

Abstract
Mouse mammary C127 cells responded to hypotonic stimulation with activation of the volume-dependent ATP-conductive large conductance (VDACL) anion channel and massive release of ATP. Arachidonic acid downregulated both VDACL currents and swelling-induced ATP release in the physiological concentration range with Kd of 4– 6 μm. The former effect observed in the whole-cell or excised patch mode was more prominent than the latter effect observed in intact cells. The arachidonate effects were direct and not mediated by downstream metabolic products, as evidenced by their insensitivity to inhibitors of arachidonate-metabolizing oxygenases, and by the observation that they were mimicked by cis-unsaturated fatty acids, which are not substrates for oxygenases. A membrane-impermeable analogue, arachidonyl coenzyme A was effective only from the cytosolic side of membrane patches suggesting that the binding site is localized intracellularly. Non-charged arachidonate analogues as well as trans-unsaturated and saturated fatty acids had no effect on VDACL currents and ATP release, indicating the importance of arachidonate's negative charge and specific hydrocarbon chain conformation in the inhibitory effect. VDACL anion channels were inhibited by arachidonic acid in two different ways: channel shutdown (Kd of 4– 5 μm) and reduced unitary conductance (Kd of 13–14 μm) without affecting voltage dependence of open probability. ATP4--conducting inward currents measured in the presence of 100 mm ATP in the bath were reversibly inhibited by arachidonic acid. Thus, we conclude that swelling-induced ATP release and its putative pathway, the VDACL anion channel, are under a negative control by intracellular arachidonic acid signalling in mammary C127 cells.
Extracellular ATP is a well-recognized autocrine and paracrine regulator of a multitude of physiological functions at cellular as well as at organ level (Bodin & Burnstock, 2001). Mechanical stress and osmotic swelling are among the most effective physiological stimuli for ATP release (Burnstock, 1999; Bodin & Burnstock, 2001). Although purinergic receptor proteins located on the plasma membrane are well characterized, the molecular nature of the ATP-releasing pathway remains poorly understood at present. As most ATP molecules exist in anionic forms at physiological pH, it is plausible that some anion channels can conduct ATP, thereby serving as a pathway for ATP release. Indeed, ATP-conducting currents associated with the expression of CFTR (Reisin et al. 1994; Schwiebert et al. 1995; Cantiello et al. 1997,1998; Pasyk & Foskett, 1997; Lader et al. 2000) or MDR1 (Abraham et al. 1993; Bosch et al. 1996; Roman et al. 1999), or independent of CFTR and MDR1 expression (Grygorczyk & Hanrahan, 1997; Sugita et al. 1998; Bodas et al. 2000) have so far been observed. We recently demonstrated that in mouse mammary C127 cells (Hazama et al. 2000b) and human Intestine 407 cells (Hazama et al. 1999) neither CFTR nor volume-sensitive outwardly rectifying (VSOR) Cl− channels were responsible for swelling-induced release of ATP. On the other hand, we demonstrated (Sabirov et al. 2001) that cell swelling activated not only conventional VSOR channels but also another type of anion channel which exhibits a large unitary conductance (≈400 pS), bell-shaped voltage dependence and ATP permeability. This volume- (and voltage-) dependent ATP-conductive large conductance (VDACL) anion channel had a pharmacological profile distinct from that of the VSOR channel, but strikingly similar to that of swelling-induced ATP release, and thus the VDACL channel was proposed to be an ATP-releasing conductive pathway in mammary C127 cells (Sabirov et al. 2001).
At present, little information is available on physiological regulators of swelling-induced ATP release machinery in general and the VDACL channel as a putative pathway of ATP release in particular. Arachidonic acid is an unsaturated fatty acid liberated from membrane phospholipids by phospholipases (Irvine, 1982; Holtzman, 1991; Meves, 1994; Brash, 2001) upon stimulation by a variety of stimuli, including hypotonic stress (Thoroed et al. 1997; Tinel et al. 1997; Basavappa et al. 1998; Pedersen et al. 2000; Hoffmann, 2000). Thus, we tested the hypothesis that the arachidonic acid signalling pathway is involved in the regulation of swelling-induced ATP release mediated by the VDACL channel. Here we report that arachidonic acid, at physiologically relevant concentrations, downregulates both VDACL channel activity and swelling-induced ATP release in C127 cells, a fact providing new evidence that the VDACL channel serves as a pathway for swelling-induced ATP release in C127 cells. Also, we provide evidence that anionic arachidonate exerts a downregulatory effect from the intracellular side of the VDACL anion channel, not only by reducing the number of active channels but also by reducing the unitary conductance of open channels. A negative charge and the cis-conformation of the hydrophobic chain constitute the structural determinants essential for the inhibitory activity of arachidonic acid.
Methods
Cells
A cell line of mouse mammary tissue origin, C127, was cultured as reported previously (Sabirov et al. 2001). For single-channel patch-clamp experiments, the cells were grown on glass coverslips. For whole-cell recordings, cells attached to a plastic substrate were resuspended by mechanical detachment, as reported previously (Kubo & Okada, 1992), and cultured with agitation for 15–300 min. Then cells were placed in a chamber and washed with bathing solution after they attached to the chamber's bottom. For ATP assays, C127 cells were cultured to confluence in 24-well plates.
Solutions
The standard Ringer solution contained (mm): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 Na-Hepes, 6 Hepes, 5 glucose (pH 7.4, 290 mosmol (kg H2O)−1). The hypotonic solution for whole-cell recording was made by reducing the concentration of NaCl in Ringer solution to 100 mm (210 mosmol (kg H2O)−1). Isotonic bath solution for whole-cell experiments was made from this hypotonic solution by adding 80 mm mannitol. The bath (intracellular) solution for inside-out experiments contained (mm): 130 KCl, 10 NaCl, 1 MgCl2, 6 Hepes, 5 Na-Hepes, 4.16 CaCl2, 5 EGTA (pH 7.4 adjusted with KOH, osmolality 302 mosmol (kg H2O)−1). In the present experiments, pCa was buffered at 6.3, although Ca2+ in a range from 15 nm up to 2 mm had no significant effect on VDACL channel behaviour and its inhibition by arachidonic acid (data not shown). The pipette (intracellular) solution for whole-cell experiments contained (mm): 125 CsCl, 2 CaCl2, 1 MgCl2, 5 Hepes, 10 EGTA (pH 7.4 adjusted with CsOH, pCa 7.6, osmolality 275 mosmol (kg H2O)−1). The pipette (extracellular) solution for inside-out experiments was standard Ringer solution. The pipette (intracellular) solution for outside-out experiments contained (mm): 120 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 Hepes, 10 EGTA (pH 7.4 adjusted with NaOH, pCa 7.5, osmolality 275 mosmol (kg H2O)−1). ATP was omitted from intracellular solutions (pipette solutions for whole-cell and outside-out and bath solution for inside-out experiments) in order to avoid the activation of VSOR Cl− currents in whole-cell measurements and to minimize the modulatory effect of ATP on VDACL channels in excised patches (Sabirov et al. 2001).
For ATP-mediated current measurements, 100 mm Na2ATP solution was used after the pH was adjusted to 7.4 with NaOH. The ATP solution was kept on ice and warmed to room temperature immediately before experiments.
Fatty acids were added to their final concentrations just before the experiments from stock solutions (40 mm or 200 mm in DMSO). Indomethacin, nordihydroguaiaretic acid (NDGA) and clotrimazole (all from Sigma) were added from a stock solution of 20 mm in DMSO to their final concentrations just before the experiments. DMSO did not have any effects, when added alone, at the concentrations employed (< 0.1 %).
The osmolality of all solutions was measured using a freezing-point depression osmometer (OM802, Vogel, Germany).
Electrophysiology
Whole-cell, macro-patch and single-channel current recordings were performed, as described in our previous paper (Sabirov et al. 2001), using an Axopatch 200A patch-clamp amplifier coupled to a DigiData 1200 or DigiData 1322A interface (Axon Instruments, Union City, CA, USA). Unless otherwise specified, currents were filtered at 1 kHz and sampled at 1–5 kHz. Data acquisition and analysis were done using pCLAMP 6, Clampex 8.1 (Axon Instruments) and WinASCD software (provided by Dr G. Droogmans, KU Leuven, Belgium). Whenever the bath Cl− concentration was changed, a salt bridge containing 3 m KCl in 2 % agarose was used. All experiments were performed at room temperature (20-25 °C).
Luciferin-luciferase ATP assay
The bulk extracellular ATP concentration was measured by the luciferin-luciferase assay (ATP Luminescence Kit; AF-2L1, DKK-TOA Co., Tokyo, Japan) at 37 °C as described previously (Hazama et al. 1999, 2000b; Sabirov et al. 2001), with slight modifications. Briefly, C127 cells were cultured to confluence in 24-well plates. Culture medium was totally replaced with 425 μl of normal Ringer solution. Cells were incubated for 60 min at 37 °C, and 100 μl of isotonic extracellular solution was removed and used as a control sample for background ATP release measurements. A hypotonic challenge was then applied by gently removing 300 μl of supernatant and adding 400 μl of a solution of the desired tonicity. Hypotonic solutions were made by mixing Ringer solution with an appropriate volume of a solution of the following composition (mm): 5 KCl, 2 CaCl2, 1 MgCl2, 5 Na-Hepes, 6 Hepes, 5 glucose (pH 7.4, 40 mosmol (kg H2O)−1). The cells were then incubated for 15 min at 37 °C, and extracellular solution samples (100 μl) were collected for the luminometric ATP assay of swelling-induced ATP release. ATP concentration was measured by mixing 50 μl of sample solution with 500 μl normal Ringer solution and 50 μl of luciferin-luciferase reagent. At this ratio, ionic salt sensitivity of the luciferase reaction (Boudreault & Grygorczyk, 2002) was negligible. When required, arachidonic acid, fatty acids and metabolic inhibitors were added during the 60 min preincubation and 15 min hypotonic stimulation periods. Arachidonic acid and other fatty acids tested had no appreciable effect on the luciferin-luciferase reaction.
Data analysis
Single-channel amplitudes were measured by manually placing cursors at the open and closed channel levels. Mean patch currents were measured at the beginning (first 25–30 ms) of current traces in order to minimize the contribution of voltage-dependent current inactivation.
Concentration-response data for arachidonic acid inhibition of macro-patch currents and single-channel currents were fitted to the following equation:
| (1) |
where Io is the current in the absence of arachidonic acid, I is the current in the presence of arachidonic acid at the concentration X, Kd is the apparent dissociation constant, and h is the Hill coefficient.
Dose-response data for arachidonic acid inhibition of swelling-induced ATP release were fitted to the following equation:
| (2) |
where A0 is the released ATP level in the absence of arachidonic acid, A is that in the presence of arachidonic acid at the concentration X, A1 is the non-inhibited fraction of released ATP, Kd is the apparent dissociation constant, and h is the Hill coefficient.
Data were analysed using Origin 5.0 (OriginLab Corp., Northampton, MA, USA). Pooled data are given as means ± s.e.m. of observations (n). Statistical differences in the data were evaluated by Student's paired or unpaired t test and considered significant at P < 0.05 (or P < 0.01 when indicated).
In all figures, membrane potential (Vm) is indicated according to the convention: Vm = Vp (the pipette potential) for whole-cell and outside-out experiments, and Vm = -Vp for inside-out experiments.
Results
Arachidonic acid sensitivity of whole-cell VDACL anion currents
In our previous study, we demonstrated that C127 cells express the VDACL anion channel, which shares the same pharmacology with swelling-induced ATP release measured at 25 or 37 °C (Sabirov et al. 2001). In the whole-cell recording mode, VDACL currents could selectively be recorded when conventional VSOR chloride currents were suppressed by omitting ATP from the pipette solution and by supplementing the hypotonic bath solution with phloretin, a relatively selective blocker of VSOR Cl− channels (Fan et al. 2001). Figure 1A shows the activation of hypotonicity-induced phloretin-insensitive whole-cell VDACL anion currents which exhibited characteristic voltage-dependent inactivation at both positive and negative potentials greater than ±20 mV (Fig. 1Ba). The reversal potential of about +5 mV was close to the Nernst potential for Cl− (+4.2 mV). Bath application of 20 μm arachidonic acid significantly reduced the whole-cell VDACL current (Fig. 1A and Bb). Arachidonic acid effectively inhibited both inward and outward currents in a voltage-independent manner (Fig. 1C). In three experiments out of ten attempts we were able to recover the whole-cell current after washing out arachidonic acid. In seven other experiments, the whole-cell configuration was lost upon washout, possibly due to a destabilizing effect of arachidonate on the gigaohm contact.
Figure 1. Swelling-activated, phloretin-insensitive chloride currents were inhibited by arachidonic acid in C127 cells.
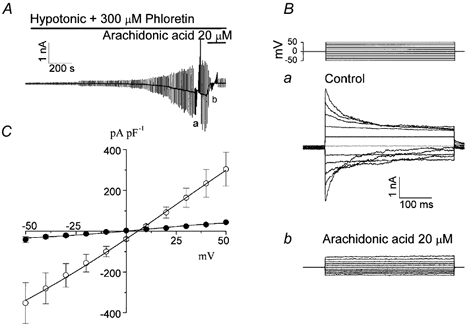
A, representative record during application of alternating pulses from 0 to ±25 mV (every 10 s) or of step pulses from −50 to +50 in 10 mV increments before (a) and after (b) application of arachidonic acid. Horizontal bars indicate the time of application of hypotonic solution containing 300 μm phloretin in the absence and presence of arachidonic acid (20 μm). B, expanded traces of current responses to step pulses recorded at the time indicated by a and b in A. The step-pulse protocol is shown in the top panel. C, current-voltage relationships measured at the beginning of the pulses in the absence (control; ○) and presence of 20 μm arachidonic acid (•). Points plotted are mean ± s.e.m. The whole-cell currents were normalized by the cell capacitance (n = 4). The remaining current in the presence of arachidonic acid had a linear I-V relationship and a reversal potential of around 3 mV, suggesting a non-selective leak current.
Arachidonic acid sensitivity of macropatch VDACL currents in inside-out and outside-out patches
We reported previously that VDACL channels could be activated in cell-attached patches by cell swelling and also could be observed in excised patches (Sabirov et al. 2001). Figure 2A shows the inhibition of mean patch current after application of 20 μm arachidonic acid to the bath in an inside-out membrane patch. The effect of arachidonic acid was reversible. Arachidonic acid inhibited VDACL currents dose-dependently with Kd of 3.9 ± 0.21 μm and 4.44 ± 0.23 μm for outward and inward currents measured at +25 mV and −25 mV, respectively (Fig. 2Aa).
Figure 2. Macro-patch currents activated in inside-out (A) and outside-out patches (B) were reversibly suppressed by arachidonic acid in a dose-dependent manner.
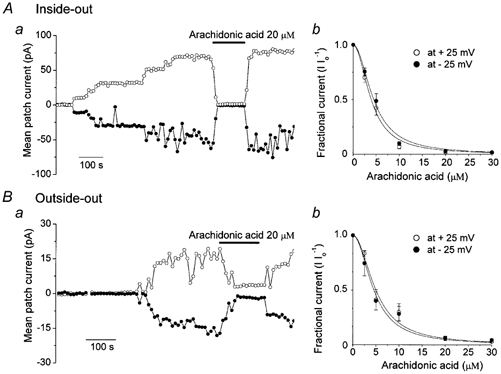
a, representative mean macropatch currents during application of alternating pulses from 0 to ±25 mV (every 10 s) are presented. Horizontal bars indicate the time of application of arachidonic acid (20 μm). b, concentration dependence of arachidonate effects on VDACL currents recorded at ±25 mV, plotted as mean ± s.e.m. All currents measured in the presence of arachidonate were normalized to the respective currents measured before arachidonate application (Io). The inside-out data were fitted to eqn (1) with Kd / 3.9 ± 0.21 μm and 4.44 ± 0.23 μm for outward and inward currents, respectively (n = 5-11). The outside-out data were fitted to eqn (1) with Kd = 4.99 ± 0.44 μm and 4.54 ± 0.37 μm for outward and inward currents, respectively (n = 5-7). The Hill coefficient was 2.
Similar reversible inhibition was observed when arachidonic acid was applied from the extracellular side to an outside-out patch after steady-state activation of VDACL channels (Fig. 2B). Inhibition of VDACL currents by external arachidonic acid was also dose-dependent with Kd of 4.99 ± 0.44 μm and 4.54 ± 0.37 μm for outward and inward currents measured at +25 mV and −25 mV, respectively (Fig. 2Bb). The dose-response curves (Fig. 2Ab and Bb) could be well fitted with a Hill coefficient of 2, but not 1, implying that more than one arachidonic acid molecule is needed to inhibit VDACL channels in both inside-out and outside-out patches.
Since fatty acids are known to move rapidly across phospholipid bilayers (Kamp & Hamilton, 1993), inhibition observed in the outside-out mode does not necessarily mean that arachidonic acid exerted the action from the outside of the channel. In order to clarify the sidedness of the effect, we tested a membrane-impermeant analogue arachidonyl coenzyme A (arachidonyl CoA: Smirnov & Aaronson, 1996; Denson et al. 2000). Arachidonyl CoA inhibited the macropatch VDACL currents when applied from the intracellular side in inside-out patches (Fig. 3A) to the same extent as arachidonic acid, whereas coenzyme A by itself at 20 μm had no effect on the VDACL channel (n = 5, data not shown). In contrast, arachidonyl CoA added from the extracellular side had no effect in the outside-out mode (Fig. 3B). The averaged data are summarized in Fig. 3C. This result suggests the arachidonic acid binding site of the VDACL channel is localized intracellularly.
Figure 3. Effects of a membrane-impermeant analogue of arachidonic acid, arachidonyl coenzyme A (20 μm), on VDACL currents recorded at ±25 mV from inside-out (A) and outside-out patches (B).
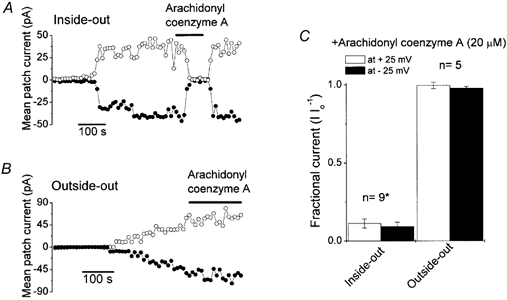
Representative mean macropatch currents during application of alternating pulses from 0 to ±25 mV (every 10 s) are presented. Horizontal bars indicate the time of application of arachidonyl coenzyme A (20 μm). C, macropatch currents in inside-out and outside-out patches in the presence of arachidonyl coenzyme A (20 μm), normalized to those measured before application of arachidonyl coenzyme A (Io). * Significantly different from control at P < 0.001.
Direct effects of arachidonic acid on VDACL currents without metabolism by oxygenases
Arachidonic acid, after liberation from membrane phospholipids, is metabolized into different biologically active compounds such as prostaglandins by the cyclooxygenase pathway, leukotrienes by the lipoxygenase pathway, and epoxides by the monooxygenase pathway (Holtzman, 1991). Therefore, in order to distinguish between a direct effect of arachidonic acid on the VDACL channel and an indirect inhibition via metabolic products of one of these oxygenases, which might have been retained on or near the excised patches, we used relatively selective blockers of these pathways: indomethacin, an inhibitor of cyclooxygenase; NDGA, a lipoxygenase inhibitor; and clotrimazole, a cytochrome P-450 monooxygenase inhibitor (Rainsford, 1988). Figure 4 shows that arachidonic acid effectively inhibited macropatch VDACL currents in the presence of these oxygenase inhibitors, whereas the inhibitors themselves did not have any effect on VDACL currents in inside-out patches. The steady-state level of arachidonate-induced inhibition in the presence of each inhibitor did not differ significantly from that in the absence of these inhibitors (Fig. 4D). These results indicate that the effect of arachidonic acid on VDACL currents was not mediated by its downstream metabolic products and was due to a direct interaction with the channel (or its accessory) protein.
Figure 4. Suppression of macropatch currents in the inside-out mode by arachidonic acid was not affected by inhibitors of oxygenases that are involved in arachidonic acid metabolism.
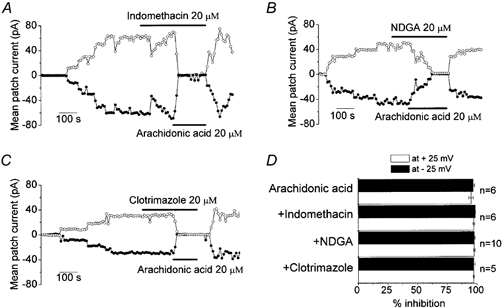
A, effect of arachidonic acid in the presence of 20 μm indomethacin. B, effect of arachidonic acid in the presence of 20 μm NDGA. C, effect of arachidonic acid in the presence of 20 μm clotrimazole. D, inhibitory effect of arachidonic acid in the absence or presence of oxygenase inhibitors (mean ± s.e.m.). Percentage inhibition of macropatch currents by arachidonic acid in the presence or absence of oxygenase inhibitors was calculated from currents measured before application of arachidonic acid.
Next we tested whether other fatty acids, which are not substrates for any oxygenases, are also able to inhibit this channel. Indeed, cis-unsaturated fatty acids, such as oleic and linoleic acid, inhibited macropatch VDACL currents albeit to a lesser degree than arachidonic acid (Fig. 5A and B). In contrast, a trans-unsaturated fatty acid, elaidic acid, and a saturated fatty acid, palmitic acid, did not noticeably affect the VDACL currents (Fig. 5A). Therefore, not only hydrophobicity but also the specific conformation of the hydrocarbon chain is important for the inhibitory effect of arachidonic acid on the VDACL channel. Non-charged analogues of arachidonic acid, arachidonyl alcohol and arachidonyl methyl ester, did not have any inhibitory effect on VDACL channels (Fig. 5C). In addition, although oleic acid was an effective VDACL inhibitor (Fig. 5A), its neutral analogue, oleyl alcohol, was ineffective (Fig. 5C). These data suggest that the negatively charged polar head is essential for the inhibitory effect of arachidonic acid. Since arachidonyl CoA does not have a carboxylic group but has phosphate residues consisting of a bulky negatively charged polar head, it is suggested that the negative charge, but not the carboxylic group per se, is essential for the inhibitory action of arachidonic acid.
Figure 5. Role of the hydrophobic tail and polar head in inhibition of VDACL currents by arachidonic acid.
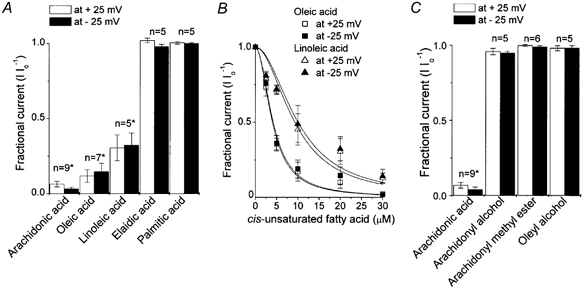
A, relative effects of cis-unsaturated (arachidonic, oleic and linoleic), trans-unsaturated (elaidic) and saturated (palmitic) fatty acids (all 20 μm) on macropatch currents recorded in the inside-out mode at ±25 mV. * Significantly different from control at P < 0.001. B, concentration-dependent inhibition of macropatch currents by oleic acid (squares) and linoleic acid (triangles). Open symbols are data for +25 mV and filled symbols are for −25 mV (mean ± s.e.m.). The data were fitted to eqn (1) with Kd / 4.02 ± 0.21 μm for +25 mV and Kd = 4.19 ± 0.34 μm for −25 mV in the presence of oleic acid, and Kd = 9.3 ± 1.2 μm for +25 mV and Kd = 10.0 ± 1.3 μm for −25 mV in the presence of linoleic acid (n = 5-7). The Hill coefficient was 2. C, effects of non-charged arachidonic acid analogues, arachidonyl alcohol and methyl ester, and a non-charged oleate analogue, oleyl alcohol, on VDACL macropatch currents in inside-out patches. Data were normalized to the mean current measured before application of drugs (Io).
Unitary conductance-reducing effect of arachidonic acid on VDACL channels
Arachidonic acid not only inhibited macropatch currents but also reduced the amplitude of the VDACL channel unitary current. Figure 6A shows inside-out single-channel records in the absence and presence of 10 μm arachidonic acid in the bath. Even in the presence of 10 μm arachidonic acid, the single-channel activity could be observed in a small number of remaining active channels, after the activity of most channels was abolished. The single-channel amplitude of both outward and inward events was diminished by arachidonic acid (Fig. 6B), while the current-voltage relationship remained linear and symmetrical (Fig. 6C), suggesting a voltage-independent block. The single-channel conductance obtained from the linear fit to I-V curves (Fig. 6C) decreased from 405.6 ± 6.4 pS to 249.2 ± 7.2 pS in the presence of 10 μm arachidonic acid. The arachidonate-induced inhibition of single-VDACL channel activity was dose-dependent with a Kd of 13.8 ± 1.6 μm for outward currents and a Kd of 14.0 ± 1.1 μm for inward currents (Fig. 6D). The Hill coefficient was estimated to be 2, but not 1, by fitting all the single-channel dose-response data, implying that this effect of arachidonic acid was also mediated by more than one molecule.
Figure 6. Effect of arachidonic acid added to the intracellular side on single VDACL channel currents recorded from inside-out patches.
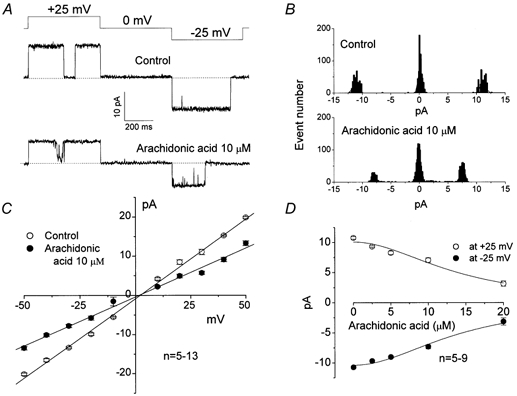
A, representative current traces recorded during application of a step pulse from 0 to ±25 mV in the absence (control) and presence of 10 μm arachidonic acid. The pulse protocol is shown at the top of the traces. B, amplitude histograms of current traces presented in A. C, unitary current-voltage relationships in the absence (control; ○) and presence of 10 μm arachidonic acid (•). Mean single-channel conductance was 405.6 ± 6.4 pS for control and 249.2 ± 7.2 pS with 10 μm arachidonic acid (P < 0.05, n = 5-13). D, concentration-response curves of arachidonic acid effects on VDACL single-channel amplitude measured at ±25 mV. The data were fitted to eqn (1) with Kd = 13.8 ± 1.6 μm and 14.0 ± 1.1 μm for outward and inward currents, respectively (n = 5-9). The Hill coefficient was 2. Points plotted are mean ± s.e.m.
No remarkable increase in open-channel noise could be detected in the presence of 10 μm arachidonic acid at +25 mV even when inside-out single-channel recordings were performed at a 5 kHz bandwidth (Fig. 7). This result suggests that voltage-independent single-channel inhibition by arachidonic acid was induced by reduction of single-channel conductance due to either fast (faster than 5 kHz) open-channel block or a conformational change of the open channel pore. Although slightly flickering events were observed at −25 mV in the presence of arachidonic acid (Fig. 7A), this did not contribute significantly to the observed inhibition of single-channel amplitude, as the Kd values for single-channel amplitudes were voltage-independent.
Figure 7. Fast reducing effect of arachidonic acid on single-channel current amplitude.
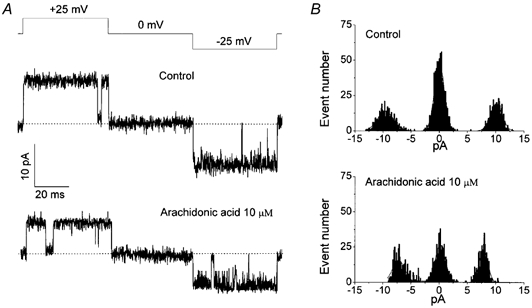
A, representative current traces recorded from an inside-out patch during application of a test pulse from 0 to ±25 mV (protocol is shown at the top of the traces) by sampling at 10 kHz and filtering at 5 kHz in the absence (control; upper record) and presence of 10 μm arachidonic acid (lower record). B, amplitude histograms for data presented in A.
Shutdown effect of arachidonic acid on VDACL channels
The inhibitory effect of arachidonic acid on macropatch currents was significantly stronger (Kd / 4–5 μm) than that on single-channel amplitudes (Kd = 13–14 μm). Figure 8A summarizes the inhibitory effect of 10 μm arachidonic acid on macropatch currents in comparison to that on single-channel amplitudes at positive and negative potentials. Both outward and inward currents were suppressed much more prominently in macropatches. Results that were essentially the same were observed when arachidonic acid was applied in the presence of a cocktail containing three different oxygenase inhibitors (indomethacin, NDGA and clotrimazole) (Fig. 9B). These data indicate that arachidonic acid reduced not only the single-channel amplitude but also the open-channel probability or the number of active channels.
Figure 8. Effect of arachidonic acid on mean macropatch currents, single-channel amplitude and open-channel probability.

Fractional macropatch and single-channel currents in inside-out patches in the presence of 10 μm arachidonic acid without (A) and with (B) a cocktail of three oxygenase inhibitors (indomethacin, NDGA and clotrimazole, each 20 μm). Data were normalized to the mean current measured before application of arachidonic acid (Io) and plotted as mean ± s.e.m.C, voltage dependence of steady-state open-channel probability. The data represent the ensemble-averaged current of 11 consecutive current responses to ramp pulses (from −50 mV to +50 mV at 10 mV s−1 rate) in the absence (control; continuous line) and presence of 5 μm arachidonic acid (○). The patch contained 5–6 channels. The Po values were calculated as Po / (I/V)/Gmax, where I is the patch current, V is voltage and Gmax is the maximal patch conductance at 0 mV.
Figure 9. Arachidonic acid sensitivity of ATP current through VDACL channels.
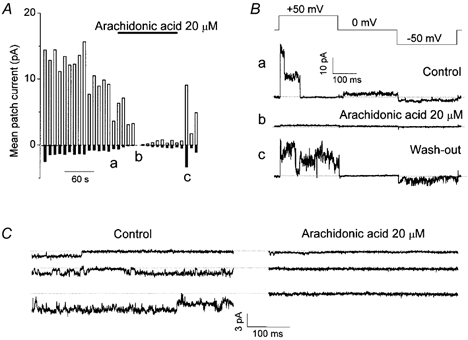
A, mean patch currents in an inside-out patch measured at ±50 mV. All anions in the bath were replaced with 100 mm ATP. Upper horizontal bar represents the time of arachidonic acid (20 μm) application. Data represent 12 similar experiments. a, b and c indicate the times when the traces in B were recorded. B, current traces recorded during application of alternating pulses from 0 to ±50 mV (protocol is shown at the top of the traces) before application of arachidonic acid (a), in the presence of 20 μm arachidonic acid (b), and after washout (c). C, representative inward ATP4- currents recorded at −50 mV from three different patches before (left: control) and during (right) application of 20 μm arachidonic acid.
In order to estimate the effect of arachidonic acid on the open-channel probability of the VDACL channel, we applied a series of slow ramp-pulses and recorded the steady-state current responses from the patches containing several channels. Channels always stayed open at potentials near 0 mV (Po / 1) and closed gradually when the membrane potential exceeded 10–20 mV yielding a bell-shaped steady-state voltage dependence of open-channel probability (Fig. 8C) similar to that observed previously (Sabirov et al. 2001). The patch shown in Fig. 8C contained six channels, as judged from the stepwise current inactivation at positive and negative voltages. Application of 5 μm arachidonic acid (sufficient to cause a 50 % reduction in macropatch currents) decreased the number of active channels from six to three, but did not have any effect on the steady-state voltage dependence of open-channel probability (Fig. 8C, ○), suggesting that arachidonic acid has a negligible effect on the open-channel probability of the VDACL channel. The same result was obtained in three other independent experiments.
Macroscopic current (I) is a product of the number of active channels (N), single-channel current amplitude (i) and open-channel probability (Po):
| (3) |
Given the absence of any effect on Po, we conclude that arachidonic acid exerts dual effects on the VDACL channel: a strong binding to the channel (or its accessory) protein with a Kd of 4–5 μm results in channel shutdown (decrease in N), whereas weaker binding to the channel lumen with a Kd of 13–14 μm causes a decrease in single-channel amplitude (i).
Arachidonic acid sensitivity of ATP currents through VDACL channels and of swelling-induced ATP release
In our previous study, we demonstrated that the VDACL channel serves as a conductive pathway for swelling-induced ATP release in C127 cells (Sabirov et al. 2001). As a natural extension, we tested whether both ATP currents through VDACL channels and swelling-induced ATP release are similarly sensitive to arachidonic acid. Figure 9A shows records of ionic current from an inside-out patch after replacing the bath (intracellular) solution with 100 mm Na4ATP. Small but sizable inward currents that correspond to electrogenic flux of anionic forms of ATP (mainly ATP4-) from bath to pipette through VDACL channels could be recorded, as reported previously (Sabirov et al. 2001). Although voltage-dependent inactivation of the ATP4- current was often detectable at −50 mV (Fig. 9C, top trace), it was not as profound as that of the Cl− current recorded in normal bath solution, suggesting a possibility that the voltage-dependent inactivation profile may somehow be affected by conducting anionic species. Both outward Cl− currents and inward ATP4- currents were simultaneously inhibited by 20 μm arachidonic acid. In 9 of 12 similar experiments, we were able to restore ATP channel activity upon washout with arachidonate-free Na4ATP solution. The current traces recorded in this experiment before (a) and during (b) application of arachidonic acid, and after recovery (c), are shown in Fig. 9B. Figure 9C shows more prolonged records of the ATP4- currents obtained in three other experiments.
Hypotonic cell swelling induced massive release of ATP from C127 monolayers in a dose-dependent manner (Fig. 10A). Arachidonic acid added at 20 μm concentration significantly inhibited swelling-induced ATP release (Fig. 10A). The inhibition was dose-dependent with a Kd of 6.2 ± 0.3 μm (Fig. 10B). This value is very close to the Kd value obtained for macropatch VDACL currents (Fig. 2Ab and Bb). It should be noted, however, that even at the highest concentrations tested, arachidonic acid inhibited the ATP release only partially. When the inhibitor cocktail containing three different oxygenase inhibitors (indomethacin, NDGA and clotrimazole, 20 μm each) was applied, the swelling-induced ATP release was suppressed by about 40 % even without addition of arachidonate. The remainder of the ATP release could be further inhibited by arachidonic acid in a dose-dependent manner with a Kd of 4.4 ± 1.2 μm. This value is close to that obtained in the absence of oxygenase inhibitors. Incomplete inhibition in the absence of oxygenase inhibitors may be largely explained by the fact that the effective arachidonic acid concentration decreases. Also, it is possible that downstream metabolic products may antagonize the direct effect of arachidonic acid on the VDACL channel.
Figure 10. Effects of arachidonic acid and other fatty acids on swelling-induced ATP release from C127 cells.
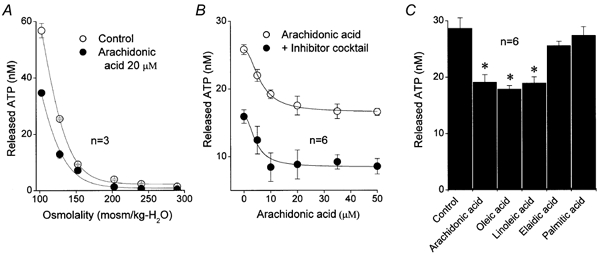
A, swelling-induced ATP release as a function of medium tonicity in the absence (control; ○) and presence of 20 μm arachidonic acid (•). B, ATP release at 127.5 mosmol (kg H2O)−1 as a function of arachidonic acid concentration in the absence (control: open circles) and presence of inhibitors of arachidonic acid metabolism (indomethacin, NDGA, clotrimazole, 20 μm each; •). The data were fitted to eqn (2) with Kd / 6.2 ± 0.3 μm for control and Kd = 4.4 ± 1.2 μm for ATP release in the presence of metabolic inhibitors (n = 6). The Hill coefficient was 2. C, relative effects of cis-unsaturated (arachidonic, oleic and linoleic), trans-unsaturated (elaidic) and saturated (palmitic) fatty acids (all 20 μm) on ATP release measured at 127.5 mosmol (kg H2O)−1. n = 6. Points plotted are mean ± s.e.m. * Significantly different from control at P < 0.01.
Cis-unsaturated fatty acids, oleic and linoleic acid, inhibited ATP release to the same degree as arachidonic acid (Fig. 10C). In contrast, a trans-unsaturated fatty acid, elaidic acid, and a saturated fatty acid, palmitic acid, did not noticeably affect the swelling-induced ATP release (Fig. 10C). The effect of each fatty acid on ATP release was qualitatively similar to the effect of each fatty acid on VDACL channel activity in macropatches (Fig. 5).
Discussion
Translocation of ATP from the intracellular compartment to the extracellular fluid is a fundamental process that provides the substrate for purinergic autocrine and paracrine cell signalling. Although the physiological importance of this process is well recognized, the cellular mechanisms are poorly understood. Existence of a conductive pathway for ATP release has been reported in a number of studies, and CFTR was suggested to be a determinant of this process (Reisin et al. 1994; Schwiebert et al. 1995; Cantiello et al. 1997, 1998; Pasyk & Foskett, 1997; Lader et al. 2000). In human epithelial Intestine 407 and mouse mammary C127 cell lines, the pathway of swelling-induced ATP release was shown to be distinct from both CFTR and VSOR chloride channels (Hazama et al. 1998, 1999, 2000a, b). In our previous study (Sabirov et al. 2001), we demonstrated that C127 cells express a large-conductance anion channel, which was silent under normal conditions but could be activated under hypotonic conditions. Based on pharmacological analysis and ATP4- current measurements, we concluded that this VDACL anion channel serves as a conductive pathway for swelling-induced ATP release from C127 cells. We found a similar channel in a previous study of kidney macula densa cells, where the channel was regulated by luminal NaCl and was suggested to mediate the ATP release-dependent tubulo-glomerular feedback mechanism (Bell et al. 2000).
Arachidonic acid is an abundant constituent of the cell. The concentration of esterified arachidonate in resting platelets, for instance, has been estimated to be as high as 5 mm (Brash, 2001). Free arachidonic acid levels are low in the plasma and cytosol, but can rise upon stimulation to 10–100 μm (Brash, 2001). Although most biological activities of arachidonic acid occur through its conversion to prostaglandins, leukotrienes and other products by the cyclooxygenase, lipoxygenase and monooxygenase pathways (Irvine, 1982; Brash, 2001), arachidonic acid itself is also an important regulator of many cellular functions, including cell volume regulation (Lambert, 1987; Kubo & Okada, 1992; Margalit et al. 1993; Civan et al. 1994; Sanchez-Olea et al. 1995; Gosling et al. 1996; Mignen et al. 1999; Hoffmann, 2000). Arachidonic acid has been shown to upregulate ClC-2 Cl− channels (Tewari et al. 2000; Cupoletti et al. 2001) but to downregulate VSOR (Kubo & Okada, 1992; Nilius et al. 1994; Sakai et al. 1996; Gosling et al. 1996; Xu et al. 1997) and other types of Cl− channel (Anderson & Welsh, 1990; Hwang et al. 1990; Zachar & Hurnak, 1994; Riquelme & Parra, 1999; Linsdell, 2000).
In the present study, we demonstrate that arachidonic acid is an effective inhibitor of the VDACL anion channel in C127 cells. This result is consistent with previous observations for maxi-Cl− channels from L6 myoblasts (Zachar & Hurnak, 1994) and large anion channels from human term placenta reconstituted in giant liposomes (Riquelme & Parra, 1999). The Kd value of 4–5 μm obtained here is similar to the values observed for a variety of ion channels (Meves, 1994) and is within the physiological range for arachidonic acid concentrations detected in different cells (Brash, 2001). Km values of 5 μm for cyclooxygenase and 3.4-28 μm for lipoxygenases (Needleman et al. 1986) also indicate that the arachidonic acid concentration in the range of 5–10 μm is physiologically relevant. Therefore, we conclude that arachidonic acid-mediated regulation of VDACL anion channel function actually takes place under physiological conditions.
Arachidonic acid effectively inhibited whole-cell and outside-out macropatch VDACL currents from the extracellular side, and inside-out patch currents from the intracellular side. However, given the rapid, diffusional movement of fatty acids across phospholipid bilayers (Kamp & Hamilton, 1993), it is reasonable to assume that arachidonic acid added to the extracellular fluid can easily reach the intracellular moiety and exert its effect from the intracellular side unless the cytosol is perfused. Consistent with this notion, an impermeant analogue, arachidonyl CoA, exerted the inhibitory action only from the intracellular side of inside-out patches. In addition, we did not observe any inhibitory change in VDACL channel activity in inside-out patches, when 20 μm arachidonic acid was added into the pipette solution (data not shown, n = 10). This observation is similar to that of Zachar & Hurnak (1994) for maxi-Cl− channels from L6 cells. Taken together with arachidonyl CoA data, it is concluded that the site of arachidonate action exists on the cytosolic side of the VDACL anion channel.
In our experiments, arachidonic acid inhibited VDACL channels in two different ways: (i) channel shutdown (decrease in number of active channels) due to a high-affinity binding with a Kd value of 4–5 μm and (ii) reduced unitary conductance due to low-affinity binding with a Kd value of 13–14 μm. The effects of arachidonic acid were direct and not mediated by downstream metabolic products as evidenced by (i) lack of an effect of inhibitors of arachidonate-metabolizing oxygenases and (ii) similar VDACL inhibition by cis-unsaturated fatty acids that could not be substrates for oxygenases. Neither trans-unsaturated nor saturated fatty acids affected VDACL currents, indicating the importance of the specific conformation of arachidonate's hydrocarbon chain in its inhibitory effect on the VDACL channel. Removing the negative charge of the carboxyl group either by substitution with hydroxyl (arachidonyl alcohol) or by esterification (arachidonyl methyl ester) completely abolished the inhibitory action of arachidonic acid, suggesting the importance of this charge. The necessity for negative charges is in apparent contradiction to voltage independence of both unitary conductance reduction and channel shutdown. We propose that the inhibitory arachidonate-binding site is located close to the internal entrance to the VDACL channel pore, where little or no voltage drop occurs. Arachidonate binding to the blocking site situated outside the electric field may lead to voltage-independent reduction of single-channel conductance due to either fast open-channel block or a conformational change of the channel pore.
According to the proposed physiological role for the VDACL channel as a conductive pathway for ATP release (Sabirov et al. 2001), arachidonic acid must block not only ATP currents but also mass release of ATP induced by cell swelling. Indeed, small inward currents carried by ATP4- were reversibly inhibited by application of arachidonic acid (Fig. 9). Swelling-induced ATP release was also suppressed by arachidonate (Fig. 10). Since arachidonic acid is constantly consumed by endogenous oxygenases, only partial inhibition by arachidonate was observed when these metabolic pathways were still active in intact non-patched cells. In whole-cell configuration, constant perfusion of the intracellular space with the pipette solution may have allowed more profound suppression of VDACL currents by arachidonate due to an increase in effective concentration of arachidonate by washout or decreased specific activity of oxygenases. Inhibition of arachidonate degradation by a cocktail of specific inhibitors for the oxygenases led to ≈40 % reduction in ATP release, presumably due to accumulation of arachidonic acid generated endogenously in response to the hypotonic stress (Thoroed et al. 1997; Tinel et al. 1997; Basavappa et al. 1998; Hoffmann, 2000; Pedersen et al. 2000). In these conditions, exogenously added arachidonate further suppressed ATP release, though still not completely. The Kd values of 4–6 μm were very close to those observed in patch-clamp experiments, providing strong evidence for the involvement of the VDACL channel in swelling-induced ATP release. This idea was further supported by the observation that only cis-unsaturated fatty acids (oleic and linoleic), but not trans-unsaturated (elaidic) or saturated (palmitic) fatty acids, could noticeably affect both swelling-induced ATP release and VDACL channel activity.
Arachidonic acid failed to completely suppress the mass ATP release from intact cells even in the presence of an inhibitory cocktail, whereas arachidonic acid nearly completely eliminated VDACL currents recorded in the whole-cell or excised patch mode. This discrepancy may be related to a complex regulation of ATP-releasing pathways in intact cells compared to excised patches or cells in the whole-cell mode. Swelling-induced activation of phospholipase A2 was reported to lead to an arachidonate-mediated increase in intracellular Ca2+ (Oike et al. 1994). On the other hand, a Ca2+-mobilizing mitogen, bombesin, and a Ca-ionophore, A23187, were shown to activate the maxi-Cl− channel (Kawahara & Takuwa, 1991). Thus, we may suppose that, in intact cells, a direct inhibiting effect of arachidonic acid on VDACL channel might be attenuated or even counteracted by the Ca2+-mediated enhancing effect on the VDACL activation system. Another possible explanation for the above discrepancy would be that arachidonate at high concentrations can form hydrophobic micelles (see Meves, 1994). A residual whole-cell current apparent in the presence of arachidonate (Fig. 1) and an arachidonate-insensitive component of macropatch current in Fig. 2B might represent a non-specific leak induced by the arachidonate micelles. We may suppose that a similar leak can be induced by arachidonate in cell membranes in ATP release experiments and might be responsible for the arachidonate-insensitive component of ATP release. On the other hand, we cannot exclude an alternative possibility that the VDACL channel is not the sole ATP-releasing pathway in C127 cells.
Cell swelling is a strong stimulus for arachidonic acid liberation from membrane phospholipids (Thoroed et al. 1997; Tinel et al. 1997; Basavappa et al. 1998; Hoffmann, 2000; Pedersen et al. 2000). However, ATP released simultaneously with cell swelling can also trigger activation of phospholipase A2 and subsequent arachidonic acid generation (Zambon et al. 2000; Scholz-Pedretti et al. 2001; Teixeira et al. 2001). Therefore, downregulation of the ATP-releasing VDACL channel may constitute a negative feedback in the coupling mechanism of the two powerful signalling pathways mediated by cytosolic arachidonate and extracellular ATP.
In summary, arachidonic acid at micromolar concentrations was found to directly downregulate both the VDACL anion channel and swelling-induced ATP release before being metabolized by oxygenases, in C127 cells. This fact provides new evidence for our previous conclusion that the VDACL anion channel serves as a pathway for swelling-induced ATP release, both being under a negative control by intracellular arachidonic acid signalling.
Acknowledgments
The authors are grateful to E. Lee, S. Tanaka and K. Shigemoto for technical assistance and to T. Okayasu for secretarial assistance. This work was supported by Grant-in-Aid for Scientific Research (to R. Z. Sabirov) and Grant-in-Aid for Scientific Research for Priority Areas of ‘ABC Proteins’ (to Y. Okada) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by grants from Houansha Foundation and Salt Science Foundation (to Y. Okada).
References
- Abraham EH, Prat AG, Gerweck L, Seneveratne T, Arceci RJ, Kramer R, Guidotti G, Cantiello HF. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proceedings of the National Academy of Sciences of the USA. 1993;90:312–316. doi: 10.1073/pnas.90.1.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MP, Welsh MJ. Fatty acids inhibit apical membrane chloride channels in airway epithelia. Proceedings of the National Academy of Sciences of the USA. 1990;87:7334–7338. doi: 10.1073/pnas.87.18.7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavappa SS, Pedersen F, Jorgensen NK, Ellory JC, Hoffmann EK. Swelling-induced arachidonic acid release via the 85-kDa cPLA2 in human neuroblastoma cells. Journal of Neurophysiology. 1998;79:1441–1449. doi: 10.1152/jn.1998.79.3.1441. [DOI] [PubMed] [Google Scholar]
- Bell PD, Lapointe J-Y, Sabirov R, Hayashi S, Okada Y. Maxi-chloride channel in macula densa cells: possible pathway for ATP release. FASEB Journal. 2000;14:A134. [Google Scholar]
- Bodas E, Aleu J, Pujol G, Martin-Satue M, Marsal J, Solsona C. ATP crossing the cell plasma membrane generates an ionic current in Xenopus oocytes. Journal of Biological Chemistry. 2000;275:20268–20273. doi: 10.1074/jbc.M000894200. [DOI] [PubMed] [Google Scholar]
- Bodin P, Burnstock G. Purinergic signalling: ATP release. Neurochemical Research. 2001;26:959–969. doi: 10.1023/a:1012388618693. [DOI] [PubMed] [Google Scholar]
- Boudreault F, Grygorczyk R. Cell swelling-induced ATP release and gadolinium-sensitive channels. American Journal of Physiology – Cell Physiology. 2002;282:C219–226. doi: 10.1152/ajpcell.00317.2001. [DOI] [PubMed] [Google Scholar]
- Bosch I, Jackson GR, Jr, Croop JM, Cantiello HF. Expression of Drosophila melanogaster P-glycoproteins is associated with ATP channel activity. American Journal of Physiology. 1996;271:C1527–1538. doi: 10.1152/ajpcell.1996.271.5.C1527. [DOI] [PubMed] [Google Scholar]
- Brash AR. Arachidonic acid as a bioactive molecule. Journal of Clinical Investigation. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Release of vasoactive substances from endothelial cells by shear stress and purinergic mechanosensory transduction. Journal of Anatomy. 1999;194:335–342. doi: 10.1046/j.1469-7580.1999.19430335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantiello HF, Jackson GR, Jr, Grosman CF, Prat AG, Borkan SC, Wang Y, Reisin IL, O,Riordan CR, Ausiello FA. Electrodiffusional ATP movement through the cystic fibrosis transmembrane conductance regulator. American Journal of Physiology. 1998;274:C799–809. doi: 10.1152/ajpcell.1998.274.3.C799. [DOI] [PubMed] [Google Scholar]
- Cantiello HF, Jackson GR, Jr, Prat AG, Gazley JL, Forrest JN, Jr, Ausiello DA. cAMP activates an ATP-conductive pathway in cultured shark rectal gland cells. American Journal of Physiology. 1997;272:C466–475. doi: 10.1152/ajpcell.1997.272.2.C466. [DOI] [PubMed] [Google Scholar]
- Civan MM, Coca-Prados M, Peterson-Yantorno K. Pathways signaling the regulatory volume decrease of cultured nonpigmented ciliary epithelial cells. Investigative Ophthalmology and Visual Science. 1994;35:2876–2886. [PubMed] [Google Scholar]
- Cuppoletti J, Tewari KP, Sherry AM, Kupert EY, Malinowska DH. ClC-2 Cl− channels in human lung epithelia: activation by arachidonic acid, amidation, and acid-activated omeprazole. American Journal of Physiology – Cell Physiology. 2001;281:C46–54. doi: 10.1152/ajpcell.2001.281.1.C46. [DOI] [PubMed] [Google Scholar]
- Denson DD, Wang X, Worrell RT, Eaton DC. Effects of fatty acids on BK channels in GH(3) cells. American Journal of Physiology – Cell Physiology. 2000;279:C1211–1219. doi: 10.1152/ajpcell.2000.279.4.C1211. [DOI] [PubMed] [Google Scholar]
- Fan HT, Morishima S, Kida H, Okada Y. Phloretin differentially inhibits volume-sensitive and cyclic AMP-activated, but not Ca-activated, Cl− channels. British Journal of Pharmacology. 2001;133:1096–1106. doi: 10.1038/sj.bjp.0704159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosling M, Poyner DR, Smith JW. Effects of arachidonic acid upon the volume-sensitive chloride current in rat osteoblast-like (ROS 17/2. 8) cells. Journal of Physiology. 1996;493:613–623. doi: 10.1113/jphysiol.1996.sp021408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grygorczyk R, Hanrahan JW. CFTR-independent ATP release from epithelial cells triggered by mechanical stimuli. American Journal of Physiology. 1997;272:C1058–1066. doi: 10.1152/ajpcell.1997.272.3.C1058. [DOI] [PubMed] [Google Scholar]
- Hazama A, Ando-Akatsuka Y, Fan H-T, Tanaka S, Okada Y. CFTR-dependent and -independent ATP release induced by osmotic swelling. In: Suketa Y, Carafoli E, Lazduvski M, Mikoshiba K, Okada Y, Wright EM, editors. Control and Disease of Sodium Dependent Transportation Proteins and Ion Channels. Amsterdam: Elsevier; 2000a. pp. 429–431. [Google Scholar]
- Hazama A, Fan H-T, Abdullaev I, Maeno E, Tanaka S, Ando-Akatsuka Y, Okada Y. Swelling-augmented ATP release and Cl− conductances in murine C127 cells. Journal of Physiology. 2000b;523:1–11. doi: 10.1111/j.1469-7793.2000.t01-6-00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazama A, Miwa A, Miyoshi T, Shimizu T, Okada Y. ATP release from swollen or CFTR-expressing epithelial cells. In: Okada Y, editor. Cell Volume Regulation: The Molecular Mechanism and Volume Sensing Machinery. Amsterdam: Elsevier; 1998. pp. 93–98. [Google Scholar]
- Hazama A, Shimizu T, Ando-Akatsuka Y, Hayashi S, Tanaka S, Maeno E, Okada Y. Swelling-induced, CFTR-independent ATP release from a human epithelial cell line: lack of correlation with volume-sensitive Cl− channels. Journal of General Physiology. 1999;114:525–533. doi: 10.1085/jgp.114.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK. Intracellular signaling involved in volume regulatory decrease. Cellular Physiology and Biochemistry. 2000;10:273–288. doi: 10.1159/000016356. [DOI] [PubMed] [Google Scholar]
- Holtzman MJ. Arachidonic acid metabolism. Implications of biological chemistry for lung function and disease. The American Review of Respiratory Disease. 1991;143:188–203. doi: 10.1164/ajrccm/143.1.188. [DOI] [PubMed] [Google Scholar]
- Hwang TC, Guggino SE, Guggino WB. Direct modulation of secretory chloride channels by arachidonic and other cis unsaturated fatty acids. Proceedings of the National Academy of Sciences of the USA. 1990;87:5706–5709. doi: 10.1073/pnas.87.15.5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF. How is the level of free arachidonic acid controlled in mammalian cells? Biochemical Journal. 1982;204:3–16. doi: 10.1042/bj2040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara K, Takuwa N. Bombesin activates large-conductance chloride channels in Swiss 3T3 fibroblasts. Biochemical and Biophysical Research Communications. 1991;177:292–298. doi: 10.1016/0006-291x(91)91981-h. [DOI] [PubMed] [Google Scholar]
- Kubo M, Okada Y. Volume-regulatory Cl− channel currents in cultured human epithelial cells. Journal of Physiology. 1992;456:351–371. doi: 10.1113/jphysiol.1992.sp019340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lader AS, Xiao YF, O'Riordan CR, Prat AG, Jackson GR, Jr, Cantiello HF. cAMP activates an ATP-permeable pathway in neonatal rat cardiac myocytes. American Journal of Physiology – Cell physiology. 2000;279:C173–187. doi: 10.1152/ajpcell.2000.279.1.C173. [DOI] [PubMed] [Google Scholar]
- Lambert IH. Effect of arachidonic acid, fatty acids, prostaglandins and leukotrienes on volume regulation in Ehrlich ascites tumor cells. Journal of Membrane Biology. 1987;98:207–221. doi: 10.1007/BF01871184. [DOI] [PubMed] [Google Scholar]
- Linsdell P. Inhibition of cystic fibrosis transmembrane conductance regulator chloride channel currents by arachidonic acid. Canadian Journal of Physiology and Pharmacology. 2000;78:490–499. [PubMed] [Google Scholar]
- Margalit A, Livne AA, Funder J, Granot Y. Initiation of RVD response in human platelets: mechanical-biochemical transduction involves pertussis-toxin-sensitive G protein and phospholipase A2. Journal of Membrane Biology. 1993;136:303–311. doi: 10.1007/BF00233669. [DOI] [PubMed] [Google Scholar]
- Meves H. Modulation of ion channels by arachidonic acid. Progress in Neurobiology. 1994;43:175–186. doi: 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Mignen O, Le Gall C, Harvey BJ, Thomas S. Volume regulation following hypotonic shock in isolated crypts of mouse distal colon. Journal of Physiology. 1999;515:501–510. doi: 10.1111/j.1469-7793.1999.501ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman P, Turk J, Jakschik BA, Morrison AR, Lefkowith JB. Arachidonic acid metabolism. Annual Review of Biochemistry. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- Nilius B, Oike M, Zahradnik I, Droogmans G. Activation of a Cl− current in hypotonic volume increase in human endothelial cells. Journal of General Physiology. 1994;103:787–805. doi: 10.1085/jgp.103.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike M, Droogmans G, Nilius B. Mechanosensitive Ca2+ transients in endothelial cells from human umbilical vein. Proceedings of the National Academy of Sciences of the USA. 1994;91:2940–2944. doi: 10.1073/pnas.91.8.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasyk EA, Foskett JK. Cystic fibrosis transmembrane conductance regulator-associated ATP and adenosine 3′-phosphate 5′-phosphosulfate channels in endoplasmic reticulum and plasma membranes. Journal of Biological Chemistry. 1997;272:7746–7751. doi: 10.1074/jbc.272.12.7746. [DOI] [PubMed] [Google Scholar]
- Pedersen S, Lambert IH, Thoroed SM, Hoffmann EK. Hypotonic cell swelling induces translocation of the alpha isoform of cytosolic phospholipase A2 but not the gamma isoform in Ehrlich ascites tumor cells. European Journal of Biochemistry. 2000;267:5531–5539. doi: 10.1046/j.1432-1327.2000.01615.x. [DOI] [PubMed] [Google Scholar]
- Rainsford KD. Inhibitors of eicosanoid metabolism. In: Curtis-Prior PB, editor. Prostaglandins: Biology and Chemistry of Prostaglandins and Related Eicosanoids. Edinburgh: Churchill Livingstone; 1988. pp. 52–68. [Google Scholar]
- Reisin IL, Prat AG, Abraham EH, Amara JF, Grygory RJ, Ausiello DA, Cantiello HF. The cystic fibrosis transbembrane conductance regulator is a dual ATP and chloride channel. Journal of Biological Chemistry. 1994;269:20584–20591. [PubMed] [Google Scholar]
- Riquelme G, Parra M. Regulation of human placental chloride channel by arachidonic acid and other cis unsaturated fatty acids. American Journal of Obstetrics and Gynecology. 1999;180:469–475. doi: 10.1016/s0002-9378(99)70234-6. [DOI] [PubMed] [Google Scholar]
- Roman RM, Feranchak AP, Davison AK, Schwiebert EM, FitZ JG. Evidence for Gd3+ inhibition of membrane ATP permeability and purinergic signaling. American Journal of Physiology. 1999;277:G1222–1230. doi: 10.1152/ajpgi.1999.277.6.G1222. [DOI] [PubMed] [Google Scholar]
- Sabirov RZ, Dutta AK, Okada Y. Volume-dependent ATP-conductive large-conductance anion channel as a pathway for swelling-induced ATP release. Journal of General Physiology. 2001;118:251–266. doi: 10.1085/jgp.118.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Kakinoki B, Diener M, Takeguchi N. Endogenous arachidonic acid inhibits hypotonically-activated Cl− channels in isolated rat hepatocytes. Japanese Journal of Physiology. 1996;46:311–318. doi: 10.2170/jjphysiol.46.311. [DOI] [PubMed] [Google Scholar]
- SancheZ-Olea R, Morales-Mulia M, Moran J, Pasantes-Morales H. Inhibition by polyunsaturated fatty acids of cell volume regulation and osmolyte fluxes in astrocytes. American Journal of Physiology. 1995;269:C96–102. doi: 10.1152/ajpcell.1995.269.1.C96. [DOI] [PubMed] [Google Scholar]
- ScholZ-Pedretti K, Pfeilschifter J, Kaszkin M. Potentiation of cytokine induction of group IIA phospholipase A2 in rat mesangial cells by ATP and adenosine via the A2A adenosine receptor. British Journal of Pharmacology. 2001;132:37–46. doi: 10.1038/sj.bjp.0703774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiebert EK, Egan ME, Hwang T-H, Fulmer SB, Allen SS, Cutting GR, Guggino WB. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 1995;81:1063–1073. doi: 10.1016/s0092-8674(05)80011-x. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Aaronson PI. Modulatory effects of arachidonic acid on the delayed rectifier K+ current in rat pulmonary arterial myocytes. Structural aspects and involvement of protein kinase C. Circulation Research. 1996;79:20–31. doi: 10.1161/01.res.79.1.20. [DOI] [PubMed] [Google Scholar]
- Sugita M, Yue Y, Foskett JK. CFTR Cl− channel and CFTR-associated ATP channel: distinct pores regulated by common gates. EMBO Journal. 1998;17:898–908. doi: 10.1093/emboj/17.4.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira M, Bernard C, Ferrary E, Butlen D. Purine and pyrimidine nucleotide-sensitive phospholipase A2 in ampulla from frog semicircular canal. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2001;280:R519–526. doi: 10.1152/ajpregu.2001.280.2.R519. [DOI] [PubMed] [Google Scholar]
- Tewari KP, Malinowska DH, Sherry AM, Cuppoletti J. PKA and arachidonic acid activation of human recombinant ClC-2 chloride channels. American Journal of Physiology – Cell Physiology. 2000;279:C40–50. doi: 10.1152/ajpcell.2000.279.1.C40. [DOI] [PubMed] [Google Scholar]
- Thoroed SM, Lauritzen V, Lambert IH, Hansen HS, Hoffmann EK. Cell swelling activates phospholipase A2 in Ehrlich ascites tumor cells. Journal of Membrane Biology. 1997;160:47–58. doi: 10.1007/s002329900294. [DOI] [PubMed] [Google Scholar]
- Tinel H, Wehner F, Kinne RK. Arachidonic acid as a second messenger for hypotonicity-induced calcium transients in rat IMCD cells. Pflügers Archiv. 1997;433:245–253. doi: 10.1007/s004240050274. [DOI] [PubMed] [Google Scholar]
- Xu WX, Kim SJ, So I, Kang TM, Rhee JC, Kim KW. Volume-sensitive chloride current activated by hyposmotic swelling in antral gastric myocytes of the guinea pig. Pflügers Archiv. 1997;435:9–19. doi: 10.1007/s004240050478. [DOI] [PubMed] [Google Scholar]
- Zachar J, Hurnak O. Arachidonic acid blocks large-conductance chloride channels in L6 myoblasts. General Physiology and Biophysics. 1994;13:193–213. [PubMed] [Google Scholar]
- Zambon AC, Hughes JR, Meszaros V, Wu V, Torres V, Brunton V, Insel PA. P2Y2 receptor of MDCK cells: cloning, expression, and cell-specific signaling. American Journal of Physiology – Renal Physiology. 2000;279:F1045–1052. doi: 10.1152/ajprenal.2000.279.6.F1045. [DOI] [PubMed] [Google Scholar]