

Abstract
The role of endothelium-derived hyperpolarizing factor (EDHF) in the regulation of blood flow in vivo was examined in the mesenteric and hindlimb circulations of anaesthetized rats. Basal mesenteric conductance decreased from 57 ± 5 to 20 ± 6 μl min−1 mmHg−1 when nitric oxide (NO) production was inhibited, and combined blockade of intermediate- and small-conductance Ca2+-activated K+ (KCa) channels with charybdotoxin (ChTx) and apamin had no further effect. Basal hindlimb conductance was reduced from 39 ± 3 to 22 ± 2 μl min−1 mmHg−1 by NO synthesis inhibition, with no effect of the KCa channel blockers. Endothelial stimulation with acetylcholine (ACh) infusion directly into the mesenteric bed increased conductance by 20 ± 2 μl min−1 mmHg−1. Blockade of NO synthesis decreased this conductance to 15 ± 1 μl min−1 mmHg−1, leaving the response attributable to EDHF. This was reduced to 2 ± 1 μl min−1 mmHg−1 by ChTx plus apamin but not by iberiotoxin, which selectively blocks large-conductance KCa channels. Similar results were obtained when bradykinin (BK) was used to stimulate the endothelium. Nitroprusside, which directly relaxes smooth muscle, evoked an increase in conductance that was resistant to all blockers tested. ACh-induced increases in hindlimb conductance were reduced from 19 ± 1 to 12 ± 1 μl min−1 mmHg−1 by NO synthesis inhibition and further reduced to 2 ± 2 μl min−1 mmHg−1 by ChTx plus apamin. In contrast to NO, ChTx- and apamin-sensitive EDHF appears to contribute little to basal conductance in rat mesenteric and hindlimb beds. However, EDHF accounts for a significant component of the conductance increase during endothelial stimulation by ACh and BK. In these beds, intermediate- and small-conductance KCa channels underpin EDHF-mediated vasodilatation.
When NO and prostaglandin synthesis are blocked, stimulation of the vascular endothelium is still capable of eliciting vasodilatation in various vascular preparations. A hallmark of this response is the occurrence in the smooth muscle of membrane hyperpolarization, spawning the term endothelium-derived hyperpolarizing factor (EDHF) to describe the event (Taylor & Weston, 1988), since the nature of the causative agent was, and remains, controversial. Evidence is emerging to suggest that EDHF cannot be attributed to one ‘factor’ alone but may be a different entity depending on the vascular bed. In some vascular beds EDHF has been identified as products of cytochrome P-450-dependent arachidonate metabolism, the epoxyeicosatrienoic acids (EETs) (Hecker et al. 1994; Campbell et al. 1996; Fleming, 2001), mediated by the opening of large-conductance KCa channels (Campbell et al. 1996; Zou et al. 1996; Baron et al. 1997; Eckman et al. 1998; Fleming, 2001) as it is blocked by iberiotoxin (IbTx). In many vascular beds EDHF is not blocked by IbTx, but is abolished by a cocktail of toxins that blocks intermediate- (charybdotoxin, ChTx) and small-conductance (apamin) KCa channels on the endothelial cells (Zygmunt & Hogestatt, 1996; Plane et al. 1997; Vanheel & Van de Voorde, 1997; Eckman et al. 1998; Edwards et al. 1998; Coleman et al. 2001a). It has been suggested that the K+ exiting the endothelial cells through these channels acts to open inwardly rectifying K+ channels and to activate Na+-K+-ATPase pumps in the smooth muscle cell membrane, giving rise to EDHF-attributed hyperpolarization and relaxation (Edwards et al. 1998). An alternative view suggests that the ChTx- plus apamin-sensitive hyperpolarization arising in the endothelial cells spreads to the underlying smooth muscle via myoendothelial gap junctions to give rise to EDHF-attributed responses (Edwards et al. 1998; Yamamoto et al. 1998; Sandow & Hill, 2000; Coleman et al. 2001a, b). Recent evidence using peptide gap junction inhibitors supports this possibility in intact rats (De Vriese et al. 2002).
It is clear that NO plays an important role in determining basal tone in vivo (Gardiner et al. 1989; Vallance et al. 1989), but the relative contribution of NO to agonist-induced vasodilatation is heterogeneous, depending on the vascular bed (Gardiner et al. 1991) and on the calibre of the vessel (Widmann et al. 1998; Nishikawa et al. 1999), confirming observations in vitro. The role of EDHF in vascular physiology has been established mainly from studies of isolated vessels, with few studies on its role in vivo. The contribution of EDHF to ACh-induced dilatation of hamster skeletal muscle arterioles about equals that of NO (De Wit et al. 1999). In dog coronary microvessels, either EDHF or NO alone are capable of eliciting maximal dilatation in response to ACh in vivo (Widmann et al. 1998; Nishikawa et al. 1999). These EDHF-attributed responses, and those occurring in mouse (Hungerford et al. 2000) and hamster (De Wit et al. 1999) skeletal muscle arterioles, are mediated by large-conductance KCa channels, suggesting that EDHF may be a product of the cytochrome P-450 enzyme in these vascular beds.
In mesenteric arteries of rat, amongst the most thoroughly studied vessels in vitro, EDHF contributes prominently to endothelium-dependent vasodilatation and, on its own, can achieve complete relaxation of an agonist-induced contraction (Nagao et al. 1992; Zygmunt et al. 1995; Plane et al. 1997; Edwards et al. 1998; Doughty et al. 1999; Wigg et al. 2001). This EDHF cannot be attributed to products of the cytochrome P-450 enzyme, rather, its actions are mediated by intermediate- and small-conductance KCa channels (Zygmunt et al. 1995; Plane et al. 1997; Doughty et al. 1999). The aim of the present work was to investigate the role of this particular EDHF in resting and induced vascular tone in vivo, using the mesenteric bed, and to determine the nature of the KCa channels mediating its actions in vivo. This bed accounts for some 20 % of cardiac output, contributing significantly to the overall peripheral resistance. The endothelial regulation of hindlimb blood flow was also investigated because unlike the mesenteric, NO, rather than EDHF, appears dominant in vitro (Zygmunt et al. 1995; Wigg et al. 2001).
Methods
The preparation
The procedures described here were approved by the Animal Ethics Committees of Monash University (PHY/2000/47). Male Wistar rats (350-500 g) were used. Most of the rats were anaesthetized with thiobutabarbital (100 mg kg−1i.p., Inactin, Research Biochemicals, Inc., MA, USA); however, urethane (1 g kg−1i.p.) was the anaesthetic used for 10 animals. The rats were placed on a thermostatically controlled pad and fitted with a rectal thermometer. The trachea was cannulated. Arterial pressure was recorded via a catheter, filled with heparinized (12 IU ml−1) saline, in the carotid artery. Saline containing bovine serum albumin (2 %) was continuously infused at 3 ml h−1 via the jugular vein.
Recording blood flow
Mesenteric blood flow (BF) was measured via a perivascular transit-time ultrasound flow probe (0.7 mm, Transonic Systems, Inc., NY, USA) around the main mesenteric artery, ≈10 mm from its emergence from the abdominal aorta. To facilitate stimulation of the endothelium, a branch of the mesenteric artery emerging just distal to the flow probe was located, ligated distally and a polyethylene cannula (outside diameter, ≈250 μm) was inserted proximal to the ligature with the tip facing the main mesenteric artery. Saline solution was continuously delivered through this cannula at 50 μl min−1. In a different group of animals, hindlimb BF was recorded by placing the flow probe around the abdominal aorta, about 10 mm proximal to the bifurcation. The local infusion site in this instance was the rectal artery which branches off the aorta immediately proximal to the bifurcation. The testicular vessels were ligated and the reproductive organs removed. Basal mean arterial pressure (MAP), heart rate (HR) and BF reached steady values within 10–15 min of completing these procedures.
Stimulation of the endothelium
The endothelium was stimulated by bolus administration (Nishikawa et al. 1999) of ACh or bradykinin (BK) via the local mesenteric or rectal cannula. Similarly, bolus application of sodium nitroprusside (SNP) was used to directly relax the smooth muscle, and 1-ethyl-2-benzimidazolinone (1-EBIO) was used to open intermediate-conductance KCa channels on the endothelial cells. The stock solutions of these agents were adjusted such that the volume injected was constant at 100 μl, which was injected over 10 s followed by prompt resumption of saline infusion at 50 μl min−1. Care was taken such that the dose of the agent infused was kept below that which changed MAP or HR (see Fig. 2B), suggesting no or minimal overflow into the general circulation at the concentrations used.
Figure 2. Mean arterial pressure and blood flow in the mesenteric bed.
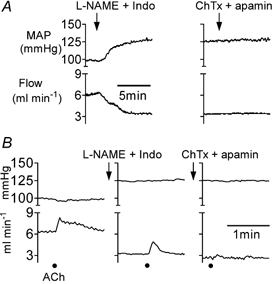
A, basal mean arterial pressure (MAP, upper traces) and blood flow (lower traces) in the mesenteric bed following administration of l-NAME plus indomethacin (Indo) and then charybdotoxin (ChTx) plus apamin. B, MAP (upper traces) and blood flow (lower traces) in the mesenteric bed in response to bolus application of acetylcholine (ACh, 3 nmol kg−1) in the control period, and following administration of l-NAME plus indomethacin, and then ChTx plus apamin. Filled circles show addition of ACh.
Blockers
NO production was blocked by 10 mg kg−1 bolus injection of Nω-nitro-l-arginine methylester (l-NAME) followed by continuous infusion at 3 mg kg−1 h−1, both via the jugular vein (Gardiner et al. 1991). In the first five animals, 30 min of l-NAME infusion was followed by endothelial testing before inhibition of prostaglandin production by infusion of indomethacin at 10 mg kg−1 h−1 for 30 min. While l-NAME increased basal MAP and decreased basal mesenteric BF (see Results), there was no additional effect of indomethacin on these parameters. Infusion of indomethacin alone for 30 min had no effect on MAP or basal BF. Hence, a bolus injection of l-NAME, and infusion of l-NAME and indomethacin were administered together for 30 min in all subsequent experiments. To test that the dose of l-NAME was effective in blocking NO production, the bolus dose was increased to 50 mg kg−1 and this had no additional effect on the remaining increase in mesenteric conductance.
The KCa channel blockers ChTx, apamin and IbTx were administered by close arterial infusion into the mesenteric or hindlimb beds at 50 μl min−1. Taking into account the prevailing flow rate in the individual animals, the stock concentrations of toxins used were calculated to achieve concentrations at the endothelium equivalent to those shown to be effective in vitro (ChTx, 3 × 10−8 mol l−1; apamin, 2.5 × 10−7 mol l−1; IbTx, 5 × 10−8 mol l−1) (Zygmunt & Hogestatt, 1996; Doughty et al. 1999; Coleman et al. 2001a). These blockers were applied for 10 min before re-testing BF responses. In experiments on four rats the endothelium was damaged or removed by including the detergent 3-[(3-cholamidopropyl)dimethylammonio]-1-propane sulfonate (CHAPS, 5 %) (Woodman et al. 2000) in the saline of the close infusion line for 3–4 min. Infusion of CHAPS was stopped and BF responses were re-tested every 15 min. Initially, the increases in BF evoked by both ACh and SNP were reduced by CHAPS. However, the response to SNP was fully restored within 1 h while the response to ACh had not returned after 2 h.
All of the drugs used were obtained from Sigma (St Louis, MO, USA), except for ChTx, IbTx, apamin and BK, which were synthesized by and purchased from Auspep (Melbourne, Australia).
Data analysis
BF, MAP and HR were recorded digitally at 0.5 Hz (Evans et al. 2000) and stored for later analysis using the software packages Origin (Microcal Software Inc., MA, USA) and Prism (GraphPad Software Inc., CA, USA). Instat (GraphPad) was used to test for statistical significance of treatments using two-way ANOVA followed by Student's t test. Paired t tests were used to assess the effects of blockers and toxins, since the measurements were made on the same animals. The data were tested for normality (Kolmogorov, Smirnov test) and for equivalence of standard errors (Bartlett's test) using Instat. Values from n animals are given as mean ± s.e.m. and P values of < 0.05 were accepted as statistically significant.
Results
Effect of blockade of NO and EDHF on MAP and basal mesenteric bed conductance
The initial values of MAP and HR in 30 rats were 117 ± 3 mmHg and 429 ± 20 beats min−1, respectively. Basal mesenteric BF was 6.7 ± 0.5 ml min−1 (n = 22) and, taking into account MAP, the conductance of the mesenteric bed was 57 ± 5 μl min−1 mmHg−1 (Fig. 1). Administration of l-NAME plus indomethacin resulted in a prompt decrease in mesenteric BF (Fig. 2A) and this was closely followed by an increase in MAP which reached a peak of 148 ± 4 mmHg (n = 22) in about 10 min. Thereafter MAP slowly declined and attained a stable value of 132 ± 3 mmHg after 20–30 min (Table 1). It has been demonstrated previously that the ion channels that mediate EDHF in isolated rat mesenteric artery are blocked by ChTx plus apamin (Plane et al. 1997; Edwards et al. 1998, 1999; Doughty et al. 1999). In this study we found that ChTx plus apamin had no further effect on MAP, HR or on mesenteric BF or conductance (Fig. 1 (left-hand bars), Fig. 2A and Table 1).
Figure 1. Basal conductance in the mesenteric bed.
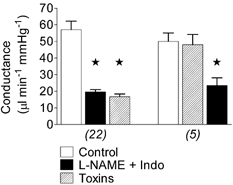
Basal conductance in the control period and following administration of l-NAME plus indomethacin (Indo), and then the toxins (charybdotoxin plus apamin) (left-hand bars; n = 22). In a second series of experiments, the order of blocker application was reversed, with toxins added first followed by l-NAME plus indomethacin (right-hand bars; n = 5). * Significantly different from control.
Table 1.
Basal values of mean arterial pressure, heart rate, blood flow and conductance in the mesenteric and femoral beds of anaesthetized rats
| MAP(mmHg) | HR(beats min−1) | Mes BF(ml min−1) | MesC(μl min−1 mmHg−1) | Fem BF(ml min−1) | FemC (μl min−1 mmHg−1) | |
|---|---|---|---|---|---|---|
| Control | 117 ± 3(22) | 429 ± 19(22) | 6.7 ± 0.5(22) | 57 ± 5(22) | 4.8 ± 0.2(5) | 39 ± 3(5) |
| +N+I | 132 ± 3*(22) | 382 ± 11*(22) | 2.6 ± 0.2*(20) | 20 ± 6*(20) | 2.9 ± 0.3*(5) | 22 ± 2*(5) |
| +Toxins | 135 ± 5*(22) | 393 ± 12(22) | 2.3 ± 0.8*(20) | 17 ± 2*(20) | 2.5 ± 0.4*(5) | 20 ± 2*(5) |
| Control | 121 ± 6(5) | 404 ± 23(5) | 6.9 ± 0.7(5) | 52 ± 5(5) | ||
| +Toxins | 128 ± 5(5) | 389 ± 15(5) | 6.6 ± 1.0(5) | 48 ± 6(5) | ||
| +N+I | 140 ± 6(5) | 376 ± 23(5) | 3.4 ± 0.9*† (5) | 23 ± 5*† |
MAP, mean arterial pressure; HR, heart rate; BF, blood flow; C, conductance; Mes, mesenteric bed; Fem, femoral bed.
Signficantly different from control.
Significant difference between l-NAME plus indomethacin (N+I) and toxins (charybdotoxin plus apamin).
Reversal of the order of administration of blockers afforded the same result in five rats: ChTx plus apamin did not influence MAP or mesenteric conductance, while subsequent addition of l-NAME plus indomethacin, this time via close infusion through the mesenteric cannula, increased MAP (Table 1) and decreased mesenteric conductance (Fig. 1, right-hand bars), though more slowly.
Effect of endothelium stimulation on mesenteric bed conductance
Stimulation of the endothelium with 10 s bolus applications of ACh evoked concentration-dependent increases in mesenteric BF and conductance (Fig. 3, n = 7). At 3 nmol kg−1 or less, ACh did not change MAP (Fig. 2B, left-hand upper trace) or HR, although it induced a robust increase in BF (from 6.2 ± 0.7 to 9.4 ± 0.8 ml min−1, n = 7; Fig. 2B, left-hand lower trace) and hence this dose was used in all subsequent tests.
Figure 3. Dose-dependent effects of acetylcholine on conductance changes in the mesenteric bed.
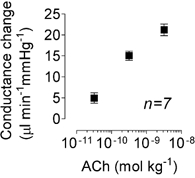
Conductance change (peak minus basal) in response to three doses of acetylcholine (ACh) in the absence of any blockers.
In the presence of l-NAME plus indomethacin, the duration of the increase in BF evoked by ACh was shortened (Fig. 2B, middle lower trace); the area under the conductance curve was reduced by 68 ± 3 % (n = 10). However, addition of l-NAME plus indomethacin reduced the peak amplitude of the conductance change by only about 24 % (Fig. 4).
Figure 4. Effects of l-NAME plus indomethacin and toxins on conductance changes induced by acetylcholine, bradykinin, sodium nitroprusside and 1-EBIO.
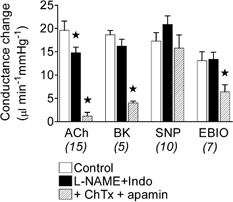
Conductance changes in the mesenteric bed in response to bolus applications of acetylcholine (ACh, 3 nmol kg−1), bradykinin (BK, 0.3 nmol kg−1), sodium nitroprusside (SNP, 3 nmol kg−1) and 1-EBIO (30 nmol kg−1) in the control period and following administration of l-NAME plus indomethacin (Indo), and then after addition of the toxins (charybdotoxin (ChTx) plus apamin). Number of animals in parentheses. * Significantly different from control.
Addition of ChTx, in the presence of l-NAME and indomethacin, reduced the amplitude of the ACh-evoked increase in conductance by 53 ± 4 % (n = 3), while in the presence of apamin it was reduced by 72 ± 5 % (n = 3) (data not shown). In vitro studies have found that the combination of both ChTx and apamin is required to achieve blockade of this EDHF. Here, the combination reduced the NO- and prostanoid-independent increase in conductance by more than 90 % (Fig. 2B and Fig. 4). BK (0.3 nmol kg−1) induced a similar increase in mesenteric conductance (Fig. 4), again, with no effect on MAP or HR.
The endothelial dependence of the response to ACh was verified by its sensitivity to CHAPS (Fig. 5, n = 4), which removes or damages endothelial cells. The endothelium-independent vasodilator SNP, at 3 nmol kg−1, increased mesenteric BF and conductance to levels that were similar in amplitude to those elicited by ACh and BK (Fig. 4), and also without changes in MAP or HR. These responses to SNP were resistant to the effects of l-NAME, indomethacin, ChTx and apamin (Fig. 4) or to CHAPS (Fig. 5).
Figure 5. Effects of l-NAME plus indomethacin and CHAPS on conductance changes induced by acetylcholine and sodium nitroprusside.
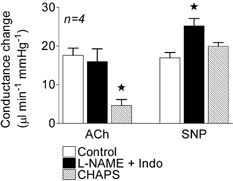
Conductance changes in the mesenteric bed in response to bolus applications of acetylcholine (ACh, 3 nmol kg−1) and sodium nitroprusside (SNP, 3 nmol kg−1) in the control period and following administration of l-NAME plus indomethacin (Indo) and then after addition of CHAPS (5 % for 3–4 min). * Significantly different from control.
Reversing the order of blocker application, so that ChTx plus apamin were administered first, markedly reduced the peak amplitude of the conductance changes in response to ACh and BK (Fig. 6) with only a small further reduction induced by l-NAME plus indomethacin.
Figure 6. Effect of order of administration of l-NAME plus indomethacin, and charybdotoxin plus apamin on conductance changes induced by acetylcholine and bradykinin.
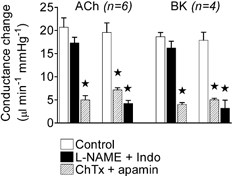
Conductance changes in the mesenteric bed in response to bolus applications of acetylcholine (ACh, 3 nmol kg−1) and bradykinin (BK, 0.3 nmol kg−1) in the control period and following administration of l-NAME plus indomethacin (Indo) and then after addition of the toxins (charybdotoxin (ChTx) plus apamin) (left-hand bars for ACh and BK). The right-hand bars for ACh and BK show blocker administration in the reverse order, first the toxins, followed by l-NAME plus indomethacin. * Significantly different from control.
The protocols used were long: (1) 30 min to ensure that l-NAME plus indomethacin had fully blocked the production of NO and prostacyclin; (2) 20–30 min required for re-testing the effects of ACh and/or BK and/or SNP on BF; and (3) 10 min for the application of ChTx plus apamin. The possibility of deterioration of the responses over this time was considered in five rats. MAP, HR and the increase in conductance evoked in the mesenteric bed by ACh and SNP were unchanged over 2–3 h in these animals (data not shown).
Iberiotoxin and 1-EBIO
The selectivity of ChTx is such that it blocks both intermediate- and large-conductance KCa channels, the latter being involved in the hyperpolarization evoked by products of cytochrome P-450 and in response to stimulation of the endothelium in some arteries in vitro (Hecker et al. 1994; Campbell et al. 1996; Baron et al. 1997; Eckman et al. 1998) and in vivo (Widmann et al. 1998; De Wit et al. 1999; Nishikawa et al. 1999; Hungerford et al. 2000). The large-conductance KCa channels are more selectively blocked by IbTx, but this agent had no effect on the increases in mesenteric conductance evoked by ACh or BK. However, subsequent exposure of the bed to ChTx plus apamin in the same animals markedly reduced the responses (Fig. 7, n = 5). There was no effect of any of these blockers on the response to SNP (Fig. 7).
Figure 7. Effect of l-NAME plus indomethacin, iberiotoxin and charybdotoxin plus apamin on conductance changes induced by acetylcholine, bradykinin and sodium nitroprusside.
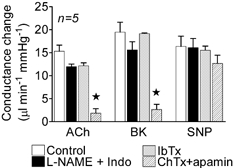
Conductance changes in the mesenteric bed in response to bolus applications of acetylcholine (ACh, 3 nmol kg−1), bradykinin (BK, 0.3 nmol kg−1), and sodium nitroprusside (SNP, 3 nmol kg−1) in the control period and following administration of l-NAME plus indomethacin (Indo), then iberiotoxin (IbTx), and finally charybdotoxin (ChTx) plus apamin. * Significantly different from control.
The intermediate-conductance channels that underpin part of the response attributable to EDHF are opened by 1-EBIO (Edwards et al. 1999; Walker et al. 2001). In the present study, close application of 1-EBIO increased conductance in the mesenteric bed, a response that was approximately halved by addition of ChTx, a blocker of these channels (Fig. 4).
Hindlimb bed
Endothelium-dependent vasodilatation of larger branches (diameter, 300–500 μm) of the rat femoral/popliteal artery appears to involve NO release, with little or no contribution of EDHF (Zygmunt et al. 1995; Wigg et al. 2001). Basal conductance in the hindlimb bed was 39 ± 3 μl min−1 mmHg−1. Similar to observations in the mesenteric bed, basal conductance was reduced by addition of l-NAME plus indomethacin but administration of the toxins (ChTx plus apamin) had no further effect (Table 1).
ACh increased conductance in the hindlimb bed, as in the mesenteric bed. The amplitude of this response was reduced by around 40 % in the presence of l-NAME plus indomethacin, and then by 90 % in the presence of ChTx plus apamin (Fig. 8, n = 5). The increase in conductance evoked by SNP (3 nmol kg−1) was proportionately smaller than that evoked by ACh in the hindlimb (Fig. 8) compared with observations in the mesenteric bed (see Figs 4, 5 and 7).
Figure 8. Effect of l-NAME plus indomethacin, and charybdotoxin plus apamin on conductance changes induced by acetylcholine and sodium nitroprusside in the femoral bed.
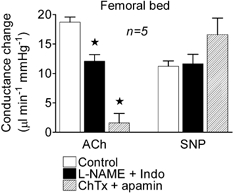
Conductance changes in the femoral bed in response to bolus applications of acetylcholine (ACh, 3 nmol kg−1) and sodium nitroprusside (SNP, 3 nmol kg−1) in the control period and following administration of l-NAME plus indomethacin (Indo) and then the toxins (charybdotoxin (ChTx) plus apamin). * Significantly different from control.
The anaesthetic
It has been suggested that barbiturate anaesthetics may suppress EDHF (De Wit et al. 1999). In the present study 10 rats were anaesthetized with urethane. Mesenteric conductance changes evoked by ACh, BK and SNP, and the effects of blockers (Fig. 7) were similar to those in which the animals were anaesthetized using Inactin (responses that have been shown in all other figures). The stability of the responses with time was verified in five of the rats anaesthetized with urethane (data not shown).
Discussion
The results presented here demonstrate a major involvement of EDHF in the peak increase in conductance induced by stimulating the endothelium in both the mesenteric and hindlimb beds of anaesthetized rats. This EDHF-dependent vasodilatation is mediated by intermediate- and small-conductance KCa channels. Thus, we have demonstrated a functional role for the ChTx plus apamin-sensitive EDHF in vivo.
The EDHF that is blocked by ChTx plus apamin has been well documented in isolated vessels, especially in rat mesenteric arteries (Nagao et al. 1992; Zygmunt et al. 1995; Plane et al. 1997; Edwards et al. 1998; Doughty et al. 1999; Wigg et al. 2001), and the consensus of the results in vitro corroborates our present observations in the rat mesenteric bed in vivo. This EDHF has been variously attributed to K+ and to the involvement of myoendothelial gap junctions. Recent in vitro studies have demonstrated the correlation between EDHF-attributed hyperpolarization in endothelial and smooth muscle cells and the existence of myoendothelial pentalaminar gap junctions (Sandow et al. 2002). Use of peptide inhibitors suggests the involvement of gap junctions in the EDHF response in the rat renal bed in vivo (De Vriese et al. 2002). Previous studies of other vascular beds in vivo have implicated the EETs as EDHF (Widmann et al. 1998; De Wit et al. 1999; Nishikawa et al. 1999; Hungerford et al. 2000), while the profile of toxin sensitivity in our study suggests that these agents are unlikley to be involved in the vascular beds investigated here. This difference supports the notion that ‘EDHF’ is not homogeneous, with regional/species variations in distribution. For example, it appears that the EETs play a significant role in vasodilatation in coronary microvessels of dog (Widmann et al. 1998; Nishikawa et al. 1999) and pig (Hecker et al. 1994), but not in guinea-pig coronary arteries (Eckman et al. 1998; Coleman et al. 2001a). In previous in vivo studies the EETs were found to mediate the EDHF response in skeletal muscle beds of mice (Hungerford et al. 2000) and hamsters (De Wit et al. 1999), whereas in the hindlimb of the rat the ChTx plus apamin-sensitive EDHF predominates (present study). The hindlimb bed supplies both skeletal muscle and skin and the relative contribution of EDHF and NO in each to the overall response recorded has not been determined.
From the evidence to hand, including the data presented here, it is tempting to suggest the possibility that the sensitivity or otherwise to barbiturates may be a means of identifying the nature of an observed EDHF-mediated vasodilatation. The barbiturate thiopentone is capable of abolishing the response to EETs in vivo (De Wit et al. 1999) and in vitro (Lischke et al. 1995) by inhibiting the activity of cytochrome P-450 enzymes (Lischke et al. 1995). The EDHF observed here in the mesenteric bed was entirely resistant to inhibition by thiobutabarbital (Inactin), providing further evidence against an involvement of EETs in EDHF in this vascular bed.
The EDHF described in this study was insensitive to blockade by IbTx and it might be argued that the dose of IbTx used was insufficient to block the large-conductance KCa channels. The total amount of the toxin delivered to each rat was 20 μg kg−1, exactly the same concentration as was found to be effective at abolishing the EET-attributed EDHF in the dog coronary microcirculation (Nishikawa et al. 1999). Of additional interest in that study, was the fact that IbTx was injected directly into the coronary circulation of the dogs, which is similar to the intramesenteric application in our study. Use of imidazole antimycotics to inhibit cytochrome P-450 enzyme activity was not attempted in the present study due to their potent ability to suppress KCa channels (Alvarez et al. 1992; Vanheel & Van de Voorde, 1997).
Our study has revealed the relative importance of NO and EDHF in the response to agonist-induced stimulation of the endothelium in the mesenteric and hindlimb beds of rats in vivo. The responses evoked by ACh and BK were biphasic in appearance, with an initial large, but brief component followed by a second more prolonged component of smaller amplitude. EDHF was largely responsible for the initial peak vasodilatation, accounting for 76 % of its amplitude. The toxins (ChTx plus apamin) were effective in reducing the amplitude of this initial component, leaving the smaller amplitude second component intact. This second component, which accounted for 68 % of the total area of vasodilatation, is mediated by NO since it was abolished by l-NAME. This is similar to observations in hamster microvessels (De Wit et al. 1999). There was no evidence for the involvement of prostanoids (including prostacyclin) in either of these vascular beds and this is in keeping with observations in these vessels in vitro (Nagao et al. 1992; Zygmunt et al. 1995; Wigg et al. 2001).
In contrast with the responses to endothelial stimulation, there was little evidence for a contribution of EDHF to basal flow in the mesenteric or hindlimb vascular beds. Similarly, the EDHF attributed to EETs also does not appear to contribute to basal tone in vivo (Jackson & Blair, 1998; Nishikawa et al. 1999). However, in the renal bed of rats, peptide gap junction inhibitors decreased basal blood flow (De Vriese et al. 2002). NO plays an important role in determining basal flow in hamster cremaster arterioles (Jackson & Blair, 1998), dog coronary arteries (Nishikawa et al. 1999) and rat mesenteric and hindlimb beds (present study). This is in keeping with the longstanding evidence that basal NO release contributes to resting vascular tone. There is no evidence for an involvement of prostanoids in determining basal tone in rat mesenteric or hindlimb beds. Indomethacin alone or the presence of l-NAME was completely without effect on basal BF and conductance. Thus, although prostacyclin is a potent anti-thrombogenic agent and may very well be basally released from the vascular endothelium, there is no evidence for an effect on smooth muscle tone here. This may reflect polarity (i.e. dominant luminal or abluminal) in its release from endothelial cells or the lack of receptors on the smooth muscle cells in these vessels in the rat.
The contribution of EDHF can vary with vessel diameter and, in general, it plays a more prominent role in smaller muscular arteries and arterioles than in larger muscular or conduit vessels in vivo (Lefroy et al. 1993; Widmann et al. 1998; Nishikawa et al. 1999) and in vitro (Hwa et al. 1994; Garland et al. 1995; Shimokawa et al. 1996; Hill et al. 2001). In the mesenteric bed, EDHF is present at all levels, but is graded in importance from larger to smaller branches (Hwa et al. 1994; Garland et al. 1995; Shimokawa et al. 1996; Hill et al. 2001). In contrast, in the hindlimb, EDHF appears to be absent in branches of 200–300 μm diameter, where NO is responsible for endothelium-dependent relaxation in vitro (Zygmunt et al. 1995; Wigg et al. 2001). It is interesting that our in vivo results have revealed an involvement of EDHF in the whole bed, and this is likely to be present at the level of the small arteries/arterioles in this vascular bed. The more extreme distribution of endothelium-derived vasodilators in the hindlimb (i.e. regional absence of EDHF) may explain the greater role of NO in the overall conductance in this bed. Thus, l-NAME reduced the amplitude of the evoked vasodilator response by about 50 % in the hindlimb bed compared with only 25 % in the mesenteric bed.
Intermediate-conductance KCa channels make an important contribution to the EDHF-mediated vasodilatation in rat mesenteric arteries in vitro (Plane et al. 1997; Vanheel & Van de Voorde, 1997; Edwards et al. 1998) and here we show that this also holds in vivo. These ion channels can be opened by 1-EBIO (Edwards et al. 1999; Coleman et al. 2001b; Wigg et al. 2001; Walker et al. 2001). We have now demonstrated that it is possible to activate these channels in vivo using this compound. In vitro studies suggest that this agent may possess additional actions, in particular, activation of cyclic AMP pathways (Adeagbo, 1999; Walker et al. 2001). This probably explains why ChTx blocked only 50 % of the increase in mesenteric conductance in response to 1-EBIO in the present study.
In conclusion, this study has demonstrated a role for the ChTx- and apamin-sensitive EDHF in the regulation of blood flow in vivo in the mesenteric and hindlimb circulations of the rat. The peak endothelium-dependent vasodilatation evoked by agonists is dominated by this EDHF, with NO contributing a second, lower-amplitude, prolonged phase of vasodilatation. The ChTx- and apamin-sensitive EDHF appears to play little or no role in the regulation of basal blood flow in these vascular territories, whereas NO appears to be important in this role. However, while NO may also be the major contributor to vasodilatation during ‘chemical’ stimulation of the endothelium in larger distributing vessels, the prominence of EDHF in smaller arteries/arterioles confers upon it a pivotally important role in determining tissue perfusion.
Acknowledgments
This work was supported by the National Heart Foundation of Australia, the Monash Small Grants Scheme and the National Health and Medical Research Council of Australia.
References
- Adeagbo ASO. 1-Ethyl-2-benzimidazolinone stimulates endothelial KCa channels and nitric oxide formation in rat mesenteric vessels. European Journal of Pharmacology. 1999;379:151–159. doi: 10.1016/s0014-2999(99)00489-6. [DOI] [PubMed] [Google Scholar]
- AlvareZ J, Montero M, Garcia-Sancho J. High affinity inhibition of Ca2+-dependent K+ channels by cytochrome P-450 inhibitors. Journal of Biological Chemistry. 1992;267:11789–11793. [PubMed] [Google Scholar]
- Baron A, Frieden M, Bény J-L. Epoxyeicosatrienoic acids activate a high-conductance, Ca2+-dependent K+ channel on pig coronary artery endothelial cells. Journal of Physiology. 1997;504:537–543. doi: 10.1111/j.1469-7793.1997.537bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Research. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Coleman HA, Tare M, Parkington HC. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. Journal of Physiology. 2001a;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HA, Tare M, Parkington HC. EDHF is not K+ but may be due to spread of current from the endothelium in guinea pig arterioles. American Journal of Physiology. 2001b;280:H2478–2483. doi: 10.1152/ajpheart.2001.280.6.H2478. [DOI] [PubMed] [Google Scholar]
- De Vriese AS, Van de Voorde J, Lameire NH. Effects of connexin-mimetic peptides on nitric oxide synthase- and cyclooxygenase-independent renal vasodilatation. Kidney International. 2002;61:177–185. doi: 10.1046/j.1523-1755.2002.00122.x. [DOI] [PubMed] [Google Scholar]
- De Wit C, Esser N, Lehr H, BolZ S, Pohl U. Pentobarbital-sensitive EDHF comediates ACh-induced arteriolar dilatation in the hamster microcirculation. American Journal of Physiology. 1999;276:H1527–1534. doi: 10.1152/ajpheart.1999.276.5.H1527. [DOI] [PubMed] [Google Scholar]
- Doughty JM, Plane F, Langton PD. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. American Journal of Physiology. 1999;276:H1107–1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- Eckman DM, Hopkins N, McBride C, Keef KD. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. British Journal of Pharmacology. 1998;124:181–189. doi: 10.1038/sj.bjp.0701778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Edwards G, Gardener MJ, Félétou M, Brady G, Vanhoutte PM, Weston AH. Further investigation of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. British Journal of Pharmacology. 1999;128:1064–1070. doi: 10.1038/sj.bjp.0702916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RG, Madden AC, Denton KM. Diversity of responses of renal cortical and medullary blood flow to vasoconstrictors in conscious rabbits. Acta Physiologica Scandinavica. 2000;169:297–308. doi: 10.1046/j.1365-201x.2000.00741.x. [DOI] [PubMed] [Google Scholar]
- Fleming I. Cytochrome P-450 enzymes in vascular homeostasis. Circulation Research. 2001;89:753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- Gardiner SM, Compton AM, Bennett T, Palmer RMJ, Moncada S. NG-monomethyl-l-arginine does not inhibit the hindquarters vasodilator action of endothelin-1 in conscious rats. European Journal of Pharmacology. 1989;171:237–240. doi: 10.1016/0014-2999(89)90113-1. [DOI] [PubMed] [Google Scholar]
- Gardiner SM, Kemp PA, Bennett T. Effect of NG-nitro-l-arginine methyl ester on vasodilator responses to acetylcholine, 5′-N-ethylcarboxamidoadenosine or salbutamol in conscious rats. British Journal of Pharmacology. 1991;103:1725–1732. doi: 10.1111/j.1476-5381.1991.tb09854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends in Pharmacological Science. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. Journal of Physiology. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CE, Phillips JK, Sandow SL. Heterogeneous control of blood flow amongst different vascular beds. Medical Research Reviews. 2001;21:1–60. doi: 10.1002/1098-1128(200101)21:1<1::aid-med1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Hungerford JE, Sessa WC, Segal SS. Vasomotor control in arterioles of the mouse cremaster muscle. FASEB Journal. 2000;14:197–207. doi: 10.1096/fasebj.14.1.197. [DOI] [PubMed] [Google Scholar]
- Hwa J, Ghibaudi L, Williams P, Chatterjei M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. American Journal of Physiology. 1994;266:H952–958. doi: 10.1152/ajpheart.1994.266.3.H952. [DOI] [PubMed] [Google Scholar]
- Jackson WF, Blair KL. Characterization and function of Ca2+-activated K+ channels in arteriolar muscle cells. American Journal of Physiology. 1998;274:H27–34. doi: 10.1152/ajpheart.1998.274.1.H27. [DOI] [PubMed] [Google Scholar]
- Lefroy DC, Crake T, Uren NG, Davies GJ, Maseri A. Effect of inhibition of nitric oxide synthesis on epicardial coronary artery calibre and coronary blood flow in humans. Circulation. 1993;88:43–54. doi: 10.1161/01.cir.88.1.43. [DOI] [PubMed] [Google Scholar]
- Lischke V, Busse R, Hecker M. Selective inhibition by barbiturates of the synthesis of endothelium-derived hyperpolarizing factor in the rabbit carotid artery. British Journal of Pharmacology. 1995;115:969–974. doi: 10.1111/j.1476-5381.1995.tb15905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T, Illiano S, Vanhoutte PM. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-l-arginine in rats. American Journal of Physiology. 1992;263:H1090–1094. doi: 10.1152/ajpheart.1992.263.4.H1090. [DOI] [PubMed] [Google Scholar]
- Nishikawa Y, Stepp DW, Chilian WM. In vivo location and mechanism of EDHF-mediated vasodilatation in canine coronary microcirculation. American Journal of Physiology. 1999;277:H1252–1259. doi: 10.1152/ajpheart.1999.277.3.H1252. [DOI] [PubMed] [Google Scholar]
- Plane F, Holland M, Waldron GJ, Garland CJ, Boyle JP. Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. British Journal of Pharmacology. 1997;121:1509–1511. doi: 10.1038/sj.bjp.0701361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circulation Research. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circulation Research. 2002;90:1108–1113. doi: 10.1161/01.res.0000019756.88731.83. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Yasutake H, Fujii K, Owada K, Nakaike R, Fukumoto Y, Takayanagi T, Nagao T, Egashira K, Fujishima M, Takeshita A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. Journal of Cardiovascular Pharmacology. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- Taylor SG, Weston AH. Endothelium-derived hyperpolarizing factor: a new endogenous inhibitor from the vascular endothelium. Trends in Pharmacological Sciences. 1988;9:272–274. doi: 10.1016/0165-6147(88)90003-x. [DOI] [PubMed] [Google Scholar]
- Vallance P, Collier J, Moncada S. Effect of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;ii:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- Vanheel B, Van de Voorde J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat main mesenteric artery. Journal of Physiology. 1997;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SD, Dora KA, Ings NT, Crane GL, Garland CJ. Activation of endothelial cell IKCa with 1-ethyl-2-benzimidazolinone evokes smooth muscle hyperpolarization in rat isolated mesenteric artery. British Journal of Pharmacology. 2001;134:1548–1554. doi: 10.1038/sj.bjp.0704415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann MD, Weintraub NL, Fudge JL, Brooks LA, Dellsperger KC. Cytochrome P-450 pathway in acetylcholine-induced canine coronary microvascular vasodilatation in vivo. American Journal of Physiology. 1998;274:H283–289. doi: 10.1152/ajpheart.1998.274.1.H283. [DOI] [PubMed] [Google Scholar]
- Wigg SJ, Tare M, Tonta MA, O'Brien RC, Meredith IT, Parkington HC. Comparison of effects of diabetes mellitus on an EDHF-dependent and an EDHF-independent artery. American Journal of Physiology. 2001;281:H232–240. doi: 10.1152/ajpheart.2001.281.1.H232. [DOI] [PubMed] [Google Scholar]
- Woodman OL, Wongsawatkul O, Sobey CG. Contribution of nitric oxide, cyclic GMP and K+ channels to acetylcholine-induced dilatation of rat conduit and resistance arteries. Clinical and Experimental Pharmacology and Physiology. 2000;27:34–40. doi: 10.1046/j.1440-1681.2000.03199.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Fukuta H, Nakahira Y, Suzuki H. Blockade by 18β-glycyrrhetinic acid of intercellular electrical coupling in guinea-pig arterioles. Journal of Physiology. 1998;511:501–508. doi: 10.1111/j.1469-7793.1998.501bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou A, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K+-channel activity. American Journal of Physiology. 1996;270:F822–832. doi: 10.1152/ajprenal.1996.270.5.F822. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Högestätt ED. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. British Journal of Pharmacology. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Ryman T, Högestätt ED. Regional differences in endothelium-dependent relaxation in the rat – contribution of nitric oxide and nitric oxide-independent mechanisms. Acta Physiologica Scandinavica. 1995;155:257–266. doi: 10.1111/j.1748-1716.1995.tb09972.x. [DOI] [PubMed] [Google Scholar]