

Abstract
Exposure to reactive oxygen species (ROS) is associated with tissue damage in the lung and may be a common element in the pathogenesis of all inflammatory lung diseases. Exposure to the ROS hydrogen peroxide (H2O2) evoked a rapid increase in transepithelial anion secretion across monolayers of the human submucosal gland serous cell line Calu-3. This increase was almost entirely abolished by the addition of diphenylamine-2-carboxylate (DPC), implicating the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel in the response. The response was also reduced by inhibitors of basolateral K+ channels. Studies of electrically isolated apical and basolateral membranes revealed that H2O2 stimulated both apical Cl− and basolateral K+ conductances (GCl and GK). Apical GCl was sensitive to DPC, but unaffected by 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), suggesting that CFTR is the major anion conduction pathway mediating the response to H2O2. Additionally, H2O2 had no effect on GCl in the presence of the adenylate cyclase inhibitor SQ22536 or following maximal stimulation of GCl with forskolin, implicating the cAMP-dependent protein kinase pathway in the apical response to H2O2. Basolateral GK was reduced by the K+ channel inhibitors clotrimazole and clofilium, indicating roles for KCNN4 and KCNQ1 in the H2O2-stimulated response. We propose that ROS-stimulated anion secretion from serous cells plays an important role in keeping the airways clear from damaging radicals that could potentially initiate tissue destruction. Our finding that this response is CFTR dependent suggests that an important host defence mechanism would be dysfunctional in the cystic fibrosis (CF) lung. Loss of this compensatory protective mechanism could expose the CF lung to ROS for extended periods, which could be important in the pathogenesis of CF lung disease.
The human respiratory tract is constantly exposed to transient instances of oxidant stress resulting from the inhalation of a variety of foreign material including atmospheric pollutants and microorganisms. Furthermore, the production of reactive oxygen species (ROS) during episodes of infection and inflammation has been implicated in the pathogenesis of a number of pulmonary diseases (Rahman & MacNee, 2000; van der Vliet & Cross, 2000). Recruitment of activated inflammatory cells such as eosinophils, neutrophils and macrophages results in the generation of highly cytotoxic ROS, which may lead to tissue damage via direct oxidation of proteins, DNA or lipids. In order to combat potential incidents of oxidant stress, the airways possess a number of anti-oxidant defence mechanisms to neutralize ROS (van der Vliet et al. 1997).
The submucosal gland serous cell is actively involved in the maintenance of a sterile environment within the airways (Basbaum et al. 1990) and serous cell secretions play a multitude of roles in primary host defence. In addition to secreting potent antimicrobial agents such as lysozyme, lactoferrin, and protease inhibitors, serous cells also control glandular secretion of salt and water, critical for mucus hydration and controlling the depth of airway surface liquid present on the surface epithelial cells, required for efficient mucociliary clearance (reviewed in Wine, 1999). The serous cell is also the principal site of expression in the lung of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel (Engelhardt et al. 1992), the channel which dysfunctions in cystic fibrosis (CF), which has led to the proposal that the normal physiological activity of these cells is highly disrupted in CF (Pilewski & Frizzell, 1999).
The Calu-3 cell line has become a widely used and accepted model of the human serous cell (Shen et al. 1994; Cowley & Linsdell, 2002). In the present study, we investigate how this model cell line responds to incidents of acute oxidant stress and propose a novel mechanism by which the airway may be protected from exposure to ROS, but which may be significantly impaired in the CF lung.
Methods
Measurement of transepithelial short-circuit current
Calu-3 cells (American Type Culture Collection, Rockville, MD, USA) were maintained and plated on Snapwell inserts (Corning Costar, Cambridge, MA, USA), as previously described (Cowley & Linsdell, 2002). Cells were grown at an air-liquid interface with medium present only on the basolateral side and experiments performed 10–20 days after the establishment of this interface. Inserts were mounted in an Ussing chamber (World Precision Instruments (WPI), Sarasota, FL, USA), and the transepithelial potential difference was clamped to zero using a DVC-1000 voltage-clamp apparatus (WPI). The transepithelial short-circuit current (Isc) was recorded using Ag-AgCl electrodes in agar bridges. Apical and basolateral solutions were maintained at 37 °C by heated water jackets, and were separately perfused and oxygenated with a 95 % O2-5 % CO2 mixture. Bath solutions for intact monolayers consisted of (mm): 120 NaCl, 25 NaHCO3, 3.3 KH2PO4, 0.8 K2HPO4, 1.2 MgCl2, 1.2 CaCl2, 10 glucose (basolateral) or mannitol (apical), pH 7.4 at 37 °C, when gassed with 95 % O2-5 % CO2. For the HCO3−-free experiments, buffers contained (mm): 145 NaCl, 3.3 KH2PO4, 0.8 K2HPO4, 1.2 MgCl2, 1.2 CaCl2, 10 Hepes and 10 glucose (basolateral) or mannitol (apical). The pH was adjusted to 7.4 with NaOH and experiments were gassed with air.
Permeabilized monolayers
To investigate the activity of apical Cl− and basolateral K+ channels in isolation, the opposite membrane was permeabilized by addition of 180 μg ml−1 nystatin (Sigma, Oakville, ON, Canada) in the presence of appropriate buffers. Thus, apical Cl− conductance (GCl) was studied in the presence of a serosal to mucosal Cl− gradient with the following bath solutions. Apical (mm): 145 sodium gluconate, 3.3 NaH2PO4, 0.8 Na2HPO4, 1.2 magnesium gluconate, 4 calcium gluconate, 10 glucose, 10 Hepes; basolateral (mm): 145 NaCl, 3.3 NaH2PO4, 0.8 Na2HPO4, 1.2 MgCl2, 1.2CaCl2, 10 mannitol, 10 Hepes. For experiments involving the adenylate cyclase inhibitor SQ22536, cells were pre-incubated for 60 min at 37 °C prior to the beginning of the experiment.
Basolateral K+ channels were studied by permeabilization of the apical membrane in the presence of a mucosal to serosal K+ gradient established by the following bath solutions. Apical (mm): 145 potassium gluconate, 3 KH2PO4, 0.8 K2HPO4, 1.2 magnesium gluconate, 4 calcium gluconate, 10 glucose, 10 Hepes; basolateral (mm): 145 sodium gluconate, 3.3 NaH2PO4, 0.8 Na2HPO4, 1.2 magnesium gluconate, 4 calcium gluconate, 10 glucose, 10 Hepes. All solutions were pH 7.4 at 37 °C.
Chemicals
Hydrogen peroxide (H2O2), diphenylamine-2-carboxylate (DPC), nystatin, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), forskolin and clotrimazole were obtained from Sigma Aldrich. Clofilium was purchased from RBI (Natick, MA, USA). SQ22536 was obtained from Calbiochem (San Diego, CA, USA) and made up in buffer. Stock solutions of DPC and clotrimazole were made up in ethanol, while nystatin, DIDS and clofilium were made up in DMSO so that the final bath concentration of solvent was ≤ 0.1 %. Application of DMSO or ethanol alone had no effect upon the monolayers. H2O2 was made up immediately prior to use in appropriate buffer.
Statistics
All data are presented as means ± standard error of the mean (s.e.m.). Student's t test or one-way analysis of variance followed by Bonferroni's t test were used to evaluate the significance of differences as appropriate. P < 0.05 was considered significant.
Results
Effect of H2O2 on Isc across intact Calu-3 monolayers
The basal Isc in intact monolayers of Calu-3 cells was 28.9 ± 1.5 μA cm−2 (range, 11.1-57.1 μA cm−2) with an initial resistance of 138.8 ± 5.4 Ω cm2 (n = 55), similar to previously reported values (Cowley & Linsdell, 2002). Basal Isc in these cells has previously been demonstrated to be almost exclusively accounted for by anion secretion (Singh et al. 1997). Basal Isc was increased by the addition of H2O2 to the apical face of intact Calu-3 monolayers (Fig. 1A) and this increase was dose dependent (Fig 1B). One millimolar H2O2 was chosen as the dose for all subsequent investigations since this caused a significant, but mid-range, effect (increase = 31.7 ± 2.4 μA cm−2, n = 17). The increase in Isc caused by the addition of 1 mm H2O2 could be reversed by washing. Subsequent addition of 1 mm H2O2 again increased Isc, causing an increase similar to the initial response (Fig. 1A). Indeed, the response to all the concentrations of H2O2 examined (up to 8 mm) were fully reversible (results not shown). Addition of 1 mm H2O2 to the basolateral side of the monolayers also increased Isc, although to a significantly lesser extent (10.2 ± 0.7 μA cm−2, n = 4, versus 31.7 ± 2.4 μA cm−2, n = 17, significance determined using Student's t test).
Figure 1. H2O2 stimulates short-circuit current (Isc) across Calu-3 monolayers.
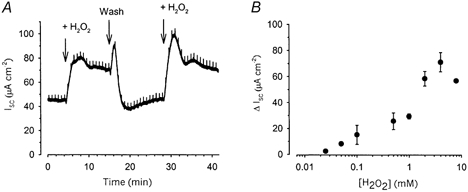
Application of H2O2 (1 mm) to the apical face of Calu-3 monolayers increased Isc in a reversible manner (A). This increase in Isc was dose dependent (B; each point represents the mean of between 3 and 17 experiments).
A transient activation of stimulation following the wash-out of H2O2 can be seen in Fig. 1A, and was a commonly seen phenomenon. Furthermore, close examination of the effect of addition of H2O2 to intact monolayers sometimes revealed a transient inhibition prior to the much larger, sustained stimulatory response. One possible explanation for these effects is that addition of H2O2 produces an inhibitory effect prior to the more sustained stimulatory response we see. Indeed, H2O2 has previously been reported to inhibit cAMP-induced Cl− secretion across colonic epithelial cells (DuVall et al. 1998) and it is possible that H2O2 has distinct effects upon Calu-3 cells. Thus when the H2O2 is washed out it may be that the inhibitory effect is removed more rapidly, permitting the larger transient stimulatory effect to be seen. However, this phenomenon was not explored in any further detail.
Pharmacological inhibition of H2O2-stimulated anion secretion
Basal and stimulated anion secretion from Calu-3 cells has previously been shown to be dependent upon the activity of CFTR Cl− channels (Shen et al. 1994; Singh et al. 1997; Devor et al. 1999). Therefore, we investigated whether the increased Isc seen in response to H2O2 was also mediated via CFTR-dependent anion secretion. To do this, we examined the effect of the CFTR inhibitor DPC on the response to H2O2. When 0.5 mm DPC was added prior to 1 mm H2O2, it inhibited the subsequent oxidant-mediated increase in Isc by 88.3 ± 5.1 % (n = 5; Fig. 2).
Figure 2. H2O2-stimulated Isc across Calu-3 monolayers is inhibitable by DPC.
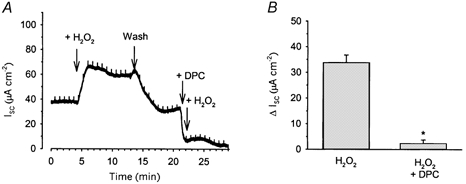
The increase in Isc seen when 1 mm H2O2 was applied to the apical face of Calu-3 monolayers could be almost completely inhibited by 0.5 mm DPC (A and B). * Significantly different from control (H2O2) as determined by Student's t test (P < 0.05).
Since the rate of transepithelial anion secretion in Calu-3 cells is dependent upon the activity of basolateral K+ channels (Devor et al. 1999; Cowley & Linsdell, 2002), which generate the driving force for anion efflux through open apical membrane channels, we investigated the effect of the K+ channel inhibitors clofilium and clotrimazole upon H2O2-stimulated anion secretion. Our previous work has shown that, at the concentrations used in this study, clotrimazole and clofilium can be used as specific inhibitors to effectively distinguish between two distinct populations of basolateral K+ channels in Calu-3 cells: a clotrimazole-sensitive Ca2+-activated K+ channel (KCNN4) and a clofilium-sensitive cAMP-activated K+ channel (probably KCNQ1; Cowley & Linsdell, 2002). To test whether activation of either of these K+ channels was involved in the H2O2-stimulated increase in secretion, we applied either 100 μM clofilium or 30 μM clotrimazole prior to the H2O2 stimulus. Clofilium (100 μM) significantly reduced the magnitude of the increase in Isc in response to 1 mm H2O2 (8.6 ± 0.3 μA cm−2, n = 4, versus control 31.7 ± 2.4 μA cm−2, n = 17; Fig. 3A and C). Similarly, application of 30 μM clotrimazole also significantly reduced the size of the subsequent H2O2-mediated increase in Isc compared with control (13.7 ± 1.2 μA cm−2, n = 3; Fig. 3B and C). Therefore, both KCNN4 and KCNQ1 appear to play a role in the increase in Isc due to H2O2.
Figure 3. H2O2-stimulated Isc across Calu-3 monolayers is inhibited by blockers of basolateral K+ channels.
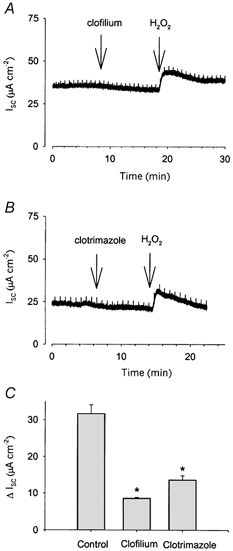
The increase in Isc seen in response to application of 1 mm H2O2 to the apical face of Calu-3 monolayers was significantly reduced when the oxidant was added following application of 100 μM clofilium (A and C; n = 4) or 30 μM clotrimazole (B and C; n = 3), applied to the basolateral face. * Significantly different from Control as determined by ANOVA, followed by Bonferroni's t test (P < 0.016).
Evidence for bicarbonate secretion
Devor et al. (1999) clearly demonstrated that Calu-3 cells are capable of secreting either Cl− or HCO3− depending upon the stimulus for secretion, although the precise mechanisms underlying this ability to switch between Cl− or HCO3− secretion is unclear. We undertook a series of experiments under HCO3−-free conditions to see if any of the secretory response we detect to H2O2 could be accounted for by HCO3−. When experiments were performed under HCO3−-free conditions, the increase in Isc in response to 1 mm H2O2 was reduced to 21.8 ± 1.2 μA cm−2 (n = 8; Fig. 4), significantly less than the response when HCO3− was present (31.7 ± 2.4 μA cm−2, n = 17).
Figure 4. The increase in Isc in response to H2O2 contains a HCO3− component.
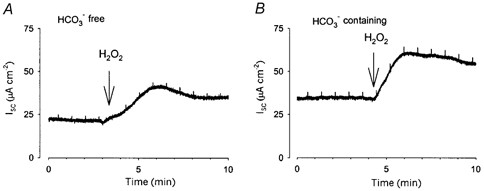
When 1 mm H2O2 was applied to Calu-3 monolayers under HCO3−-free conditions (A; n = 8), the increase in Isc was significantly smaller than that seen when bicarbonate was present (B). Significance determined using Student's t test (P < 0.05).
Effect of H2O2 on apical Cl− conductance
To further examine the mechanism of the increase in transepithelial anion secretion in response to H2O2, we isolated the apical membrane GCl by permeabilizing the basolateral membrane with the pore-forming antibiotic nystatin in the presence of a serosal to mucosal Cl− gradient. Addition of 1 mm H2O2 to the apical face of the permeabilized monolayers resulted in an increase in Isc (38.9 ± 5.2 μA cm−2, n = 8; Fig. 5A), which under these conditions represents increased efflux of Cl− ions across the apical membrane down their concentration gradient. This increase in Isc was insensitive to the disulphonic stilbene DIDS (250 μM) applied to the extracellular aspect of the membrane (n = 4; Fig. 5B). While DIDS inhibits a variety of Cl− channels including epithelial outwardly rectifying Cl− channels (Bridges et al. 1989), CFTR is insensitive to extracellularly applied DIDS. However, the H2O2-stimulated increase in Isc was sensitive to the application of the CFTR inhibitor DPC (n = 7; Fig. 5B). Such a DPC-sensitive, DIDS-insensitive response is a characteristic hallmark of CFTR (Schultz et al. 1999).
Figure 5. Apical Cl− conductance in permeabilized Calu-3 cells is increased by H2O2.
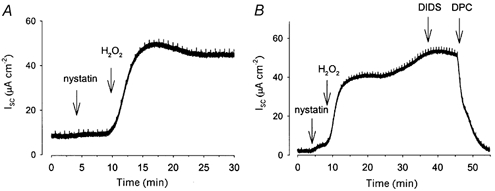
Apical GCl was isolated by permeabilizing the basolateral membrane with nystatin in the presence of a serosal to mucosal Cl− gradient, so that an increase in Isc reflects anion efflux. Addition of 1 mm H2O2 to the apical face of the membrane induced an increase in GCl (A; n = 8), which was insensitive to 250 μM DIDS (B; n = 4) but inhibited by 1 mm DPC applied apically (B; n = 7).
To investigate the mechanism responsible for the increase in apical membrane GCl, we used an inhibitor of adenylate cyclase, SQ22536. Monolayers of Calu-3 cells were pre-incubated with SQ22536 for 60 min prior to permeabilization of the basolateral membrane with nystatin. Under these conditions, application of 1 mm H2O2 to the apical membrane produced no increase in Isc (n = 3; Fig. 6A), indicating that adenylate cyclase activity is required to mediate the H2O2-induced increase in GCl.
Figure 6. Apical Cl− conductance in response to H2O2 is likely to occur via the protein kinase A pathway.
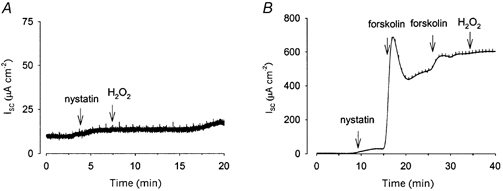
The increase in apical GCl seen in response to H2O2 (1 mm) was abolished when monolayers were pre-incubated for 60 min with 0.5 mm SQ22536, an inhibitor of adenylate cyclase (A; n = 3). In B, maximal activation of the cAMP pathway was achieved by additions of 2 μM forskolin. Under these conditions, the subsequent addition of H2O2 caused no further increase in GCl.
We also investigated whether the effects of H2O2 were occurring via the cAMP pathway by maximally stimulating that pathway with forskolin before the addition of the oxidant. Figure 6B shows that, following application of forskolin, there was no further increase in apical membrane GCl when the oxidant was applied. This result, together with the lack of response following adenylate cyclase inhibition, implicates the cAMP-dependent protein kinase pathway in the apical response to H2O2.
Effect of H2O2 on basolateral K+ conductance
To isolate the basolateral K+ conductance (GK) the apical membrane was permeabilized with nystatin in the presence of Cl−-free buffers to establish a mucosal to serosal K+ gradient. Under these conditions, application of 1 mm H2O2 to the apical aspect of permeabilized Calu-3 monolayers produced an increase in Isc of 21.5 ± 2.8 μA cm−2 (n = 6; Fig. 7A) which reflects K+ movement through basolateral K+ channels down a concentration gradient. Application of 1 mm H2O2 to the basolateral side of the monolayers produced a smaller increase in Isc (11.9 ± 1.9 μA cm−2; n = 9). Regardless of whether the H2O2 was added to the basolateral or apical face, this increase in K+ flux appeared biphasic, with a rapid onset transient increase in Isc being followed by a slower onset, more sustained response. Both phases of the response were of similar magnitude. Since application of the oxidant to the apical side of the monolayers produced a significantly larger response, all subsequent basolateral K+ conductance experiments were performed with 1 mm H2O2 applied apically.
Figure 7. Basolateral K+ conductance in permeabilized Calu-3 cells is increased by H2O2.
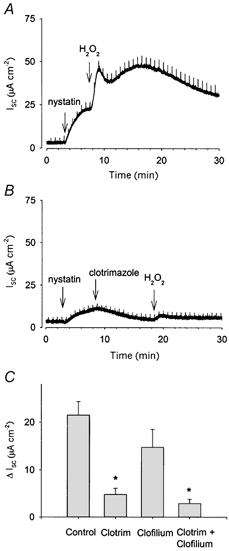
Basolateral GK was isolated by permeabilization of the apical membrane with nystatin in the presence of a mucosal to serosal K+ gradient. The application of 1 mm H2O2 to the apical membrane under these conditions caused a biphasic increase in Isc, reflecting an increase in K+ efflux (A; n = 6). B and C, addition of 30 μM clotrimazole (Clotrim) to the basolateral face caused a decrease in the size of the H2O2 response, but did not completely abolish it. Application of 100 μM clofilium basolaterally also reduced the size of the H2O2 response, though this decrease was not significant (C). Application of both clotrimazole and clofilium reduced the response further, but could not entirely abolish it (C). * Significantly different from Control as determined by ANOVA, followed by Bonferroni's t test (P < 0.008).
Since application of the basolateral K+ channel inhibitors clofilium and clotrimazole to intact monolayers of Calu-3 cells reduced the increase in Isc in response to H2O2 (Fig. 3), we further investigated the action of these inhibitors directly on basolateral membranes. Permeabilized monolayers were exposed to 1 mm H2O2 in the presence of either 30 μM clotrimazole or 100 μM clofilium applied to the basolateral face. Pre-application of 30 μM clotrimazole resulted in a large decrease in the size of the H2O2-mediated response (mean increase = 4.8 ± 1.3 μA cm−2, n = 3, Fig. 7B and C, compared with control 21.5 ± 2.8 μA cm−2, n = 6). Pre-application of 100 μM clofilium reduced the size of the H2O2-mediated response (14.7 ± 3.8 μA cm−2, n = 4), although this was not statistically significant. Addition of both clofilium and clotrimazole together almost, but not entirely, eliminated the response (mean increase in response to H2O2 = 2.3 ± 0.9 μA cm−2, n = 4; Fig. 7C). It appears therefore that the major component of this increase in GK may be mediated via the Ca2+-activated KCNN4, while KCNQ1 appears less important. Furthermore, the presence of a small oxidant-stimulated response in the presence of clotrimazole and clofilium suggests that one or more additional unidentified basolateral K+ conductances may also be stimulated by application of H2O2.
Discussion
We here present data indicating that the ROS, H2O2, stimulates transepithelial anion secretion across intact monolayers of Calu-3 cells. Furthermore, this increased anion secretion appears to be mediated via apically located CFTR Cl− channels. Basolateral K+ channels are also stimulated in response to H2O2, which may indicate the existence of a coordinated ion transport mechanism that could maximize the secretory response in Calu-3 cells.
Submucosal gland serous cells secrete a variety of antimicrobial compounds, including lysozyme, lactoferrin, peroxidase, secretory leukocyte protease inhibitor and secretory IgA (Basbaum et al. 1990). These cells also play a crucial role in maintaining effective mucociliary clearance, since they are responsible for the glandular secretion of salt and water (reviewed by Pilewski & Frizzell, 1999), hydrating mucus as it is secreted from mucous cells and regulating the volume of the airway surface liquid lining the respiratory epithelial cells. Thus, serous cell secretions play a critical role in defence of the airways from inflammatory insults and the presence of other noxious material. Serous cells also express high levels of the CFTR Cl− channel, involved in controlling salt and water secretion, but which is mutated in CF. Pulmonary disease is presently the major cause of morbidity and mortality associated with CF and the progression of CF lung disease is characterized by recurrent bacterial infections, persistent inflammation, bronchiectasis and eventual tissue destruction. The high degree of CFTR expression in the serous cell in relation to other airway cell types has led to the proposal that these cells represent the primary site of CF pathology (Pilewski & Frizzell, 1999).
Activation of inflammatory phagocytic cells leads to the induction of a respiratory burst as cells attack invading organisms. O2 is converted to O2.− (superoxide) when NADPH donates an electron, a reaction catalysed by NADPH oxidase (for review see van der Vliet & Cross, 2000). Most of the O2.− produced is converted to H2O2, which is highly toxic to bacteria. Thus, H2O2 is a physiologically relevant ROS, important in host defence mechanisms in the airways and implicated in the pathogenesis of CF lung disease (van der Vliet et al. 1997). In the present study we report that acute exposure to H2O2 stimulates transepithelial anion secretion from Calu-3 cells, a well-characterized and widely used model of human submucosal gland serous cells. Furthermore, our finding that this anion secretion occurs almost entirely via apical CFTR has significant implications for the CF lung.
CF lung disease is characterized by a persistent influx of neutrophils into the airways (Konstan & Berger, 1993). Suspensions of 3 × 105 neutrophils can generate up to 15 μM H2O2 (Test & Weiss, 1984), a concentration that can be potentiated when those neutrophils are adherent to cell surfaces (Nathan, 1987). Bronchoalveolar lavage of CF patients has recorded a count of 38 × 106 neutrophils per millilitre extracellular lining fluid (Konstan et al. 1994), which is approximately 380 times higher than in healthy, control subjects. Thus localized accumulations of activated neutrophils could transiently generate high concentrations of H2O2 equal to those we find stimulate anion secretion (Fig. 1B).
Our observation that H2O2 stimulates transepithelial anion secretion across Calu-3 cells suggests that submucosal gland serous cells dynamically respond to incidents of oxidant stress in the airways as a part of a normal defence mechanism. Recent reports indicate that Calu-3 cells are able to respond to a variety of stresses as part of an integrated host defence mechanism, since physical stimulation (Huang et al. 2001) and exposure to nitric oxide (Duszyk, 2001a) both stimulate CFTR activity. In the present manuscript we demonstrate that the ROS H2O2 stimulates anion secretion from Calu-3 cells. In the submucosal gland in vivo, anion secretion is coupled to fluid secretion (Ballard et al. 1999; Devor et al. 1999; Pilewski & Frizzell, 1999) and it is this release of fluid from the more distal serous cells that is responsible for the ‘flushing out’ of mucins from the more proximal mucous cells of the submucosal gland. Furthermore, the fluid secreted from submucosal gland serous cells is rich in endogenous antibacterial agents (Basbaum et al. 1990). Therefore, any agent stimulating secretion from the serous cell would result in the release of fluid rich in the potent antimicrobial agents such as lysozyme, lactoferrin and protease inhibitors. We propose therefore that ROS-stimulated secretion from these cells would result in release of fluid rich in endogenous antibacterials and altered airway surface liquid quantity and quality. Together these responses could act to rid the airways of the inflammatory insult that triggered the initial ROS production (Fig. 8). This secretion may also occur in parallel with the release of mucus from submucosal gland mucous cells, since ROS reportedly increase mucus secretion from respiratory epithelial cells (Adler et al. 1990). Together, the release of hydrated mucus from the submucosal gland would optimize mucociliary clearance and assist the airways in expelling bacteria.
Figure 8. Proposed mechanism of exacerbation of ROS-mediated tissue damage in the CF lung via loss of CFTR-mediated host defence mechanisms in the serous cell.
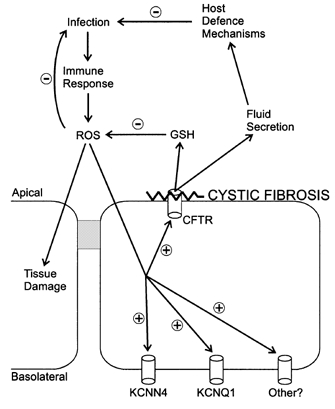
ROS are hypothesized to stimulate CFTR-mediated host defence mechanisms, via the coordinate activity of basolateral K+ and apical CFTR Cl− channels. Stimulation may be direct (for example, via protein oxidation of the channel) or indirect (via second messenger pathways such as PKA). In the CF lung, this relationship would be severed by the functional absence of CFTR, leading to an increased potential for ROS-mediated tissue damage. GSH, glutathione.
The increase in Isc seen in response to H2O2 in Calu-3 cells was almost entirely inhibited by DPC (Fig. 2). Furthermore, in permeabilized monolayers the H2O2-stimulated increase in apical membrane GCl was DIDS insensitive and DPC sensitive (Fig. 2 and Fig. 5), a pharmacological profile which strongly suggests activation of CFTR. CFTR activity in Calu-3 cells is known to be regulated by protein kinase A (PKA) (Huang et al. 2000; Cobb et al. 2002) and our finding that incubation with the adenylate cyclase inhibitor SQ22536 abolished the H2O2-stimulated increase in apical anion conductance provides evidence that the oxidant effect is also mediated via the PKA pathway (Fig. 6).
Our finding that the H2O2-stimulated increase in secretion appears to be mediated via CFTR raises the intriguing possibility that a major defence mechanism normally protecting the airway from incidents of oxidant stress is absent from the CF lung (see Fig. 8). CF is associated with recurrent cycles of bacterial infection and inflammatory responses which expose the CF airways to high levels of ROS as activated phagocytic cells migrate to the site of infection. Since the H2O2-stimulated CFTR-mediated increase in secretion we report here would be absent in the CF lung, we propose that the CF lung has a reduced capacity to secrete anions in response to oxidant stress. Thus, there would be a concomitant decrease in fluid secretion from these cells – a fluid rich in antimicrobial agents that could normally help clear the infectious agents.
In Calu-3 cells, exposure to H2O2 leads to an increase in both apical membrane GCl (Fig. 5) and basolateral membrane GK (Fig. 7). We previously determined that the net rate of secretion from Calu-3 cells is determined by the activity of basolateral K+ channels (Cowley & Linsdell, 2002). It appears that stimulation of both channel types could occur as part of a coordinated response to maximize the secretory response of the cells to the ROS insult. The Ca+-activated K+ channel KCNN4 appears stimulated by oxidant stress, although it is not apparent whether this stimulation is due to increased levels of Ca2+ directly affecting the channel, or to the effects of PKA, since Gerlach et al. (2000) determined that the Ca2+-dependent gating of KCNN4 was modulated by PKA-dependent phosphorylation. In contrast, the cAMP-activated KCNQ1 does not appear to be stimulated by H2O2 to the same extent, since the effect of clofilium on isolated basolateral GK was less dramatic. However, we determined clofilium to be a potent inhibitor of H2O2-stimulated Isc in intact monolayers (Fig. 3). Together these results suggest that it is inhibition of resting KCNQ1 activity (and the consequent decrease in the driving force for anion efflux via apical CFTR) that is responsible for the effects of clofilium in intact monolayers, since H2O2 does not appear to directly stimulate KCNQ1.
Another important airway host defence response affected by loss of functional CFTR is release of the powerful antioxidant glutathione (GSH). GSH is present in high quantities in the extracellular fluid lining the airway epithelium where it acts to directly scavenge ROS (van der Vliet & Cross, 2000). CFTR has been directly implicated in deficient GSH release from CF airway epithelial cells (Linsdell & Hanrahan, 1998; Gao et al. 1999) resulting in the much-reduced levels of GSH reported in the extracellular fluid lining the CF lung (Roum et al. 1993). Since H2O2 increases CFTR activity, it should also stimulate CFTR-mediated GSH release. This potentially compensatory oxidant-induced anti-oxidant release would be lost in the CF lung (Fig. 8). Similarly, CFTR may be directly involved in the release of antimicrobial compounds such as lysozyme from submucosal glands (Duszyk, 2001b). Thus CFTR-mediated release of antioxidants and antimicrobials could effectively limit the time course of airway exposure to ROS and ROS-producing microorganisms whilst loss of these mechanisms would further compromise the CF lung.
CFTR is permeable to both Cl− and HCO3− (Linsdell et al. 1997) and our results suggest that both Cl− and HCO3− may be secreted in response to H2O2 (Fig. 4). Devor et al. (1999) were the first to demonstrate that Calu-3 cells are capable of switching between Cl− and HCO3− secretion, depending upon the stimulus. These authors suggest that HCO3− secretion could potentially affect the clearance of mucus and antimicrobial agents from submucosal glands; therefore loss of CFTR-mediated HCO3− secretion could further compromise the CF lung and reduce its ability to carry out effective mucociliary clearance.
In conclusion, we propose the presence of ROS in the extracellular environment signals to serous cells that an infectious insult is present in the airways and that an immune response has been initiated in response to that insult (Fig. 8). ROS may act as a trigger for secretion of fluid from serous cells, a secretory response that we propose is maximized by the coordinated stimulation of apical CFTR and basolateral K+ channels. In this way, submucosal gland serous cells may dynamically respond to oxidant stress by secreting fluid rich in antibacterial compounds and antioxidants and which would also hydrate mucus. Such reactions would be central to airway host defence and help act to clear infections before ROS-mediated tissue damage can be initiated. This potentially crucial feedback mechanism would be lost in CF, predisposing the CF lung to extensive exposure to infection and inflammation, excessive contact with ROS, and consequent initiation of tissue damage.
Acknowledgments
We wish to thank Angie Lewis and Susan Burbridge for technical assistance. This work was supported by the Canadian Cystic Fibrosis Foundation (CCFF) and the Canadian Institutes for Health Research. P.L. is a CCFF scholar.
References
- Adler KB, Holden-Stauffer WJ, Repine JE. Oxygen metabolites stimulate release of high-molecular-weight glycoconjugates by cell and organ cultures of rodent respiratory epithelium via an arachadonic acid-dependent mechanism. Journal of Clinical Investigation. 1990;85:75–85. doi: 10.1172/JCI114436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard ST, Trout L, Bebök Z, Sorscher EJ, Crews A. CFTR involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. American Journal of Physiology. 1999;277:L694–699. doi: 10.1152/ajplung.1999.277.4.L694. [DOI] [PubMed] [Google Scholar]
- Basbaum CB, Berthold J, Finkbeiner WE. The serous cell. Annual Review of Physiology. 1990;52:97–113. doi: 10.1146/annurev.ph.52.030190.000525. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Worrell RT, FriELLZZ RA, Benos DJ. Stilbene disulfonate blockade of colonic secretory Cl− channels in polar lipid bilayers. American Journal of Physiology. 1989;256:C902–912. doi: 10.1152/ajpcell.1989.256.4.C902. [DOI] [PubMed] [Google Scholar]
- Cobb BR, RuiZ F, King CM, Fortenberry J, Greer H, Kovacs T, Sorscher EJ, Clancy JP. A2 adenosine receptors regulate CFTR through PKA and PLA2. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2002;282:L12–25. doi: 10.1152/ajplung.2002.282.1.L12. [DOI] [PubMed] [Google Scholar]
- Cowley EA, Linsdell P. Characterization of basolateral K+ channels underlying anion secretion in the human airway cell line Calu-3. Journal of Physiology. 2002;538:747–757. doi: 10.1113/jphysiol.2001.013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor DC, Singh AK, Lambert LC, Deluca A, FriELLZZ RA, Bridges RJ. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. Journal of General Physiology. 1999;133:743–760. doi: 10.1085/jgp.113.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duszyk M. Regulation of anion secretion by nitric oxide in human airway epithelial cells. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2001a;281:L450–457. doi: 10.1152/ajplung.2001.281.2.L450. [DOI] [PubMed] [Google Scholar]
- Duszyk M. CFTR and lysozyme secretion in human airway epithelial cells. Pflügers Archiv. 2001b;443:S45–49. doi: 10.1007/s004240100643. [DOI] [PubMed] [Google Scholar]
- DuVall MD, Guo Y, Matalon S. Hydrogen peroxide inhibits cAMP-induced Cl− secretion across colonic epithelial cells. American Journal of Physiology. 1998;275:C1313–1322. doi: 10.1152/ajpcell.1998.275.5.C1313. [DOI] [PubMed] [Google Scholar]
- Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nature Genetics. 1992;2:240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- Gao L, Kim KJ, Yankaskas JR, Forman HJ. Abnormal glutathione transport in cystic fibrosis airway epithelia. American Journal of Physiology. 1999;277:L113–118. doi: 10.1152/ajplung.1999.277.1.L113. [DOI] [PubMed] [Google Scholar]
- Gerlach AC, Gangopadhyay NN, Devor DC. Kinase-dependent regulation of the intermediate conductance, calcium-dependent potassium channel, hIK1. Journal of Biological Chemistry. 2000;275:585–598. doi: 10.1074/jbc.275.1.585. [DOI] [PubMed] [Google Scholar]
- Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proceedings of the National Academy of Sciences of the USA. 2001;98:14120–14125. doi: 10.1073/pnas.241318498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Trotter K, Boucher RC, Milgram SL, Stutts MJ. PKA holoenzyme is functionally coupled to CFTR by AKAPs. American Journal of Physiology – Cell Physiology. 2000;278:C417–422. doi: 10.1152/ajpcell.2000.278.2.C417. [DOI] [PubMed] [Google Scholar]
- Konstan MW, Berger M. Infection and inflammation in the lung in cystic fibrosis. In: Davis PB, editor. Cystic Fibrosis. New York: Marcel Dekker Inc.; 1993. pp. 219–276. [Google Scholar]
- Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. American Journal of Respiratory and Critical Care Medicine. 1994;150:448–454. doi: 10.1164/ajrccm.150.2.8049828. [DOI] [PubMed] [Google Scholar]
- Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. American Journal of Physiology. 1998;275:C323–326. doi: 10.1152/ajpcell.1998.275.1.C323. [DOI] [PubMed] [Google Scholar]
- Linsdell P, Tabacharani JA, Rommens JM, Hou YX, Chang XB, Tsui LC, Riordan JR, Hanrahan JW. Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. Journal of General Physiology. 1997;110:355–364. doi: 10.1085/jgp.110.4.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan CF. Neutrophil activation on biological surfaces. Journal of Clinical Investigation. 1987;80:1550–1560. doi: 10.1172/JCI113241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilewski JM, FriELLZZ RA. Role of CFTR in airway disease. Physiological Reviews. 1999;79:S215–255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. European Respiratory Journal. 2000;16:534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]
- Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. Journal of Applied Physiology. 1993;75:2419–2424. doi: 10.1152/jappl.1993.75.6.2419. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Singh AK, Devor DC, Bridges RJ. Pharmacology of CFTR chloride channel activity. Physiological Reviews. 1999;79:S109–144. doi: 10.1152/physrev.1999.79.1.S109. [DOI] [PubMed] [Google Scholar]
- Shen B-Q, Finkbeiner WE, Wine JJ, Mrsny RJ, Widdicombe JH. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl− secretion. American Journal of Physiology. 1994;266:L493–501. doi: 10.1152/ajplung.1994.266.5.L493. [DOI] [PubMed] [Google Scholar]
- Singh M, Krouse ME, Moon S, Wine JJ. Most basal Isc in Calu-3 human airway cells is bicarbonate-dependent Cl− secretion. American Journal of Physiology. 1997;272:L690–698. doi: 10.1152/ajplung.1997.272.4.L690. [DOI] [PubMed] [Google Scholar]
- Test ST, Weiss SJ. Quantitative and temporal characterization of the extracellular H2O2 pool generated by human neutrophils. Journal of Biological Chemistry. 1984;259:399–405. [PubMed] [Google Scholar]
- van der Vliet A, Cross CE. Oxidants, nitrosants, and the lung. American Journal of Medicine. 2000;109:398–421. doi: 10.1016/s0002-9343(00)00479-4. [DOI] [PubMed] [Google Scholar]
- van der Vliet A, Eiserich JP, Marelich GP, Halliwell B, Cross CE. Oxidative stress in cystic fibrosis: does it occur and does it matter? Advances in Pharmacology. 1997;38:491–513. doi: 10.1016/s1054-3589(08)60996-5. [DOI] [PubMed] [Google Scholar]