradrenaline was not due to a change in cell size. Under hypotonic conditions maximum cell width was 16 ± 2 μm and following application of noradrenaline the maximum cell width was 16 ± 2 μm (n = 5).
It has been stipulated that activation of ICl(swell) requires internal ATP (reviewed by Strange et al. 1996; Nilius et al. 1997; Okada, 1997). The present experiments were carried out without ATP in the patch-pipette solution since it is apparent that rabbit portal vein myocytes generate sufficient endogenous ATP to sustain phosphorylation. Thus, for example, contractile agents such as noradrenaline and caffeine induce contraction of myocytes that have been dialysed with ATP-free pipette solution for periods of up to 60 min. However we carried out a few experiments on the effect of noradrenaline with 1 mm ATP in the pipette solution. The enhancement of ICl(swell) produced by 10 μm noradrenaline, when 1 mm ATP was present in the pipette, was 66 ± 10 % (n = 5) at −50 mV and 67 ± 10 % (n = 5) at +100 mV. The inhibitory effect of 10 μm noradrenaline on the amplitude of ICl(swell) in the presence of 1 mm ATP in the pipette, was 62 ± 6 % (n = 5) at −50 mV and 62 ± 6 % (n = 5) at +100 mV. These values are not significantly different from those obtained with ATP-free pipette solution.
Noradrenaline had no effect on the resting conductance recorded under isotonic conditions. In isotonic solutions the current amplitudes at −50 mV and +100 mV were 1.0 ± 0.07 and 5.7 ± 0.2 pA pF−1 (n = 5), respectively. When 10 μm noradrenaline was added to the bathing solution the current amplitudes were 1.1 ± 0.1 and 5.7 ± 0.4 pA pF−1 (n = 5) at −50 and +100 mV, respectively. Similarly, in all further experiments none of the drugs investigated had an effect on the amplitude of ICl(swell) under isotonic conditions.
However, under isotonic conditions application of 10 μm noradrenaline sometimes activated a small ‘noisy’ current (about 10–30 pA in amplitude at −50 mV) which appeared to be the non-selective cation current Icat previously described (e.g. Helliwell & Large, 1996; Byrne & Large, 1988). Noradrenaline did not appear to evoke Icat under hypotonic solutions. Nevertheless we carried out experiments in which Icat was blocked to determine whether the increase of ICl(swell) produced by noradrenaline was due to the concomitant activation of Icat. With 1 mm Cd2+ in the bathing solution, which totally blocks Icat (A. S. Aromolaran & W. A. Large, unpublished data), 10 μm noradrenaline increased ICl(swell) by 60 ± 12 and 59 ± 11 % at, respectively, −50 and +100 mV (n = 8). In other experiments where NMDG chloride was used to replace both external NaCl and internal CsCl 10 μm noradrenaline increased ICl(swell) by 57 ± 11 and 62 ± 8 % at, respectively, −50 and +100 mV (n = 5). In the latter conditions it is probable that no cation conductance could be recorded and the quantitative data on the effect of noradrenaline on ICl(swell) are similar to the values without Cd2+ or NMDG. Consequently the enhanced conductance produced by noradrenaline is due solely to an effect on ICl(swell) and not due to co-activation of a cation conductance.
It can be concluded that the biphasic effect of noradrenaline is due to activation of α1-adrenoceptors to increase ICl(swell) whereas the inhibitory effect of noradrenaline is mediated by β-adrenoceptor stimulation. It should be noted that in three populations of cells in the presence of propranolol, noradrenaline up to 50 μm did not increase ICl(swell). We have no explanation for these negative results but these cells were not used for our studies.
Effect of U73122 and IP3 on ICl(swell)
α1-Adrenoceptor stimulation is linked to the phosphoinositide cascade via activation of phosphatidylinositol phospholipase C (PLC). This leads to the production of the second messengers diacylglycerol (DAG), with subsequent activation of protein kinase C (PKC), and the Ca2+-releasing messenger inositol 1,4,5-trisphosphate (IP3). Therefore we investigated the effect of U73122, an inhibitor of PLC, on ICl(swell). Application of 1 μm U73122 to the bathing solution produced slow inhibition of ICl(swell) which reached its maximum effect after approximately 10 min and a typical record is shown in Fig. 3A. The effect of U73122 was voltage-independent (Fig. 3B) and the inhibition produced by 1 μm U73122 was 49 ± 3 % (n = 5) at −50 mV and 50 ± 4 % (n = 5) at +100 mV. In five cells application of 2 μm U73343, the inactive analogue of U73122, had no effect on ICl(swell). A typical trace is shown in Fig. 3C and it can be seen that subsequent addition of U73122 produced its normal inhibitory effect. This result suggests that during activation of ICl(swell) there is tonic PLC activity that enhances the amplitude of this current.
Figure 3.
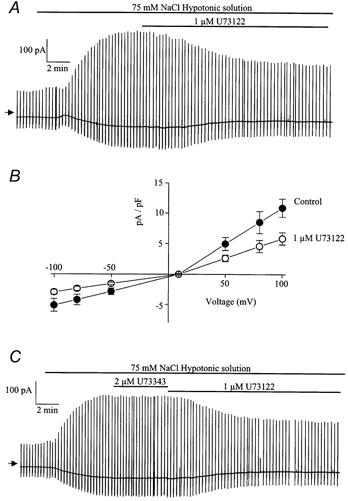
Effect of U73122 on ICl(swell)
A, representative trace illustrating inhibitory effect of 1 μm U73122 on ICl(swell). B, leak-subtracted current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 1 μm U73122 (○) shown in A. Each point is the mean ±s.e.m. of 5 cells C, representative trace illustrating the effects of 2 μm U73343 (inactive analogue) and 1 μm U73122 on ICl(swell).
In normal isotonic conditions inclusion of 100 μm IP3 in the pipette solution had no effect on the resting current. Furthermore, under hypotonic conditions the normalised amplitudes of ICl(swell) at −50 and +100 mV were, respectively, 3 ± 0.3 pA pF−1 (n = 6) and 12 ± 0.7 pA pF−1 (n = 6) in control conditions. Inclusion of 100 μm IP3 in the pipette solution had no effect on the development of evoked ICl(swell) and the mean current amplitudes at −50 and +100 mV were, respectively, 3.7 ± 0.8 pA pF−1 (n = 6) and 13 ± 0.8 pA pF−1 (n = 6). These data suggest that the IP3 arm of the signalling cascade is not responsible for the noradrenaline-dependent increase of ICl(swell).
Effect of the phorbol esters PMA and PDBu on ICl(swell)
In the next series of experiments we investigated the possible role of PKC in the stimulation of ICl(swell) by studying the effects of phorbol esters, which are known to directly activate PKC, on ICl(swell). The addition of the phorbol ester PMA (100 nm-1 μm) to the bathing solution increased the amplitude of ICl(swell) in all cells and a representative trace showing the effect of 1 μm PMA is shown in Fig. 4A. This effect was not voltage dependent (Fig. 4B) and the maximum enhancement produced by 100 nm PMA was 41 ± 6 % (n = 5) at −50 mV and 46 ± 7 % (n = 5) at +100 mV. The effect of PMA was concentration dependent and 1 μm increased ICl(swell) by 55 ± 4 % (n = 5) at −50 mV and by 67 ± 7 % (n = 5) at +100 mV. Application of another phorbol ester, PDBu (100-500 nm), to the bathing solution also produced an increase in the amplitude of the current (Fig. 4C) with a similar time course to PMA and showed no voltage dependence (Fig. 4D). The enhancement of ICl(swell) by 500 nm PDBu was 108 ± 15 % (n = 6) at −50 mV and 94 ± 7 % at +100 mV (n = 6). Interestingly, application of PMA or PDBu to the bathing solution produced a small transient reduction in the amplitude of ICl(swell) prior to the sustained enhancement of the current. This was a consistent observation but was not investigated further in this study.
Figure 4.
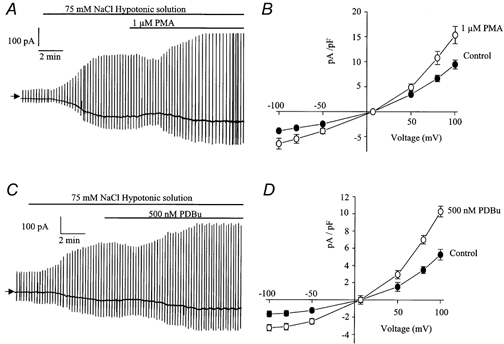
Effect of phorbol esters on ICl(swell)
A, representative trace illustrating the excitatory effect of 1 μm PMA on ICl(swell). B, leak-subtracted mean current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 1 μm PMA (○). Each point is the mean ±s.e.m. of 5 cells. C, representative trace illustrating the excitatory effect of 500 nm PDBu on ICl(swell). D, leak-subtracted mean current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 500 nm PDBu (○). Each point is the mean ±s.e.m. of 6 cells.
In the presence of 1 mm Cd2+, which abolishes Icat, application of 500 nm PDBu to the bathing solution increased the amplitude of ICl(swell) by 80 ± 9 and 82 ± 12 % (n = 4) at, respectively, −50 and +100 mV.
Inhibition of ICl(swell) by chelerythrine and calphostin-C
Since phorbol esters augmented ICl(swell) we subsequently studied the effects of the PKC inhibitors chelerythrine and calphostin-C on ICl(swell). Application of chelerythrine (1-10 μm) to the bathing solution produced slow inhibition of ICl(swell) which took approximately 15 min to reach its maximum effect. A typical effect of 10 μm chelerythrine is shown in Fig 5A. The inhibitory effect of chelerythrine was voltage-independent (Fig. 5B) and the maximum inhibition of ICl(swell) by 10 μm chelerythrine was 63 ± 10 % (n = 5) at −50 mV and 65 ± 7 % (n = 5) at +100 mV (Fig. 5C). From Fig. 5C the estimated IC50 of chelerythrine on ICl(swell) was approximately 2 μm. Similarly, calphosin-C (1-10 μm) also inhibited ICl(swell) with a similar time course to chelerythrine and a representative trace is shown in Fig. 6A. The inhibitory effect of calphostin-C was also voltage-independent (Fig. 6B) and the maximal inhibition of ICl(swell) produced by 10 μm calphostin-C was 58 ± 7 (n = 5) at −50 mV and 58 ± 6 % (n = 5) at +100 mV. The IC50 for the inhibitory effect of calphostin-C on ICl(swell) was approximately 1 μm (Fig. 6C).
Figure 5.
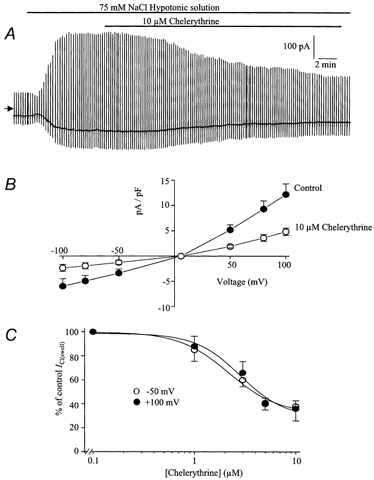
Effect of chelerythrine on ICl(swell)
A, typical cell showing the inhibition of ICl(swell) by 10 μm chelerythrine. B, leak-subtracted mean current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 10 μm chelerythrine (○). Each point is the mean ±s.e.m. of 5 cells. C, concentration-effect curve of the chelerythrine-induced inhibition of ICl(swell) at −50 mV (○) and +100 mV (•). Each point is the mean ±s.e.m. of 5 cells.
Figure 6.
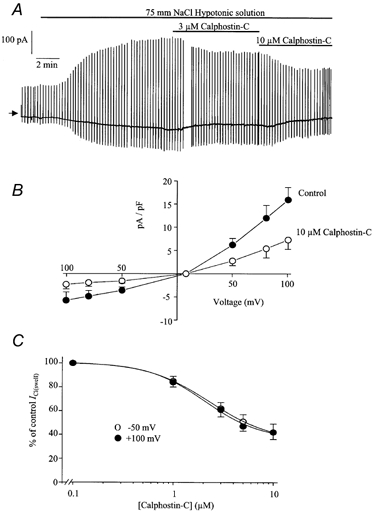
Effect of calphostin-C on ICl(swell)
A, typical cell showing the inhibition of ICl(swell) by 3 and 10 μm calphostin-C. B, leak-subtracted mean current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 10 μm calphostin-C (○). Each point is the mean ±s.e.m of 5 cells. C, concentration-effect curve of the calphostin-C-induced inhibition of ICl(swell) at −50 mV (○) and +100 mV (•). Each point is the mean ±s.e.m. of 5 cells.
Both the effects of phorbol esters and PKC inhibitors had long time courses which prevented a study of their reversibility as it was usually difficult to keep the whole-cell configuration for more than 30–45 min.
Effect of isoprenaline on ICl(swell)
The above data with pharmacological receptor antagonists imply that the noradrenaline-induced inhibition of ICl(swell) was a result of β-adrenoceptor activation. To confirm this possibility we investigated the effect of isoprenaline, a selective agonist of β-adrenoceptors, on ICl(swell). Application of isoprenaline (1-10 μm) to the bathing solution (no prazosin present) produced slow inhibition of ICl(swell) which reached a plateau within approximately 10 min (Fig. 7A) and was not voltage dependent (Fig. 7B). The maximum inhibition produced by 10 μm isoprenaline was 55 ± 6 % at −50 mV and 53 ± 5 % at +100 mV (n = 6). These results confirm that β-adrenoceptor stimulation inhibits ICl(swell).
Figure 7.
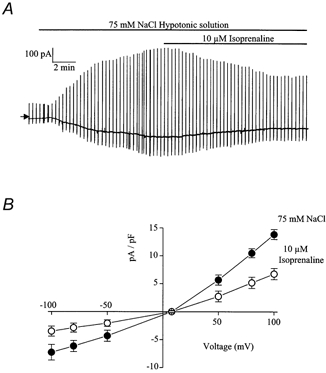
The effect of isoprenaline on ICl(swell)
A, representative trace illustrating the inhibitory effect of 10 μm isoprenaline on ICl(swell). B, leak-subtracted current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 10 μm isoprenaline (○). Each point is the mean ±s.e.m. of 6 cells.
Effect of 8-Br-cAMP and KT5720 on ICl(swell)
Many of the effects of β-adrenoceptor stimulation in smooth muscle are due to the activation of adenylate cyclase and subsequent increase in intracellular concentration of cAMP (Lincoln & Fisher-Simpson, 1984). Therefore it is possible that the inhibition of ICl(swell) by noradrenaline in the presence of prazosin and isoprenaline was cAMP dependent. We therefore investigated the effect of the membrane permeable synthetic analogue of cAMP, 8-Br-cAMP, on evoked ICl(swell). Application of 100 μm 8-Br-cAMP to the bathing solution produced slow inhibition of ICl(swell) and a representative trace of the effect of 100 μm 8-Br-cAMP is shown in Fig. 8A. The inhibitory effect of 8-Br-cAMP showed no voltage dependence (Fig. 8B) and the inhibition produced by 100 μm 8-Br-cAMP was 62 ± 6 % (n = 6) at −50 mV and 61 ± 5 % (n = 6) at +100 mV. Many of the effects of cAMP are mediated by a cAMP-dependent protein kinase and therefore we studied the effect of the selective cAMP-dependent protein kinase inhibitor, KT5720, on evoked ICl(swell). Application of 1 μm KT5720 to the bathing solution produced a marked increase in evoked ICl(swell) reaching a plateau approximately 7 min after application. An example of a typical trace is shown in Fig 8C. The effect of 1 μm KT5720 was not voltage dependent (Fig. 8D) and increased ICl(swell) by 89 ± 10 % (n = 5) at −50 mV and by 84 ± 8 % (n = 5) at +100 mV.
Figure 8.
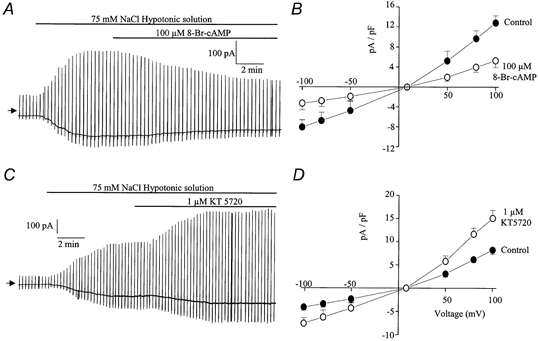
Effect of 8-Br-cAMP and KT5720 on ICl(swell)
A, representative trace illustrating the inhibitory effect of 100 μm 8-Br-cAMP on ICl(swell). B, leak-subtracted current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 100 μm 8- Br-cAMP (○). Each point is the mean ±s.e.m. of 6 cells. C, representative trace illustrating the increase of ICl(swell) induced by 1 μm KT5720. D, leak-subtracted current-voltage relationship of ICl(swell) evoked by 75 mm NaCl (•) and in the same hypotonic solution containing 1 μm KT5720 (○). Each point is the mean ±s.e.m. of 5 cells.
Role of PKC and PKA on the effects of noradrenaline
The above data suggest that the excitatory effects of noradrenaline on ICl(swell) may be mediated via PKC. Therefore we investigated the effects of chelerythrine against the action of noradrenaline in the presence of propranolol. Due to the slow time course of these agents, chelerythrine was applied to the hypotonic bathing solution during the development of ICl(swell) and noradrenaline was applied immediately the current reached the peak value. A concentration of 50 μm noradrenaline was used in order to produce a near maximal increase in the amplitude of ICl(swell). In this series of experiments application of 50 μm noradrenaline to the bathing solution increased the amplitude of ICl(swell) by 110 ± 15 % at −50 mV and by 111 ± 14 % (n = 7) at +100 mV. However, when cells were pre-treated with 10 μm chelerythrine, the increase of ICl(swell) to 50 μm noradrenaline was 9 ± 3 % at −50 mV and 10 ± 3 % at +100 mV (n = 7). Therefore chelerythrine markedly inhibited the potentiating effect of noradrenaline on ICl(swell).
We also investigated the effects of KT5720 against the inhibitory action of noradrenaline in the presence of prazosin. Application of 1 μm KT5720 to the bathing solution produced an increase in the amplitude of ICl(swell) of 77 ± 8 % at −50 mV and 75 ± 9 % at +100 mV (n = 5). Subsequent application of 10 μm noradrenaline in the presence of KT5720 produced little inhibition of ICl(swell). The inhibition produced by 10 μm noradrenaline in the presence of 1 μm KT5720 was 5 ± 2 % at −50 mV and 7 ± 2 % at +100 mV (n = 5). Therefore the inhibitory action of noradrenaline was inhibited by KT5720.
DISCUSSION
The present study shows that noradrenaline modulates ICl(swell) in rabbit portal vein smooth muscle cells by two distinct pathways. In the absence of pharmacological antagonists noradrenaline increased the amplitude of ICl(swell) in some cells and decreased the current in others. In the presence of the β-adrenoceptor antagonist propranolol, noradrenaline enhanced ICl(swell) while in the presence of the selective α1-adrenoceptor antagonist prazosin, noradrenaline always reduced ICl(swell). It is important to emphasise that the increase in current produced by noradrenaline was not due to co-activation of Icat. In the presence of 1 mm Cd2+, which totally blocks Icat, the increase of ICl(swell) was similar to that observed in the absence of Cd2+. Therefore the increase in current is solely due to an increase in ICl(swell). These results show that the increase and decrease of ICl(swell) caused by noradrenaline are mediated by, respectively, α1- and β-adrenoceptors in these vascular myocytes. The present study does not relate the changes in amplitude of ICl(swell) to single channel conductance, open channel probability and/or channel number which is the subject of future work.
Modulation of ICl(swell) by noradrenaline
The modulation of ICl(swell) by noradrenaline was dependent on the class of adrenoceptor stimulated. Stimulation of α1-adrenoceptor augmented ICl(swell) that was blocked by the PKC inhibitors chelerythrine and calphostin-C and suggests that the noradrenaline-dependent enhancement of ICl(swell) is mediated by PKC. This hypothesis is supported by the observation that in the absence of noradrenaline, inhibitors of PKC decreased ICl(swell) and activators of PKC increased the conductance. In comparison inclusion of IP3 in the pipette did not affect the amplitude of ICl(swell).
The inhibitory effect of noradrenaline was produced by β-adrenoceptor stimulation and was blocked by the PKA inhibitor KT5720 which suggests that this inhibitory action is mediated by a PKA-dependent pathway. In addition, in the absence of noradrenaline the cell permeable analogue of cAMP, 8-bromo cAMP, decreased the amplitude of ICl(swell) whereas the PKA inhibitor KT5720 increased ICl(swell). Previously we have shown that cGMP-dependent protein kinase increases the amplitude of ICl(swell) (Ellershaw et al. 2000). The present study shows that PKC also increases this conductance whereas PKA decreases the amplitude of ICl(swell). Thus in rabbit portal vein smooth muscle cells it appears that ICl(swell) is the target of complex kinase regulation. In addition, the ability of PLC, PKC and PKA inhibitors to modulate ICl(swell) in the absence of noradrenaline suggests that hypotonic swelling of rabbit portal vein myocytes increases the activity of these kinases. Moreover, in the absence of noradrenaline, the PLC inhibitor U73122 also decreased the amplitude of ICl(swell) which suggests that there is tonic PLC activity during activation of ICl(swell).
Comparison of regulation of ICl(swell) in other tisssues
There have been no previous reports on the effect of adrenoceptor stimulation on ICl(swell) in smooth muscle. However there have been numerous reports regarding phosphorylation reactions and ICl(swell) in many different cell types and there is much conflicting data on this subject (e.g. see reviews by Okada, 1997; Strange et al. 1996). For example, stimulation of PKC by phorbol esters decreases ICl(swell) in rabbit and guinea-pig cardiac cells (Duan et al. 1995, 1999) but increases ICl(swell) in canine atrial cells (Du & Sorota, 1999). More pertinently to the present work in smooth muscle, it was shown in canine colonic smooth muscle that phorbol esters reduced ICl(swell). Moreover the PKC inhibitor chelerythrine activated the current in isotonic conditions (Dick et al. 1998). Clearly these latter results are in marked contrast to the present work in vascular smooth muscle where chelerythrine had no effect in isotonic solutions and decreased ICl(swell) which illustrates the complex modulation of this conductance in different types of smooth muscle and other cell types. Overall the data on PKC modulation of native ICl(swell) appears conflicting and dependent on the cell type studied.
Similarly there are conflicting results regarding PKA modulation of ICl(swell) in other classes of cell. In agreement with the present work it was concluded that PKA-induced phosphorylation reduced ICl(swell) in chick (Hall et al. 1995) and in mammalian cardiac cells (Du & Sorota, 1997, Nagasaki et al. 2000). In contrast, increasing intracellular levels of cAMP potentiated the amplitude of ICl(swell) by a PKA-independent pathway in canine atrial cells and in human epithelial cells (Shimizu et al. 2000). At present these contradictory data cannot be explained but may be related to the different physiological functions of ICl(swell) in these diverse cell types.
Conclusions
In freshly dispersed rabbit portal vein myocytes noradrenaline stimulates α-adrenoceptors to increase ICl(swell) via a PKC-dependent mechanism whereas β-adrenoceptor activation reduces the amplitude of ICl(swell) by a PKA-dependent mechanism. The fact that different kinases modulate ICl(swell) in different ways shows that phosphorylation is a crucial determinant of ICl(swell) activity. However, comparison with other smooth muscle preparations and other cell types illustrates that there is complex regulation of this conductance by receptor transduction pathways.
Acknowledgments
This work was funded by The British Heart Foundation and The Wellcome Trust.
REFERENCES
- Byrne NG, Large WA. Membrane ionic mechanisms activated by noradrenaline in cells isolated from the rabbit portal vein. Journal of Physiology. 1988;404:557–573. doi: 10.1113/jphysiol.1988.sp017306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick GM, Bradley KK, Horowitz B, Hume JR, Sanders KM. Functional and molecular identification of a novel chloride conductance in canine colonic smooth muscle. American Journal of Physiology. 1998;275:C940–950. doi: 10.1152/ajpcell.1998.275.4.C940. [DOI] [PubMed] [Google Scholar]
- Doughty JM, Langton PD. Measurement of chloride flux associated with the myogenic response in rat cerebral arteries. Journal of Physiology. 2001;534:753–761. doi: 10.1111/j.1469-7793.2001.t01-1-00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X-Y, Sorota S. Modulation of dog atrial swelling-induced chloride current by cAMP: protein kinase A-dependent and -independent pathways. Journal of Physiology. 1997;500:111–122. doi: 10.1113/jphysiol.1997.sp022003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X-Y, Sorota S. Protein kinase C stimulates swelling-induced chloride current in canine atrial cells. Pflügers Archiv. 1999;437:227–234. doi: 10.1007/s004240050773. [DOI] [PubMed] [Google Scholar]
- Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. Journal of General Physiology. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Fermini B, Nattel S. Alpha-adrenergic control of volume-regulated Cl− currents in rabbit atrial myocytes. Characterization of a novel ionic regulatory mechanism. Circulation Research. 1995;77:379–393. doi: 10.1161/01.res.77.2.379. [DOI] [PubMed] [Google Scholar]
- Ellershaw DC, Greenwood IA, Large WA. Dual modulation of swelling-activated chloride current by NO and NO donors in rabbit portal vein myocytes. Journal of Physiology. 2000;528:15–24. doi: 10.1111/j.1469-7793.2000.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JE, Greenwood IA, Large WA. Tonic regulation of vascular tone by nitric oxide and chloride ions in rat isolated small coronary arteries. American Journal of Physiology — Heart and Circulatory Physiology. 2000;279:H2604–2611. doi: 10.1152/ajpheart.2000.279.6.H2604. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Large WA. Properties of a Cl− current activated by cell swelling in rabbit portal vein vascular smooth muscle cells. American Journal of Physiology. 1998;275:H1524–1532. doi: 10.1152/ajpheart.1998.275.5.H1524. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Large WA. Properties and role of chloride channels in smooth muscle. In: Kozlowski R, editor. Chloride Channels. Oxford: Isis Medical Media; 1999. [Google Scholar]
- Hall SK, Zhang J, Lieberman M. Cyclic AMP prevents activation of a swelling-induced chloride-sensitive conductance in chick heart cells. Journal of Physiology. 1995;488:359–369. doi: 10.1113/jphysiol.1995.sp020972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell RM, Large WA. Dual effect of external Ca2+ on noradrenaline-activated cation current in rabbit portal vein smooth muscle cells. Journal of Physiology. 1996;492:75–88. doi: 10.1113/jphysiol.1996.sp021290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman ME, Kasby CB, Suthers MD, Wilson JAF. Some properties of smooth muscle of the rabbit portal vein. Journal of Physiology. 1968;196:111–132. doi: 10.1113/jphysiol.1968.sp008498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln TM, Fisher-Simpson V. A comparison of the effects of forskolin and nitroprusside on cyclic nucleotides and relaxation in the rat aorta. European Journal of Pharmacology. 1984;101:17–27. doi: 10.1016/0014-2999(84)90026-8. [DOI] [PubMed] [Google Scholar]
- Nagasaki M, Ye L, Duan D, HorowitZ B & Hume JR. Intracellular cyclic AMP inhibits native and recombinant volume-regulated chloride channels from mammalian heart. Journal of Physiology. 2000;523:705–717. doi: 10.1111/j.1469-7793.2000.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT. Bayliss, myogenic tone and volume-regulated chloride channels in arterial smooth muscle. Journal of Physiology. 1998;507:629. doi: 10.1111/j.1469-7793.1998.629bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. Journal of Physiology. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Buyse G, Manopoulos V, Droogmans G. Properties of volume-regulated anion channels in mammalian cells. Progress in Biophysics and Molecular Biology. 1997;68:69–119. doi: 10.1016/s0079-6107(97)00021-7. [DOI] [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward rectifier Cl− channel: fresh start to the molecular identity and volume sensor. American Journal of Physiology. 1997;273:C755–789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Morishima S, Okada Y. Ca2+-sensing receptor-mediated regulation of volume-sensitive Cl− channels in human epithelial cells. Journal of Physiology. 2000;528:457–472. doi: 10.1111/j.1469-7793.2000.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. American Journal of Physiology. 1996;270:C711–730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Tolloczko B, Tao FC, Zacour ME, Martin JG. Tyrosine kinase-dependent calcium signaling in airway smooth muscle cells. American Journal of Physiology — Lung Cellular and Molecular Physiology. 2000;278:L1138–1145. doi: 10.1152/ajplung.2000.278.6.L1138. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. Journal of Physiology. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]