

Abstract
The present study determined the effects of acetylcholine (ACh) on the L-type Ca2+ current (ICa,l) stimulated by β1- or β2-adrenergic receptor (AR) agonists in cat atrial myocytes. When isoproterenol (ISO; 0.1 μm) plus the β2-AR antagonist ICI 118,551 (ISO-β1-AR stimulation) or 0.1 μm fenoterol, a β2-AR agonist (FEN-β2-AR stimulation) increased ICa,l, ACh (1 μm) inhibited ICa,l by –60 ± 4 and –63 ± 6 %, respectively. When ISO plus the β1-AR antagonist atenolol (ISO-β2-AR stimulation) or 1 μm zinterol (ZIN-β2-AR stimulation) increased ICa,l, ACh-induced inhibition of ICa,l was significantly smaller, at –21 ± 3 and −24 ± 3 %, respectively. l-N5-(1-iminoethyl)ornithine (l-NIO, 10 μm), an inhibitor of nitric oxide (NO) synthase, enhanced ACh-induced inhibition of ICa,l when stimulated by ZIN-β2-ARs, but not when stimulated by ISO-β1-ARs or FEN-β2-ARs. Haemoglobin (50 μm), a NO scavenger, also enhanced ACh-induced inhibition when ICa,l was stimulated by ZIN-β2-ARs, but not when stimulated by FEN-β2-ARs. ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs was not affected by 10 μm 1H-[1,2,4] oxadiazolo[4,3-a] quinoxaline-1-one (ODQ) a guanylate cyclase inhibitor, but was significantly enhanced by 500 μm reduced glutathione or 100 μm dithiothreitol, agents that act as sinks for S-nitrosylation. ACh-induced inhibition was smaller when ICa,l was stimulated by spermine/NO, a NO donor, than by milrinone, a phosphodiesterase type III inhibitor. ISO (ISO-β1/β2-AR stimulation) increased ICa,l and even though ISO releases NO, ACh prominently inhibited ICa,l. This inhibitory effect of ACh was enhanced by l-NIO. Stimulation of ZIN-β2-ARs increased intracellular NO, whereas ISO-β1-ARs or FEN-β2-ARs failed to increase intracellular NO. These results indicate that in atrial myocytes, NO released by selective β2-AR stimulation prevents ACh-induced inhibition of ICa,l stimulated by β2-ARs. NO acts via a cGMP-independent, S-nitrosylation mechanism. Although FEN acts via β2-ARs, it fails to stimulate Gi-/NO signalling and preferentially stimulates Gs-/adenylate cyclase signalling, similar to β1-ARs. These findings indicate that NO signalling modulates muscarinic receptor inhibition of atrial function stimulated by β2-ARs.
In general, acetylcholine (ACh) elicits an accentuated inhibition of cardiac chronotropic and inotropic activities that are stimulated by β-adrenergic receptor (β-AR) stimulation. ACh exerts its inhibitory effects through stimulation of M2 muscarinic receptors coupled via Gi-proteins to inhibit adenylate cyclase. It has been proposed that nitric oxide (NO) signalling activated by ACh may also participate in the inhibitory effects of ACh, although this idea remains controversial (Han et al. 1998; Vandecasteele et al. 1999; Belevych & Harvey, 2000; Godecke et al. 2001). Although ACh inhibits stimulation by β-ARs, cardiac muscle contains different subtypes of β-ARs and each subtype exhibits significantly different signal transduction mechanisms (Steinberg, 1999; Xiao et al. 1999). For example, β1-ARs act exclusively via Gs-proteins coupled to adenylate cyclase to catalyse the synthesis of cAMP, which in turn activates protein kinase (PK)A signalling. In contrast, in several animal species (Xiao et al. 1995; Kuschel et al. 1999; Wang et al. 2002), including humans (Kilts et al. 2000), β2-ARs couple to a variety of signalling pathways via both Gs- and Gi-proteins. In cat atrial myocytes, β2-ARs couple via Gs-proteins to adenylate cyclase and via Gi-proteins to phosphatidylinositol 3′-kinase (PI-3K) signalling to release intracellular NO (Wang et al. 2002). In cardiac muscle, NO signalling operates primarily via two basic biochemical mechanisms: (1) stimulation of guanylate cyclase/cGMP production and (2) nitrosylation of sulfhydryl groups on the cysteine residues of various proteins (Broillet, 1999). It is now recognized that S-nitrosylation (cGMP-independent) reactions can modulate a wide variety of cellular functions (Broillet, 1999; Stamler et al. 2001). Together, these considerations raise the question of how stimulation via different β-AR subtypes and agonists respond to inhibition by muscarinic receptor stimulation. In neonatal rat ventricular myocytes, muscarinic receptor stimulation inhibits cAMP accumulation and positive inotropic responses elicited by β1-ARs but not by β2-AR stimulation (Aprigliano et al. 1997). The purpose of the present study was therefore to determine whether there are differential inhibitory effects of ACh on ICa,l when ICa,l is prestimulated by either β1- or β2-AR agonists in adult cat atrial myocytes and, if so, the nature of the underlying mechanism.
Methods
Adult cats of either gender were anaesthetized with sodium pentobarbital (50 mg kg−1; i.p.). Once anaesthetized, a midsternal thoracotomy was performed and the heart rapidly excised. The heart was mounted on a Langendorff perfusion apparatus and atrial myocytes were dispersed by digestion with collagenase (type II, Worthington Biochemical), as reported previously (Wu et al. 1991). No discernable differences were noted between left and right atrial myocytes. Cells used for studies were transferred to a small tissue bath on the stage of an inverted microscope (Nikon Diaphot) and superfused with a Hepes-buffered, modified Tyrode solution containing (mm): NaCl 145, KCl 4, MgCl2 1, CaCl2 2, Hepes 5, glucose 11, titrated with NaOH to pH 7.4. Solutions were perfused by gravity and experiments were performed at 35 ± 1°C. Atrial myocytes selected for study were elongated and quiescent. Voltage and ionic currents were recorded using a nystatin (150 μg ml−1)-perforated-patch (Horn & Marty, 1988) whole-cell recording method (Hamill et al. 1981). This method minimizes dialysis of intracellular constituents with the internal pipette solution and thereby preserves the physiological milieu and second-messenger signalling pathways. The internal pipette solution contained (mm): caesium glutamate 100, KCl 40, MgCl2 1.0, Na2-ATP 4, EGTA 0.5, Hepes 5, titrated with KOH to pH 7.2. In addition to Cs+ in the pipette solution, 5 mm CsCl was added to all solutions to block K+ conductances. A single suction pipette was used to record voltage (bridge mode) or ionic currents (discontinuous voltage-clamp mode) using an Axoclamp 2A amplifier (Axon Instruments). Computer software (pCLAMP; Axon Instruments) was used to deliver the voltage protocols and to acquire and analyse data. ICa,l was activated by depolarizing pulses from a holding potential of −40 to 0 mV for 200 ms every 10 s. Peak ICa,l amplitude was measured with respect to steady-state current and not compensated for leak currents. Increases in peak ICa,l amplitude induced by β-AR stimulation were determined with respect to basal ICa,l amplitude. Changes in ICa,l amplitude induced by ACh were determined as a percentage of the changes in ICa,l amplitude induced by each β-AR agonist (see legend to Fig. 1).
Figure 1. Effects of 1 μm ACh on L-type Ca2+ current (ICa,l) stimulated by various β-adrenergic receptor (AR) agonists.

Effect of 1 μm ACh on ICa,l stimulated by isoproterenol (ISO)-β1-ARs (A), ISO-β2-ARs (B), zinterol (ZIN)-β2-ARs (C) and fenoterol (FEN)-β2-ARs (D). Each panel shows original traces of peak ICa,l during each phase (a-c) of the experiment and a graph of consecutive measurements of peak ICa,l throughout the experiment. ACh-induced inhibition of ICa,l was relatively large when ICa,l was prestimulated by ISO-β1-ARs (A; −61 %) or FEN-β2-ARs (D; −58 %), and significantly smaller when ICa,l was prestimulated by ISO-β2-ARs (B; −19 %) or ZIN-β2-ARs (C; −22 %). The percentage inhibition of ICa,l induced by ACh was determined as the change in ICa,l induced by ACh (b - c) in relation to the stimulated change in ICa,l induced by the β-AR agonist (b - a) using the formula: % = (b - c)/(b - a) × 100. ICI = ICI 118,551, a selective β2-AR antagonist.
Direct measurements of intracellular NO concentration ([NO]i) were obtained by incubating cells with the fluorescent NO-sensitive dye 4,5-diaminofluorescein (DAF-2) (Kojima et al. 1998; Nakatsubo et al. 1998; Wang et al. 2002). Experiments were performed at room temperature. Cells were exposed to the membrane-permeant DAF-2 diacetate (5 μm; Calbiochem, San Diego, CA, USA) for 10 min at room temperature in standard Tyrode solution. Cells were subsequently washed for 10 min in Tyrode solution containing 100 μml-arginine. DAF-2 fluorescence was excited at 480 nm. Emitted cellular fluorescence was recorded at 540 nm. Single-cell fluorescence signals were recorded with a photomultiplier tube (model R2693, Hamamatsu) by masking individual cells with an iris positioned in the emission path. Changes in cellular DAF-2 fluorescence intensities (F) in each experiment were normalized to the level of fluorescence recorded prior to stimulation (F0), and changes in [NO]i are expressed as F/F0. In the experiments designed to measure [NO]i, solutions contained 100 μml-arginine. Cells were field stimulated at 1 Hz.
Selective stimulation of β1-ARs or β2-ARs was accomplished as follows: 0.1 μm isoproterenol (isoprenaline; ISO), a mixed β1-/β2-AR agonist, in the presence of 0.01 μm atenolol, a selective β1-AR antagonist (ISO-β2-AR stimulation); 0.1 μm ISO plus 0.01 μm ICI 118,551, a selective β2-AR antagonist (ISO-β1-AR stimulation); 1 μm zinterol (ZIN), a selective β2-AR agonist (ZIN-β2-AR stimulation); 0.1 μm fenoterol (FEN), another selective β2-AR agonist (FEN-β2-AR stimulation). Previous work has shown that the stimulatory effects of ISO on ICa,l are abolished by the combined exposure to atenolol plus ICI 118,551 and that BRL 37344, a specific β3-AR agonist, has no effect on ICa,l (Wang et al. 2002).
Drugs used in this study include ISO, acetylcholine chloride, FEN, atenolol, haemoglobin, l-N5-(1-iminoethyl)ornithine (l-NIO), 1H-[1,2,4] oxadiazolo[4,3-a]quinoxaline-1-one (ODQ), reduced glutathione (GSH), dithiothreitol (DTT), spermine/NO (SNO), milrinone (all from Sigma); Rp-cAMPs (LC Laboratories); ZIN (provided by Bristol-Myers Squibb, Princeton, NJ, USA) and ICI 118,551 (provided by AstraZeneca, Wilmington, DE, USA).
In general, results were obtained in cells isolated from the same hearts studied under control and test conditions. Data from two groups of cells were analysed using Student's unpaired t test with significance at P < 0.05. Data from multiple groups were analysed using a one-way analysis of variance (ANOVA) and Bonferroni test at P < 0.05.
The animal procedures used in this study were performed in accordance with the guidelines of the Animal Care and Use Committee of Loyola University Medical Center.
Results
Figure 1A–D shows the effects of ACh on ICa,l prestimulated by various β-AR agonists. Each panel shows selected recordings of peak ICa,l obtained at different times (labelled a-c) during each experiment and consecutive measurements of peak ICa,l throughout each experiment. ISO-β1-AR stimulation increased ICa,l (256 %) and the addition of ACh prominently inhibited ICa,l (-61 %; Fig. 1A). ISO-β2-AR stimulation also increased ICa,l (282 %), and yet ACh elicited a much smaller inhibition of ICa,l (-19 %; Fig. 1B) compared with the effects of ACh on ICa,l stimulated by ISO-β1-ARs. Likewise, ZIN-β2-AR stimulation increased ICa,l (182 %), and once again ACh elicited a relatively small inhibition of ICa,l (-22 %; Fig. 1C). FEN-β2-AR stimulation also increased ICa,l (233 %; Fig. 1D). However, unlike the response of ICa,l stimulated by either ISO-β2-ARs or ZIN-β2-ARs, ACh markedly inhibited ICa,l (-58 %) stimulated by FEN-β2-ARs. The effects of FEN to stimulate ICa,l were abolished by 0.1 μm ICI 118,551 (control, 320 ± 47 % vs. + ICI, 5 ± 9 %; n = 4), and abolished by 50 μm Rp-cAMPs, a specific inhibitor of cAMP-dependent PKA activity (control, 201 ± 43 % vs. + Rp-cAMPs, 4 ± 7 %; n = 5; data not shown). These latter findings confirm that FEN stimulates ICa,l specifically via β2-AR-mediated activation of cAMP/PKA activity.
Figure 2A summarizes the ability of each β-AR agonist to stimulate ICa,l (open bars) and the ability of ACh to inhibit each β-AR agonist-stimulated ICa,l (hatched bars). Each β-AR agonist induced a prominent increase in ICa,l amplitude. Note that stimulation of ICa,l by FEN-β2-ARs was essentially of the same order of magnitude as that induced by ISO-β2-AR stimulation. ACh-induced inhibition of ICa,l was largest when ICa,l was stimulated by either ISO-β1-ARs (-60 ± 4 %; n = 16) or FEN-β2-ARs (-63 ± 6 %; n = 12). These values were not significantly different from each other. The inhibitory effects of ACh were smallest when ICa,l was stimulated by either ISO-β2-ARs (-21 ± 3 %; n = 11) or ZIN-β2-ARs (-24 ± 3 %; n = 23). Again, these values were not different from each other. However, the inhibitory effects of ACh on ICa,l stimulated by either ISO-β2-ARs or ZIN-β2-ARs were significantly smaller than the inhibitory effects of ACh when ICa,l was stimulated by either ISO-β1-ARs or FEN-β2-ARs. It should be noted that the magnitude of the inhibitory effect of ACh on ICa,l was not dependent upon the basal (prestimulated) ICa,l amplitude or the absolute current level to which ICa,l was stimulated by a given β-AR agonist. Moreover, when ICa,l was stimulated via β2-ARs using either ISO or ZIN, ACh-induced inhibition of ICa,l was approximately of the same order of magnitude as the effects of ACh to inhibit basal (unstimulated) ICa,l (Wang & Lipsius, 1995; Wang et al. 1998). This suggests that ACh does not exert any significant inhibition of ICa,l that can be attributed to the stimulation of ICa,l by either ISO-β2-ARs or ZIN-β2-ARs.
Figure 2. Summary of the effects of each β-AR agonist to stimulate ICa,l (open bars) and ACh-induced inhibition of ICa,l prestimulated by each β-AR agonist (hatched bars).
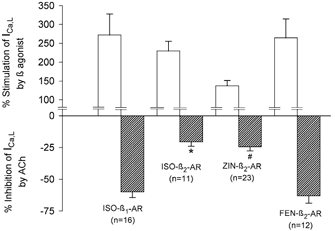
Each β-AR agonist stimulated ICa,l. ACh-induced inhibition of ICa,l was largest when ICa,l was stimulated by ISO-β1-ARs or FEN-β2-ARs, and significantly smaller when ICa,l was stimulated by ISO-β2-ARs or ZIN-β2-ARs. Note that the ability of ACh to inhibit ICa,l was not significantly different when ICa,l was stimulated by either ISO-β1-ARs or FEN-β2-ARs. The numbers in parentheses indicate the number of cells studied. * P < 0.05 comparing ISO-β2-AR vs. ISO-β1-AR or FEN-β2-AR responses. #P < 0.05 comparing ZIN-β2-AR vs. ISO-β1-AR or FEN-β2-AR responses.
Previous results indicate that in cat atrial myocytes, intracellular NO release is regulated differentially by stimulation of different β-AR subtypes (Wang et al. 2002). We therefore examined whether the NO that is activated by β-AR stimulation influences ACh-induced inhibition of stimulated ICa,l. The data shown in Fig. 2 indicate that the ability of ACh to inhibit ICa,l is essentially the same when ICa,l is stimulated by either ISO-β2-ARs or ZIN-β2-ARs. Therefore, because of its greater selectivity for β2-ARs, of the two agonists only ZIN was used in the following experiments. We tested the effects of ACh on ICa,l stimulated by ISO-β1-ARs, ZIN-β2-ARs or FEN-β2-ARs in the absence and presence of 10 μml-NIO, an inhibitor of endothelial NO synthase (eNOS; Rees et al. 1990). The graph in Fig. 3 summarizes both the stimulatory effects of each β-AR on ICa,l (upper bars) and the inhibitory effects of ACh on ICa,l stimulated by each agonist (lower bars) in the absence (open bars) and presence (hatched bars) of l-NIO. l-NIO alone had little effect on stimulation of ICa,l induced by ISO-β1-AR or FEN-β2-AR stimulation, indicating that NO signalling does not participate in the effects of these agonists to stimulate ICa,l. However, l-NIO attenuated the stimulation of ICa,l induced by stimulation of ZIN-β2-ARs (ZIN, 160 ± 31 % vs. ZIN + l-NIO, 95 ± 19 %). These findings are consistent with the idea that ZIN acts via NO signalling to partially stimulate ICa,l (Wang et al. 2002). Moreover, these results agree with previous findings that ZIN-β2-ARs stimulate ICa,l via two cAMP-dependent signalling pathways; Gs-/adenylate cyclase signalling and Gi-/PI-3K/NO signalling (Wang et al. 2002). l-NIO had little effect on ACh-induced inhibition of ICa,l stimulated by either ISO-β1-ARs or FEN-β2-ARs. However, l-NIO significantly enhanced ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs (ACh, −14 ± 3 % vs. ACh + l-NIO, −65 ± 7 %; P < 0.001). Note that in the presence of l-NIO, ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs was similar in magnitude to that obtained when ICa,l was stimulated by ISO-β1-ARs or FEN-β2-ARs. These findings are consistent with the idea that NO signalling induced by ZIN-β2-ARs is responsible for the inability of ACh to effectively inhibit ICa,l stimulated by ZIN-β2-ARs. In contrast, stimulation of ISO-β1-ARs or FEN-β2-ARs does not induce NO signalling, and therefore the inhibitory effects of ACh are unaffected by inhibition of eNOS. This latter finding also indicates that NO signalling is not involved in ACh-induced inhibition of ICa,l stimulated by these β-AR agonists.
Figure 3. Summary of the effects of each β-AR agonist to stimulate ICa,l and ACh-induced inhibition of ICa,l prestimulated by each β-AR agonist in the absence (open bars) and presence (hatched bars) of 10 μml-N5-(1-iminoethyl)ornithine (l-NIO).
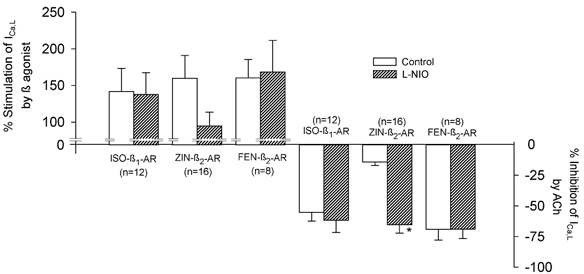
l-NIO had no significant effect on ISO-β1-AR- or FEN-β2-AR-induced stimulation of ICa,l, and l-NIO attenuated ZIN-β2-AR-induced stimulation of ICa,l. Likewise, l-NIO had no significant effect on ACh-induced inhibition of ICa,l stimulated by either ISO-β1-ARs or FEN-β2-ARs, but l-NIO significantly enhanced the ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs. The numbers in parentheses indicate the number of cells studied. * P < 0.05.
To examine further the role of endogenous NO release, we tested the effects of haemoglobin, a potent NO scavenger, on ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs. Our previous studies have shown that in cat atrial myocytes extracellular haemoglobin abolishes the regulation of ICa,l mediated by endogenous NO signalling (Wang et al. 1998). As shown in Fig. 4A, in the absence of haemoglobin, 1 μm ACh elicited a typical inhibition of ICa,l (-36 %) stimulated by ZIN-β2-ARs. In another atrial cell from the same heart (Fig. 4B), the presence of 50 μm haemoglobin markedly enhanced the ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs (-112 %). In this experiment, the presence of haemoglobin allowed ACh to completely inhibit the ZIN-β2-AR-mediated stimulation of ICa,l as well as some basal ICa,l. The data shown in Fig. 4A and B were chosen from two cells in which the amplitudes of initial basal ICa,l and the ZIN-β2-AR-stimulated ICa,l were approximately the same in the absence and presence of haemoglobin. This was done to illustrate that the greater inhibitory effect of ACh on ICa,l in the presence of haemoglobin was not due to any differences in basal ICa,l or ZIN-stimulated ICa,l amplitude. In total, the ACh-induced inhibition of ZIN-stimulated ICa,l was significantly larger in the presence (-85 ± 9 %; n = 8) than in the absence (-36 ± 5 %; n = 5) of haemoglobin (P < 0.05). In contrast, additional experiments showed that ACh-induced inhibition of ICa,l stimulated by FEN-β2-ARs was not significantly different in the absence (-58 ± 11 %; n = 4) or presence (-54 ± 3 %; n = 4) of 50 μm haemoglobin (data not shown). These findings further establish that stimulation of ZIN-β2-ARs but not FEN-β2-ARs acts via NO signalling to prevent ACh-induced inhibition of ICa,l.
Figure 4. ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs in the absence (A) and presence (B) of 50 μm haemoglobin.
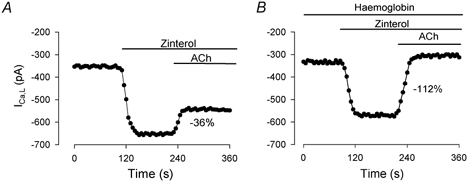
A, in the absence of haemoglobin, ACh induced a typical inhibition of ICa,l (-36 %) stimulated by ZIN-β2-ARs. B, in another atrial cell, the presence of haemoglobin enhanced the ACh-induced inhibition of ICa,l (-112 %) stimulated by ZIN-β2-ARs. Data from these two cells were selected to illustrate that the effects of ACh cannot be attributed to significant differences in basal ICa,l or ZIN-β2-AR-stimulated ICa,l amplitudes.
A primary target of NO is guanylate cyclase (and thus the production of cGMP). Therefore, to determine the potential role of downstream cGMP signalling, we tested the effects of ACh on ICa,l stimulated by ZIN-β2-ARs in the absence and presence 10 μm ODQ, an inhibitor of guanylate cyclase (Brunner et al. 1996). Our previous work has shown that in cat atrial myocytes, ODQ inhibits NO signalling mechanisms that are mediated via cGMP (Wang et al. 1998). As shown in Fig. 5A, ODQ alone significantly decreased ZIN-mediated stimulation of ICa,l (ZIN, 206 ± 33 % vs. ZIN + ODQ, 80 ± 12 %). Again, this is consistent with previous findings that the stimulatory effects of ZIN on ICa,l are mediated partially via NO-cGMP-induced inhibition of phosphodiesterase (PDE) type III with the resulting increase in cAMP (Wang et al. 2002). However, there was no significant difference between the inhibitory effects of ACh on ZIN-stimulated ICa,l in the absence (-22 ± 3 %) or presence (-26 ± 5 %) of ODQ. These findings suggest that NO signalling induced by ZIN is operating to prevent the effects of ACh via a cGMP-independent mechanism.
Figure 5. ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs in the absence (open bars) and presence (hatched bars) of 1H-[1,2,4] oxadiazolo[4,3-a]quinoxaline-1-one (ODQ; A) and glutathione (B).
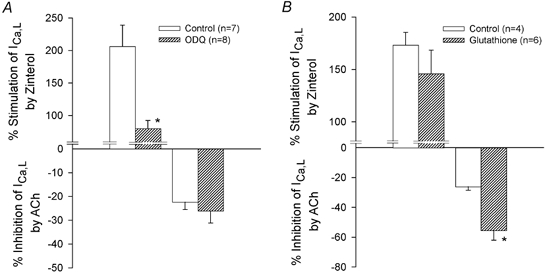
A, compared with control, 10 μm ODQ attenuated the stimulation of ICa,l by ZIN-β2-ARs. However, ODQ did not significantly affect the ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs. B, compared with control, 500 μm glutathione had no significant effect on ICa,l stimulated by ZIN-β2-ARs. However, glutathione significantly enhanced ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs. * P < 0.05.
Alternatively, NO can alter a wide variety of cellular functions via S-nitrosylation (cGMP-independent) reactions (Broillet, 1999; Stamler et al. 2001). To examine this possibility, we tested the effects of ACh to inhibit ICa,l stimulated by ZIN-β2-ARs in the absence and presence of reduced glutathione (GSH), a sink for S-nitrosylation by NO. Exposure to 500 μm GSH alone had no effect on basal ICa,l and no significant effect on ZIN-stimulated ICa,l (Fig. 5B). However, as shown in Fig. 5B, GSH significantly enhanced ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs (ACh, −26 ± 2 % vs. ACh + GSH, −56 ± 6 %; P < 0.05). Similar results were obtained with 100 μm dithiothreitol (DTT), another agent that acts as a sink for S-nitrosylation (ACh, −20 ± 2 %, n = 5, vs. ACh + DTT, −57 ± 3 %, n = 8; P < 0.05). These findings suggest that the NO released by ZIN-β2-AR stimulation is acting primarily via an S-nitrosylation mechanism to prevent ACh-induced inhibition of ICa,l.
In the following experiments, we tested the ability of ACh to inhibit ICa,l when ICa,l was stimulated by exogenous NO (i.e. without β2-AR stimulation). In cat atrial myocytes, inhibition of PDE activity by milrinone elicits a prominent activation of cAMP-dependent stimulation of ICa,l (Wang & Lipsius, 1995). In both cat (Wang et al. 1998) and human (Kirstein et al. 1995) atrial myocytes, NO stimulates ICa,l via increases in endogenous cAMP generated by cGMP-mediated inhibition of PDE III. Therefore, ICa,l was stimulated by inhibiting PDE III using either 100 μm SNO, an NO donor, or 5 μm milrinone, a specific PDE III inhibitor. As shown in Fig. 6A, when ICa,l was stimulated by milrinone (89 %), ACh elicited a prominent inhibition of ICa,l (-86 %). However, in another atrial myocyte from the same heart (Fig. 6B), when ICa,l was stimulated by SNO (98 %), ACh-induced inhibition was markedly smaller (-32 %). In total, stimulation of ICa,l elicited by SNO (98 ± 8 %; n = 8) and milrinone (112 ± 5 %; n = 8) was similar. However, ACh-induced inhibition of ICa,l was significantly smaller when ICa,l was stimulated by SNO (-29 ± 5 %) compared with milrinone (-73 ± 8 %; P < 0.001). These experiments could be interpreted as further evidence that NO is responsible for preventing the inhibitory effects of ACh. However, an alternative explanation is that ACh is simply not able to effectively inhibit the stimulatory effects of NO signalling on ICa,l (see Discussion).
Figure 6. ACh-induced inhibition of ICa,l when ICa,l is stimulated by either 5 μm milrinone (A) or 100 μm spermine/nitric oxide (NO; B).

Graphs show consecutive measurements of peak ICa,l amplitude throughout each experiment. During stimulation of ICa,l by milrinone, 1 μm ACh induced a prominent inhibition of ICa,l (-86 %). In another atrial cell from the same heart, although spermine/NO elicited a comparable stimulation of ICa,l, exposure to 1 μm ACh elicited a markedly smaller inhibition of ICa,l (-32 %).
So far, the results indicate that NO signalling prevents the ACh-induced inhibition of ICa,l selectively stimulated by ISO-β2-ARs or ZIN-β2-ARs. In addition, ACh-induced inhibition of ISO-β1-AR stimulation is prominent because this signalling pathway fails to release NO. This raises the question of whether the NO released by ISO-β2-ARs can prevent the ability of ACh to inhibit ISO-β1-AR stimulation. This question was approached by testing the effects of ACh on ICa,l stimulated by ISO, a mixed β1/β2-AR agonist. Our previous work has shown that ISO elicits NO release via activation of β2-ARs (Wang et al. 2002). As expected, 0.1 μm ISO markedly increased ICa,l (213 %), and 1 μm ACh prominently inhibited ICa,l (-57 %; Fig. 7A). Based on the present results, this prominent ACh-induced inhibition of ICa,l stimulated by ISO-β1/β2-ARs is due primarily to the effect of ACh on ISO-β1-AR signalling. Moreover, as shown in Fig. 7B, in another atrial myocyte, exposure to 10 μml-NIO attenuated ISO-β1/β2-AR stimulation of ICa,l (127 %). This confirms previous findings that ISO acts via NO signalling to partially stimulate ICa,l (Wang et al. 2002). In addition, ACh-induced inhibition of ICa,l was enhanced (-78 %) when NO release was blocked. As summarized in Fig. 7C, compared with the control, l-NIO decreased stimulation of ICa,l by ISO-β1/β2-ARs (ISO, 355 ± 69 % vs. ISO + l-NIO, 183 ± 21 %), and ACh-induced inhibition of ICa,l was significantly enhanced (ACh, −45 ± 5 % vs. ACh + l-NIO, −74 ± 5 %; P < 0.002). These findings suggest that the NO released by ISO-β2-AR stimulation does not prevent the ability of ACh to inhibit ISO-β1-AR stimulation. Moreover, they support the idea that NO acts preferentially to prevent ACh-induced inhibition of ICa,l stimulated by β2-ARs. In addition, the fact that ACh could still inhibit β1-AR stimulation even though ISO releases NO, makes it unlikely that NO is acting to inhibit muscarinic receptor function.
Figure 7. Effects of ACh on ISO-stimulated ICa,l in the absence (A) and presence (B) of 10 μml-NIO.
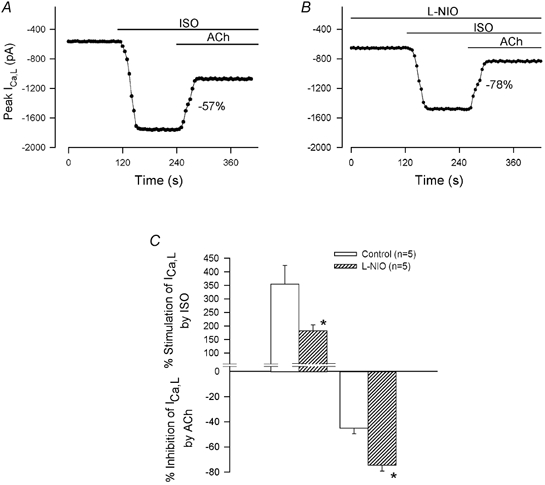
A, in the absence of l-NIO, ACh elicited a prominent inhibition of ISO-stimulated ICa,l (-57 %). B, in another atrial myocyte from the same heart, l-NIO attenuated ISO-induced stimulation of ICa,l and enhanced ACh-induced inhibition of ISO-stimulated ICa,l (-78 %). C, summary of the data shows that l-NIO significantly decreased ISO-induced stimulation of ICa,l and significantly enhanced ACh-induced inhibition of ISO-stimulated ICa,l. * P < 0.05.
The present experiments also indicate that although FEN acts via β2-ARs, it preferentially activates a signalling pathway similar to that activated by β1-ARs (i.e. Gs-/adenylate cyclase). If this is correct, then the combined stimulation of FEN-β2-ARs plus ZIN-β2-ARs should behave like ISO, a mixed β1/β2-AR agonist. In other words, ACh should elicit a prominent inhibition of FEN-induced stimulation of ICa,l even though ZIN-β2-AR stimulation releases NO. In fact, simultaneous exposure to 0.1 μm FEN plus 1 μm ZIN increased ICa,l (188 ± 75 %), and ACh prominently inhibited ICa,l (-47 ± 6 %, n = 2; data not shown). In cells from the same heart, l-NIO attenuated the stimulation of ICa,l by FEN + ZIN (107 ± 26 %) and enhanced the ACh-induced inhibition of ICa,l (-75 ± 7 %; n = 3).
The present results indicate that different β-AR subtypes, as well as different β-AR agonists acting on the same β2-AR subtype, differentially regulate NO release. To examine this idea directly, we measured endogenous [NO]i to determine the effect of each type of β-AR agonist. The effect of 10 μm ZIN-β2-AR stimulation was to increase [NO]i (Fig. 8A), whereas 1 μm ISO-β1-AR (Fig. 8B) and 1 μm FEN-β2-AR stimulation (Fig. 8C) each failed to increase [NO]i. Previous work has shown that ISO alone releases NO and that the ability of ISO and ZIN to release NO is abolished by inhibition of eNOS (l-NIO) or by blocking β2-ARs (ICI 118,551; Wang et al. 2002). Note also that although the concentrations of FEN and ISO used in these experiments were 10 times higher than those used in the electrophysiology experiments, they failed to release NO. Figure 8D summarizes the effects of each β-AR agonist on [NO]i. Additional experiments showed that simultaneous exposure to FEN + ZIN increased [NO]i essentially the same as ZIN-β2-AR stimulation alone (n = 3; data not shown). Together, the present findings support the idea that the differential ability of ACh to inhibit ICa,l depends upon whether the β-AR agonist used to stimulate ICa,l is capable of releasing NO.
Figure 8. Measurements of intracellular NO ([NO]i) in response to stimulation by ZIN-β2-ARs (A), ISO-β1-ARs (B) and FEN-β2-ARs (C).
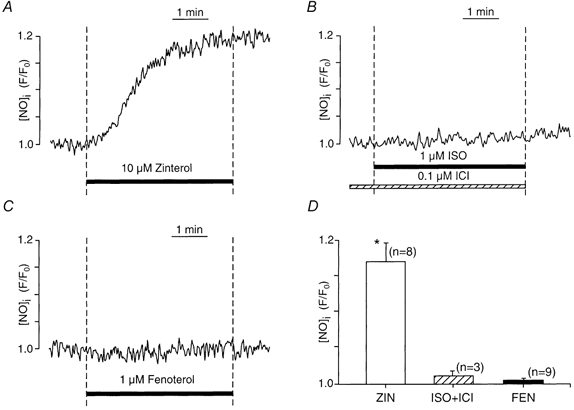
A, stimulation of ZIN-β2-ARs increased [NO]i. B, stimulation of ISO-β1-ARs failed to increase [NO]i. C, stimulation of FEN-β2-ARs also failed to increase [NO]i. D, summary showing the effects of each β-AR agonist on [NO]i. ZIN-β2-AR (open bar); ISO-β1-AR (hatched bar); FEN-β2-AR (filled bar). Numbers in parentheses indicate the number of cells tested. * P < 0.05.
Discussion
In general, stimulation of muscarinic receptors elicits an accentuated inhibition of β-AR stimulation of the heart. The present results indicate that in cat atrial myocytes, the magnitude of ACh-induced inhibition of β-AR-stimulated ICa,l differs significantly depending upon which β-AR subtype and agonist stimulates ICa,l. In general, ACh induces a significantly greater inhibition when ICa,l is stimulated by β1-AR agonists compared with β2-AR agonists. These findings basically agree with those reported in neonatal rat ventricular myocytes in which β1-AR stimulation of cAMP and inotropic activity is more susceptible than β2-AR stimulation to inhibition by muscarinic receptor stimulation (Aprigliano et al. 1997). A major finding of the present study is that in adult atrial myocytes, the mechanism responsible for these disparate effects of ACh-induced inhibition is the release of NO by selective β2-AR stimulation. In other words, the magnitude of ACh-induced inhibition correlated directly with whether a specific β-AR agonist released NO. Indeed, ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs or ISO-β2-ARs were comparably small, and as shown here and in our previous work (Wang et al. 2002), both of these β2-AR agonists release NO. On the other hand, ACh-induced inhibition of ICa,l was prominent when ICa,l was stimulated by either ISO-β1-ARs or FEN-β2-ARs, and both of these agonists fail to release NO. It is worth emphasizing that the inhibitory effects of ACh on ICa,l differed markedly when ICa,l was stimulated via the same β2-AR subtype but by different β2-AR agonists. The fact that stimulation of ZIN-β2-ARs releases NO and FEN-β2-ARs does not release NO strongly supports the idea that the primary mechanism responsible for the differential inhibitory effects of ACh is not stimulation of β2-ARs per se, but rather whether a particular β2-AR agonist releases NO. The functional role of endogenous NO release was further demonstrated by the findings that inhibition of NO release (l-NIO) enhanced the ability of ACh to inhibit ICa,l stimulated by ZIN-β2-ARs but not when ICa,l was stimulated by either ISO-β1-ARs or FEN-β2-ARs. Likewise, when NO signalling was prevented by haemoglobin, ACh-induced inhibition of ICa,l was enhanced when ICa,l was stimulated by ZIN-β2-ARs but not when stimulated by FEN-β2-ARs. We therefore conclude that NO release by specific β2-AR agonists is responsible for preventing the inhibitory effects of ACh on β2-AR stimulation of ICa,l.
Our previous work indicates that ZIN-β2-AR stimulation acts via two parallel signalling pathways to generate cAMP-dependent stimulation of ICa,l; Gs-/adenylate cyclase signalling and Gi-/PI-3K/NO signalling (Wang et al. 2002; see Fig. 9). The present results indicate that when ICa,l was stimulated by ZIN-β2-ARs, NO released via the Gi-signalling pathway prevented ACh-induced inhibition of ICa,l stimulated by the Gs-signalling pathway (Fig. 9). Thus, when ZIN-β2-AR-mediated NO signalling was blocked, the enhanced inhibitory effect of ACh now resulted from inhibition of the remaining Gs-/adenylate cyclase signalling. This is consistent with the prominent ACh-induced inhibition of β1-AR stimulation, which is mediated exclusively via Gs-/adenylate cyclase signalling. The present results also indicate that NO released by β2-AR stimulation preferentially prevents the ability of ACh to inhibit β2-AR but not β1-AR stimulation. Thus, ISO stimulates both β1- and β2-ARs and releases NO via β2-AR signalling (Wang et al. 2002). Even though ISO releases NO, ACh elicited a prominent inhibition of ISO-stimulated ICa,l. Based on the present study, this prominent effect of ACh is mediated primarily via inhibition of β1-AR signalling. This idea is supported by the finding that when NO signalling was blocked the inhibitory effects of ACh were enhanced, presumably via the additional ACh-induced inhibition of β2-AR signalling. In addition, the fact that ACh could elicit a prominent inhibition of ISO-stimulated ICa,l even though ISO releases NO suggests that NO is not exerting a general inhibition of muscarinic receptor function. Rather, the fact that NO preferentially prevents ACh-induced inhibition of β2-AR signalling and not β1-AR signalling suggests that endogenous NO released by β2-ARs acts locally near the β2-AR to modulate the inhibitory effect of muscarinic receptor signalling (Fig. 9). Our previous work also has shown that in atrial myocytes, NO signalling either induced by β2-ARs or by exposure to exogenous NO acts locally to regulate ACh-induced activation of K+ channels (Wang et al. 2002). Both of these findings are consistent with local regulation by NO signalling (Dittrich et al. 2001). In addition, β2-ARs (but not β1-ARs) and eNOS are both localized within caveolae (Steinberg & Brunton, 2001), providing a possible substrate for the present findings that β2-AR, but not β1-AR function may be regulated locally by NO signalling.
Figure 9. Schematic diagram of the proposed signalling mechanisms responsible for muscarinic receptor-mediated inhibition of β1-AR- and β2-AR-stimulated ICa,l.
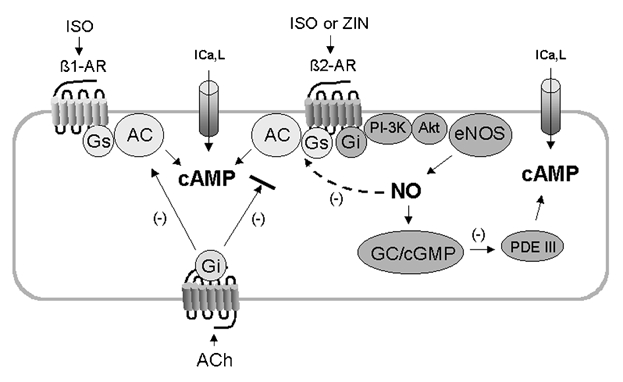
ISO stimulates both β1-ARs and β2-ARs via Gs-adenylate cyclase (AC)/cAMP signalling to increase ICa,l. In addition, ISO or ZIN act via β2-ARs and Gi-/PI-3K/Akt signalling to activate endothelial nitric oxide synthase (eNOS) and NO release (Wang et al. 2002). NO acts via guanylate cyclase (GC)/cGMP-inhibition of phosphodiesterase (PDE) III to raise endogenous cAMP and stimulate ICa,l. ACh acts via muscarinic receptors coupled to Gi-protein to inhibit AC activated by β1-AR/Gs-signalling. However, ACh is unable to effectively inhibit β2-AR/Gi-NO signalling. In addition, NO released by β2-AR stimulation acts locally via S-nitrosylation to decrease β2-AR/Gs-/AC signalling. As a result, β2-AR signalling is due primarily to Gi-NO signalling, which is not effectively inhibited by ACh. Not shown in this diagram is the effect of FEN. Although FEN acts via β2-ARs, it preferentially stimulates only the Gs-/AC signalling pathway and therefore does not release NO. As a result, β2-AR/Gs-/AC signalling is not depressed and therefore ACh-induced inhibition of ICa,l stimulated by FEN is prominent, similar to that obtained with stimulation of ICa,l via β1-ARs.
Several of the present results indicate that FEN acts specifically via β2-ARs to activate a cAMP signalling pathway similar to β1-ARs. First, the effects of ISO-β1-AR and FEN-β2-AR stimulation were essentially the same (i.e. both increased ICa,l to the same extent, both failed to activate NO release and both of their stimulatory effects on ICa,l were prominently inhibited by ACh). In addition, FEN-mediated stimulation of ICa,l was abolished by specific blockade of β2-ARs, and by specific inhibition of cAMP-dependent PKA activity. As mentioned earlier, β1-ARs act exclusively via Gs-/adenylate cyclase signalling. In cat atrial myocytes, β2-ARs act via Gs-/adenylate cyclase signalling and via Gi-/PI-3K signalling to release NO (Wang et al. 2002). This explains why β1-AR stimulation fails to release NO. Moreover, the present results lead to the conclusion that FEN is unable to stimulate NO release because it fails to activate Gi-protein signalling and acts preferentially to stimulate Gs-/adenylate cyclase signalling. The present findings also show that when ICa,l was stimulated simultaneously by FEN + ZIN, ACh-induced inhibition was prominent even though NO was released by ZIN. This suggests that FEN-β2-AR signalling is not regulated locally by the NO released by ZIN-β2-ARs. Similar results were obtained when ICa,l was stimulated by ISO, a mixed β1/β2-AR agonist. The idea that FEN stimulates a β2-AR exclusively via Gs-/adenylate cyclase signalling can be explained by either: (1) two separate subclasses of β2-ARs or (2) preferential activation of Gs- over Gi-protein signalling via a single class of β2-ARs. The latter idea is consistent with agonist-directed trafficking of receptor signalling (Kenakin, 1995). In general, agonist trafficking of receptor responses is defined as the ability of different agonists, acting on a single receptor that couples to more than one G-protein, to preferentially activate a particular G-protein signalling pathway. The underlying theory is that different types of agonist can induce and/or select different receptor conformations (see also Kukkonen et al. 2001). This phenomenon has been reported for a variety of receptor-signalling mechanisms, some of which include 5-HT receptor coupling to phospholipases C and A2 (Berg et al. 1998), differential activation of Gs- and Gi-signalling by different cannabinoid receptor agonists (Bonhaus et al. 1998), and differential coupling of α2A-ARs to Gs- and Gi-mediated regulation of adenylate cyclase (Brink et al. 2000).
A primary target of NO signalling is guanylate cyclase (and thus the production of cGMP). The present results, however, indicate that ACh-induced inhibition of ICa,l stimulated by ZIN-β2-ARs was unaffected by blocking guanylate cyclase with ODQ. Alternatively, reduced glutathione or dithiothreitol, two agents that act as a sink for S-nitrosylation by NO, significantly enhanced ACh-induced inhibition of ICa,l when stimulated by ZIN-β2-ARs. These findings suggest that NO is preventing the inhibitory effects of ACh primarily via a cGMP-independent, S-nitrosylation reaction. Because S-nitrosylation can modulate a wide variety of cellular functions (Broillet, 1999; Stamler et al. 2001) its specific site of action remains unclear. However, as mentioned earlier, the fact that ACh can prominently inhibit ISO-stimulated ICa,l even though ISO releases NO, makes it unlikely that muscarinic receptor signalling is the site of NO action. Alternatively, NO can act via S-nitrosylation to decrease β2-AR/Gs-signalling (Adam et al. 1999) and to inhibit adenylate cyclase activity (McVey et al. 1999). Therefore, based on the present data, we propose the following: stimulation of β2-ARs acts via both Gs- and Gi-signalling pathways. ACh is unable to effectively inhibit the β2-AR/Gi-pathway that acts via NO/ cGMP/cAMP signalling (Fig. 6). Moreover, NO released via β2-AR/Gi-signalling acts via S-nitrosylation to inhibit the β2-AR/Gs-/adenylate cyclase signalling pathway (Fig. 9). Therefore, the cAMP signalling pathway activated by β2-AR stimulation is mediated primarily via Gi-/NO signalling. As a result, ACh is unable to effectively inhibit β2-AR stimulation of ICa,l (Fig. 9). However, when NO signalling is blocked, β2-AR stimulation is now mediated primarily via Gs-adenylate cyclase signalling, which is effectively inhibited by muscarinic receptor stimulation.
In the present experiments, the inhibitory effects of ACh were determined as a percentage of the ICa,l amplitude stimulated by each β-AR agonist. By this method, a smaller or larger β-AR-stimulated increase in ICa,l amplitude could result in a larger or smaller calculated percentage inhibition induced by ACh. Several results, however, argue against the idea that this can account for the effects of ACh presented here. For example, ZIN elicited the smallest stimulation of ICa,l and yet ACh exerted the smallest (rather than the largest) inhibition of ICa,l stimulated by ZIN-β2-ARs (Fig. 2). Conversely, stimulation of either ISO-β1-ARs or FEN-β2-ARs elicited the largest stimulation of ICa,l and yet the effects of ACh also were largest (rather than smallest) when ICa,l was stimulated by these agonists. Although ODQ significantly decreased ZIN-mediated stimulation of ICa,l, the inhibitory effects of ACh were not different in the presence or absence of ODQ (Fig. 5). On the other hand, stimulation of ICa,l by SNO and milrinone were similar, and yet the effects of ACh were significantly smaller when ICa,l was stimulated by SNO compared with milrinone (Fig. 6). We also selected data in which the stimulated amplitude of ICa,l between two different cells was approximately the same and yet the effects of ACh in the presence of haemoglobin were markedly enhanced compared with those in absence of haemoglobin (Fig. 4). Finally, the relationship between ICa,l amplitudes stimulated by each β-AR agonist and the percentage changes induced by ACh showed no significant correlation.
Functionally, the present results suggest that the NO released by specific β2-AR agonists prevents ACh-induced inhibition of β2-AR stimulation. Of course, noradrenaline is the primary neurotransmitter of sympathetic nerve activity and it acts primarily via β1-AR signalling, with limited β2-AR activity. In addition, the heart, including cat atria (Hedberg et al. 1980), exhibit a lower density of β2-ARs than β1-ARs, although the density of β2-ARs in human atria is higher than in ventricular muscle (Stiles et al. 1983). β2-AR signalling may therefore be a normal second line of sympathetic cardiac support. However, the relative contribution of different receptor subtypes to β-AR signalling in the heart is not static. In cat atrial myocytes, stimulation of integrin-mediated signalling dramatically decreases β1-AR and increases β2-AR signalling mechanisms, which regulate ICa,l (Wang et al. 2000). In addition, the failing human heart exhibits a pronounced decrease in the ratio of β1: β2-ARs (Bristow et al. 1986). These considerations make it possible that under various pathological conditions, β2-AR signalling assumes significantly greater importance. Moreover, evidence is accumulating that β2-AR stimulation is beneficial. For example, β2-AR stimulation acts via Gi-/PI-3K signalling to inhibit apoptosis (Communal et al. 1999; Chesley et al. 2000), while β1-AR stimulation promotes apoptosis of cardiac myocytes (Communal et al. 1999). Interestingly, stimulation of muscarinic receptors inhibits the ability of β1-AR stimulation to induce apoptosis (Communal et al. 1999). Endogenous NO release also exerts cardioprotective effects (Ping et al. 1999). It should be noted that in cat atrial myocytes, stimulation of β2-ARs acts via Gi-/PI-3K signalling to elicit endogenous NO release (Wang et al. 2002). We therefore speculate that the mechanisms presented here may prevent muscarinic receptor stimulation from inhibiting the potentially beneficial effects of β2-AR stimulation, while the potentially deleterious effects of tonic or chronic β1-AR stimulation may be inhibited by muscarinic receptor stimulation. As a corollary, this mechanism may contribute to the beneficial effects of high vagal tone on the heart. In addition, the finding that FEN acts via β2-ARs to preferentially activate a β1-AR-like signalling pathway may be relevant to the development of pharmacological β2-AR agonists that can preferentially activate specific signalling mechanisms.
Acknowledgments
We thank Ms Holly Gray for her technical assistance with these experiments. Financial support was provided by National Institutes of Health grants HL27652 and HL63753 (S.L.L.) and HL51941 and HL62231 (L.A.B.).
References
- Adam L, Bouvier M, Jones TLZ. Nitric oxide modulates β2-adrenergic receptor palmitoylation and signaling. Journal of Biological Chemistry. 1999;274:26337–26343. doi: 10.1074/jbc.274.37.26337. [DOI] [PubMed] [Google Scholar]
- Aprigliano O, Rybin VO, Pak E, Robinson RB, Steinberg SF. β1- and β2-adrenergic receptors exhibit differing susceptibility to muscarinic accentuated antagonism. American Journal of Physiology. 1997;272:H2726–2735. doi: 10.1152/ajpheart.1997.272.6.H2726. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Harvey RD. Muscarinic inhibitory and stimulatory regulation of the L-type Ca2+ current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. Journal of Physiology. 2000;528:279–289. doi: 10.1111/j.1469-7793.2000.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Molecular Pharmacology. 1998;54:94–104. [PubMed] [Google Scholar]
- Bonhaus DW, Chang LK, Kwan J, Martin GR. Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. Journal of Pharmacology and Experimental Therapeutics. 1998;287:884–888. [PubMed] [Google Scholar]
- Brink CB, Wade SM, Neubig RR. Agonist-directed trafficking of porcine α2A-adrenergic receptor signaling in Chinese hamster ovary cells: I-isoproterenol selectively activates Gs. Journal of Pharmacology and Experimental Therapeutics. 2000;294:539–547. [PubMed] [Google Scholar]
- Bristow MR, Ginsberg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S, Stinson EB. β1- and β2-adrenergic-receptor subpopulation in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circulation Research. 1986;59:297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- Broillet MC. S-Nitrosylation of proteins. Cellular and Molecular Life Sciences. 1999;55:1036–1042. doi: 10.1007/s000180050354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner F, Schmidt K, Nielsen EB, Mayer B. Novel guanylyl cyclase inhibitor potently inhibits cyclic GMP accumulation in endothelial cells and relaxation of bovine pulmonary artery. Journal of Pharmacology and Experimental Therapeutics. 1996;277:48–53. [PubMed] [Google Scholar]
- Chesley A, Lundber MS, Asai T, Xiao RP, Ohtani S, Lakatta EG, Crow MT. The β2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphatidylinositol 3′-kinase. Circulation Research. 2000;87:1172–1179. doi: 10.1161/01.res.87.12.1172. [DOI] [PubMed] [Google Scholar]
- Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- Dittrich M, Jurevicius J, Georget M, Rochias F, Fleischmann J, Hescheler J, Fischmeister R. Local response of L-type Ca2+ current to nitric oxide in frog ventricular myocytes. Journal of Physiology. 2001;534:109–121. doi: 10.1111/j.1469-7793.2001.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godecke A, Heinicke T, Kamkin A, Kiseleva I, Strasser RH, Decking UKM, Stumpe T, Isenberg G, Schrader J. Inotropic response to β-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. Journal of Physiology. 2001;532:195–204. doi: 10.1111/j.1469-7793.2001.0195g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Han X, Kubota I, Feron O, Opel DJ, Arstall MA, Zhao YY, Huang P, Fishman MC, Michel T, Kelly RA. Muscarinic cholinergic regulation of cardiac myocyte ICa-l is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proceedings of the National Academy of Sciences of the USA. 1998;95:6510–6515. doi: 10.1073/pnas.95.11.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg A, Minneman KP, Molinoff PB. Differential distribution of beta-1 and beta-2 adrenergic receptors in cat and guinea-pig heart. Journal of Pharmacology and Experimental Therapeutics. 1980;212:503–508. [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. Journal of General Physiology. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy II: agonist trafficking of receptor signals. Trends in Pharmacological Sciences. 1995;6:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kilts JD, Gerhardt MA, Richerdson MD, Sreeram G, MacKensen GB, Grocott HP, White WD, Davis RD, Newman MF, Reves JG, Schwinn DA, Kwatra MM. β2-adrenergic and several other G protein-coupled receptors in human atrial membranes activate both Gs and Gi. Circulation Research. 2000;87:705–709. doi: 10.1161/01.res.87.8.705. [DOI] [PubMed] [Google Scholar]
- Kirstein M, Rivet-Bastide M, Hatem S, Benardeau A, Mercadier JJ, Fischmeister R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. Journal of Clinical Investigation. 1995;95:794–802. doi: 10.1172/JCI117729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Nakatsubo N, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, Hirata Y, Nagano T. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Analytical Chemistry. 1998;70:2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- Kukkonen JP, Nasman J, Akerman KEO. Modelling of promiscuous receptor-Gi/Gs-protein coupling and effector response. Trends in Pharmacological Sciences. 2001;22:616–622. doi: 10.1016/s0165-6147(00)01864-2. [DOI] [PubMed] [Google Scholar]
- Kuschel M, Zhou YY, Cheng H, Zhang SJ, Chen Y, Lakatta EG, Xiao RP. Gi protein-mediated functional compartmentalization of cardiac β2-adrenergic signaling. Journal of Biological Chemistry. 1999;274:22048–22052. doi: 10.1074/jbc.274.31.22048. [DOI] [PubMed] [Google Scholar]
- McVey M, Hill J, Howlett A, Klein C. Adenylyl cyclase, a coincidence detector for nitric oxide. Journal of Biological Chemistry. 1999;274:18887–18892. doi: 10.1074/jbc.274.27.18887. [DOI] [PubMed] [Google Scholar]
- Nakatsubo N, Kojima H, Kikuchi K, Nagoshi H, Hirata Y, Maeda D, Imai Y, Irimura T, Nagano T. Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: diaminofluoresceins. FEBS Letters. 1998;427:263–266. doi: 10.1016/s0014-5793(98)00440-2. [DOI] [PubMed] [Google Scholar]
- Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, Banerjee S, Dawn B, Balafnova Z, Bolli R. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circulation Research. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- Rees DD, Palmer RMJ, SchulZ R, Hodson H, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. British Journal of Pharmacology. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. The molecular basis for distinct β-adrenergic receptor subtype actions in cardiomyocytes. Circulation Research. 1999;85:1101–1111. doi: 10.1161/01.res.85.11.1101. [DOI] [PubMed] [Google Scholar]
- Steinberg SF, Brunton LL. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annual Review of Pharmacology and Toxicology. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- Stiles GL, Taylor S, LefkowitZ RJ. Human cardiac beta-adrenergic receptors: subtype heterogeneity delineated by direct radioligand binding. Life Sciences. 1983;33:467–473. doi: 10.1016/0024-3205(83)90796-8. [DOI] [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, ScholZ H, Stein B, Verde I, Fischmeister R. Muscarinic and β-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nature Medicine. 1999;5:331–334. doi: 10.1038/6553. [DOI] [PubMed] [Google Scholar]
- Wang YG, Dedkova EN, Steinberg SF, Blatter LA, Lipsius SL. β2-Adrenergic receptor signaling acts via NO release to mediate ACh-induced activation of ATP-sensitive K+ current in cat atrial myocytes. Journal of General Physiology. 2002;119:69–82. doi: 10.1085/jgp.119.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YG, Lipsius SL. Acetylcholine elicits a rebound stimulation of Ca2+ current mediated by pertussis toxin-sensitive G protein and cAMP-dependent protein kinase A in atrial myocytes. Circulation Research. 1995;76:634–644. doi: 10.1161/01.res.76.4.634. [DOI] [PubMed] [Google Scholar]
- Wang YG, Rechenmacher CE, Lipsius SL. Nitric oxide signaling mediates stimulation of L-type Ca2+ current elicited by withdrawal of acetylcholine in cat atrial myocytes. Journal of General Physiology. 1998;111:113–125. doi: 10.1085/jgp.111.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YG, Samarel AM, Lipsius SL. Laminin binding to β1-integrins selectively alters β1- and β2-adrenoceptor signalling in cat atrial myocytes. Journal of Physiology. 2000;527:3–9. doi: 10.1111/j.1469-7793.2000.t01-2-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Vereecke J, Carmeliet E, Lipsius SL. Ionic currents activated during hyperpolarization of single right atrial myocytes from cat heart. Circulation Research. 1991;68:1059–1069. doi: 10.1161/01.res.68.4.1059. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Cheng H, Zhou YY, Kuschel M, Lakatta EG. Recent advances in cardiac β2-adrenergic signal transduction. Circulation Research. 1999;85:1092–1100. doi: 10.1161/01.res.85.11.1092. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Ji X, Lakatta EG. Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Molecular Pharmacology. 1995;47:322–329. [PubMed] [Google Scholar]