

Abstract
Many fast excitatory synapses in the hippocampus are enriched with both AMPARs (α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate receptors) and NMDARs (N-methyl-d-aspartate receptors). Their proximity allows them to be activated simultaneously by the same neurotransmitter, l-glutamate. Activation of AMPARs leads to influx of sodium and calcium ions, which can increase or decrease NMDAR activity through sodium concentration-dependent cascades or a calcium-calmodulin-dependent inactivation process, respectively. Here we provide evidence that the activation of AMPARs inhibits NMDARs through a non-ionotropic mechanism. NMDA-induced current in isolated rat CA1 hippocampal cells and nucleated patches of cultured mouse hippocampal neurones decreased when AMPARs were activated. Conversely, when AMPARs were blocked, the NMDA component of glutamate-induced current increased. The inhibitory action of AMPAR activation on NMDAR-mediated current depends upon the open state of AMPA channels and rapidly diminishes after deactivation of AMPARs. The inhibitory action was independent of membrane voltage, univalent cation fluxes and calcium influx. The AMPA-NMDA cross-inhibition also occurred in evoked synaptic current in CA1 neurones from intact mouse hippocampal slices. This cross-talk may play a role in preventing overexcitation during bursting activities in the hippocampus.
Ionotropic glutamate receptors are the primary excitatory neurotransmitter receptors in the mammalian brain and there are three subtypes: AMPA, kainate and NMDA receptors. Two of them – the AMPA and NMDA receptors – are commonly found in excitatory synapses. AMPARs are hetero- or homo-oligomers of the four subunits GluR1-4 and are the major mediators for fast excitatory synaptic transmission. NMDARs are hetero-oligomers of NR1 and NR2a-d subunits and play an essential role in modulating synaptic plasticity, namely long-term potentiation and long-term depression (Bliss & Collingridge, 1993; Bear & Abraham, 1996; Malenka & Nicoll, 1999).
Two important characteristics of fast excitatory synaptic transmission make it possible for the AMPARs and NMDARs to interact locally. First, in the brain AMPARs and NMDARs are activated by the same neurotransmitter, l-glutamate, although other amino acids such as aspartate (Fleck et al. 1993) and glycine (as a coagonist for NMDARs) may also act on these receptors. Second, in most areas of the adult brain including the hippocampus, AMPARs colocalize with NMDARs in individual postsynaptic densities, and are estimated to be in a spatially restricted area of approximately 0.05 μm2 (Nusser et al. 1998; Takumi et al. 1999; Racca et al. 2000). Consequently, presynaptically released glutamate is equally exposed to both AMPA and NMDA receptors. It is not uncommon to see both types of receptors coactivated together under normal physiological conditions (Bekkers & Stevens, 1989; Spruston et al. 1995). As AMPARs and NMDARs are ligand-gated ion channels, research on local interactions (hetero- and auto-modulations) has focused on the consequences of cation influx. Activation of AMPARs leads primarily to Na+ and in some cases Ca2+ influx through a non-selective cation channel (Hume et al. 1991). An elevation in cytoplasmic calcium concentration within a synapse through AMPARs and NMDARs could down-regulate NMDAR function through a calcium-calmodulin-dependent inactivation mechanism (Mayer & Westbrook, 1985; MacDermott et al. 1986; Rosenmund et al. 1995). Cytoplasmic increases in sodium concentration may also modulate NMDAR function through a Src kinase (Yu & Salter 1998). On the other hand, activation of NMDARs also leads to Ca2+ as well as Na+ influx. The Ca2+ influx will not only induce calcium-calmodulin-dependent inactivation of NMDARs, but more importantly can up- and down-regulate the functions of local AMPARs through protein kinases, which then leads to long-term potentiation and long-term depression, respectively (Bliss & Collingridge, 1993; Bear & Abraham, 1996; Malenka & Nicoll, 1999).
Recently, non-ionotropic functions were reported for both AMPARs and NMDARs (Wang et al. 1997; Hayashi et al. 1999; Satake et al. 2000; Vissel et al. 2001), suggesting additional novel signalling routes exist. Here we provide evidence that AMPAR activation has an inhibitory action on NMDARs through a non-ionotropic mechanism in isolated hippocampal CA1 and nucleated patches of cultured hippocampal neurones. More importantly, it was also observed during synaptic transmission in hippocampal slices.
Methods
All animal experiments were carried out under the guidelines of the Samuel Lunenfeld Research Institute, Mount Sinai Hospital, University of Toronto.
Acute isolation of hippocampal neurones and patch clamp recording
Enzymatic digestion was used to isolate CA1 pyramidal neurones from 2≈4 week postnatal rat hippocampus (Lu et al. 1998). Briefly, Wistar rats were anaesthetized with halothane and decapitated. The brain was rapidly removed and rinsed in cold extracellular fluid (ECF). The hippocampus was surgically isolated and cut into 600 μm thick transverse slices. For digestion, the slices were incubated at room temperature (22≈24 °C) in ECF containing 2≈4 mg ml−1 papain (derived from papaya latex, Sigma) for 30 min. The CA1 region was separated from the rest of the slice and fine surgical forceps were used to tease away single CA1 pyramidal cells. Pyramidal-shaped cells were selected for recording. The ECF contained (mm): 140 NaCl, 1.3 CaCl2, 5.0 KCl, 25 Hepes, 33 glucose. The pH was adjusted to 7.3 with NaOH and the osmolarity of the solution was ≈325 mosmol l−1. Na+-free solution consisted of 10 mm CaCl2, 25 mm Hepes with equiosmotic glucose substituting for NaCl and KCl. The whole cell patch electrode had a resistance of 3≈5 MΩ and were filled with a solution that contained (mm): 140 CsF, 2 tetraethylammonium (TEA), 30 Hepes, 11 EGTA, 2 MgATP. The pH was adjusted to 7.2 using CsOH and the osmolarity was 295 mosmol l−1. A multibarrelled perfusion system (SF-77B Perfusion Fast-Step, Warner Instruments Corp., Hamden, CT, USA) was employed to rapidly switch from normal ECF to solutions that contained glutamate (0.5 mm). The time interval between glutamate applications was 30 s. Patches that displayed a ‘rundown’ greater than 2 % per minute were discarded. All recordings were performed at room temperature (22-24 °C). Recording electrodes were prepared with a puller (P-87 Flaming/Brown micropipette puller, Sutter Instrument Co., Novato, CA, USA). Voltage clamp for whole cell recordings was done with an Axopatch-200B (Axon Instruments, Inc., Union City, CA, USA). Series resistance was compensated by 85–90 %. Current signals were filtered at 2 kHz and digitized at 5–10 kHz using DigiData 1200 interface (Axon Instruments, Inc.) and pCLAMP6 software.
Cultured hippocampal neurones and patch clamp on nucleated patches
Cultures of embryonic hippocampal neurones were prepared from Swiss white mice as described previously (MacDonald et al. 1989). Briefly, fetal hippocampi were obtained from mice killed by cervical dislocation. Neurones were dissociated using mechanical trituration and plated on 35 mm collagen-coated culture dishes. Monolayers of cells were formed following 12–16 days in vitro. Prior to recording, cells were rinsed with a standard ECF. The compositions of the ECF and pipette solution were identical to those for isolated hippocampal neurones described above. In some patches we used potassium methylsulphate (KMeSO4) to replace CsF for the pipette solution. Tightly sealed whole cell recording was obtained prior to pulling out a nucleated patch. A nucleated patch was formed by slowly pulling the patch pipette away from the whole cell while applying a gentle negative pressure (Sather et al. 1992; Bai et al. 1999). To increase the signal-to-noise ratio an average of 5–10 individual current responses under each condition were routinely used for data analysis and presentation.
Patch clamp on hippocampal slice
Slice whole cell EPSC recordings were carried out on transverse hippocampal slices (400 μm) obtained from 4-9-week-old male mice (C57BL/6). Hippocampal slices were kept in a holding chamber for at least 1 h with artificial cerebrospinal fluid (ACSF) saturated with 95 % O2-5 % CO2. A single slice was then transferred to a submerged type recording chamber where it was continuously superfused with ACSF (2-3 ml min−1) at 30 °C. The composition of ACSF (mm) was: 124 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 10 d-glucose. Bicuculline (10 μM) and CGP55845A (1 μM) were added in the ACSF to block GABAA and GABAB receptors, respectively. A surgical cut was made between CA1 and CA3 regions to prevent epileptic bursting. Stimulation was delivered by bipolar twisted platinum wires located at the stratum radiatum. A short burst of stimuli (10 stimuli at 100 Hz) was delivered every 50 s. The recording pipette (resistance 3–5 MΩ) was filled with a solution that contained (mm): 130 caesium gluconate, 10 KCl, 30 Hepes, 11 EGTA, 2 MgATP. The osmolarity of the solution was 290 mosmol l−1 and the pH was brought to 7.2 with 1 m CsOH. QX314 (2 mm) was included to block voltage-dependent sodium channels and GABAB receptors. Current responses were amplified with an Axopatch-1D, filtered at 2 kHz, digitized at 10 kHz, and analysed with pCLAMP6 software (Axon Instruments, Inc.).
Chemicals
Kainic acid (Ocean Produce International, Shelburne, Nova Scotia, Canada) was disolved in 0.05 m NaOH to make a stock solution (50 mm). Chelerythrine chloride (Alexis Corporation, San Diego, CA, USA) was disolved in DMSO (20 mm). The AMPA/kainate receptor antagonists CNQX disodium salt and NBQX disodium salt, the non-competitive AMPA receptor antagonist SYM2206, the NMDA antagonist CPP, the general protein kinase inhibitor H7 and the selective GABAB receptor antagonist CGP55845 were obtained from Tocris Cookson Inc. (Ellisville, MO, USA). Lavendustin A was purchased from Calbiochem (San Diego, CA, USA). All the other chemicals are products of Sigma (St Louis, MO, USA).
Data analysis
All the data are expressed as means ± s.e.m. Student's paired t test, unpaired t test or two-way ANOVA were used to test statistical significance (*P < 0.05 or **P < 0.01) between paired, unpaired and groups of data, respectively.
Results
To investigate the possible interaction of AMPARs and the NMDARs, we used the subtype-selective agonists kainate and NMDA to activate AMPARs and NMDARs, respectively. Application of kainate (KA, 200 μM for 250 ms) with a rapid solution switching system onto a single, isolated CA1 pyramidal cell resulted in an inward current with minimum apparent desensitization (Fig. 1). Application of NMDA (100 μM for 250 ms) in the presence of 20 μM of the co-agonist glycine induced an inward current that showed a transient peak and a pronounced desensitization (Fig. 1). Co-application of kainate and NMDA at the same concentrations resulted in an inward current with an amplitude significantly less than the linear sum of the individual kainate and NMDA current (P = 0.003 Student's paired t test, n = 7, Fig. 1), indicating that the currents induced by kainate and NMDA were not completely independent.
Figure 1. Kainate and NMDA-induced currents are sub-additive in hippocampal neurones.
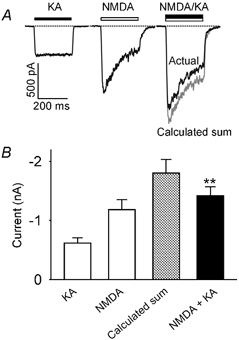
A, current traces recorded from the same acutely isolated CA1 hippocampal neurone (VH = −60 mV) in response to rapid application of kainate (KA, 200 μM for 250 ms), NMDA (100 μM for 250 ms) and both agonists (NMDA + KA at the same concentrations for 250 ms). The grey trace is a calculated sum of the two individual current traces in response to kainate alone and NMDA alone. Note the difference between the current of the calculated sum (grey trace) and the actual current response to co-application of kainate and NMDA. To obtain full-sized NMDAR-mediated currents, the bath contained 20 μM glycine and zero magnesium. B, bar graph showing the average current amplitude induced by kainate alone (KA), NMDA alone (NMDA), the calculated sum (grey bar) and co-application of the two agonists (NMDA + KA, black bar). The current amplitude for co-application of NMDA and KA was significantly less than the calculated sum of the two currents induced by these two agonists individually (P < 0.01, n = 7).
To further characterize the subadditive interactions between kainate- and NMDA-induced current, we applied 1200 ms duration pulses of different concentrations of kainate (range 0–1200 μM) and generated a dose-response curve (Fig. 2A and B). In the middle 400 ms of each kainate pulse we applied a pulse of 100 μM NMDA. The amplitude of NMDA-induced current (INMDA) decreased significantly with concentrations of kainate at 40 μM or higher (Fig. 2A and C), indicating that kainate dose-dependently inhibits NMDAR activation.
Figure 2. Kainate dose-dependently reduces NMDA-induced current in hippocampal neurones.
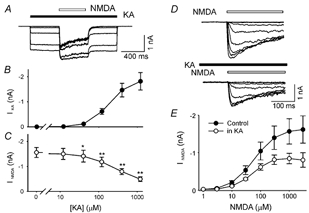
A, superimposed current records from a single isolated CA1 hippocampal neurone (VH = −60 mV) in response to a series of kainate pulses of different concentrations (KA, filled bar, 0, 12, 40, 120, 400 and 1200 μM). A 400 ms pulse of 100 μM NMDA (open bar) was delivered during the middle of each 1200 ms kainate pulse. B, dose-response curve for kainate-induced current (IKA). C, NMDAR current (INMDA) induced by 100 μM NMDA was significantly reduced by co-application of kainate at concentrations of 40 μM or higher (*P < 0.05; **P < 0.01, n = 7). INMDA is the difference between the peak of the current during NMDA application and the current before application of NMDA. D, all current recordings are from the same isolated hippocampal neurone in response to eight different concentrations of NMDA during control conditions (top traces) and in the presence of 200 μM kainate (bottom traces). E, dose-response relationships of NMDA-induced current are illustrated during control conditions (•) and in the presence of kainate (200 μM, ○). A logistic equation was used to estimate the maximum response (INMDA,max) and EC50 for each individual cell. In the presence of kainate INMDA,max was reduced from 1.66 ± 0.37 nA to 0.89 ± 0.15 nA (n = 5, P = 0.038). However, there was no difference in the estimated EC50 during control (61 ± 5 μM) and in the presence of kainate (47 ± 7 μM, n = 5, P = 0.18).
Kainate may inhibit NMDARs through its direct action on NMDARs: for example, to compete at the glutamate- and glycine-binding site of the NMDARs and shift the NMDA dose-response curve to the right. In additional experiments we tested this possibility by studying the NMDA dose- response curves in control conditions and in the presence of kainate (200 μM, Fig. 2D and E). In the presence of kainate the maximum amplitude of NMDA current was reduced from 1.66 ± 0.37 nA to 0.89 ± 0.15 nA (n = 5, P = 0.038, Fig. 2E). However, there was no right shift in the NMDA dose-response curve and the estimated EC50 for the control (61 ± 5 μM) was not increased in the presence of kainate (47 ± 7 μM, n = 5, P = 0.18). It is unlikely that kainate inhibits NMDA-induced current by competing on the glutamate- and/or glycine-binding site of the NMDA receptors. We have also studied the possibility that NMDA competes at the glutamate-binding site of AMPARs to cause the observed inhibition. We determined the dose-response curve for kainate in control conditions and in the presence of NMDA (100 μM, data not shown). The kainate EC50 in control conditions (209 ± 12 μM) was not different from that obtained in the presence of NMDA (215 ± 14 μM, n = 6, P = 0.68 with Student's paired t test), indicating NMDA is not directly competing with kainate on AMPARs.
Having recognized that the inhibitory action of kainate on NMDA current was not due to a direct action on the NMDA receptors, we next determined which of the kainate-activated receptors, i.e. the AMPAR and the kainate receptor, is the mediator for the inhibition. The AMPAR-selective antagonist GYKI53655 (20 μM) abolished the kainate-induced current (97.2 ± 1.2 % block, n = 5) and eliminated the inhibitory action of kainate on NMDA-induced current. Similar observations were also obtained for another AMPAR-selective antagonist SYM2206, indicating the inhibitory signals are mediated through the AMPARs and not kainate receptors (data not shown). Both GYKI53655 and SYM2206 are non-competitive antagonists for AMPARs (Bleakman et al. 1996; Pelletier et al. 1996), which block the AMPAR channels without affecting the binding of kainate on the AMPARs. Thus, the binding of kainate on the AMPARs is not sufficient to generate the inhibitory signal. The open state of the AMPAR channel appears to be required to produce the inhibition.
We have optimized the recording conditions to minimize space clamp escape by choosing cells that have short dendrites, use of caesium as the internal cation to block most potassium channels, and used moderate concentrations of NMDA and kainate that do not induce large amplitude currents. However, we could still not rule out completely an improper space clamp contribution to the observed interaction. To further eliminate this possibility we repeated the major experiment on nucleated patches obtained from cultured hippocampal neurones, one of the best preparations for achieving a good space clamp. In nucleated patches the INMDA, induced by 100 μM and 2000 μM NMDA under control conditions, was significantly larger than the INMDA in the presence of kainate (200 μM and 2000 μM Fig. 3, P < 0.001 and P = 0.007 Student's paired t-test, respectively), demonstrating the inhibitory interactions between AMPARs and NMDARs. In additional experiment we tested the actions of kainate (200 μM) on INMDA induced by 2000 μM NMDA. As shown in Fig. 3B the inhibitory action was very similar to that obtained when lower NMDA was used. The average current amplitudes for kainate were 44 ± 11 pA (200 μM) and 103 ± 37 pA (2000 μM) in the nucleated patches.
Figure 3. Kainate reduces NMDA-induced current in nucleated patches of hippocampal neurones.
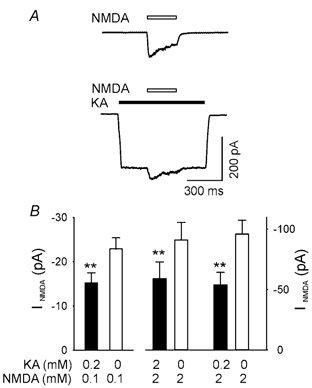
A, current traces obtained from the same nucleated patch of a cultured hippocampal neurone with KMeSO4-based pipette solution to show NMDA-induced (2 mm) current under control conditions is larger than the same concentration of NMDA-induced current in the presence of kainate (2 mm). B, currents induced by different concentrations (indicated under the bars) of NMDA are shown in control conditions (open bars) and during kainite-induced (concentrations indicated) response (filled bars). Note that the current scale is different for the left and right panels of the histogram. NMDA-induced current was significantly lower in the presence of kainate than in control conditions for all concentrations tested.
The inhibitory action of AMPARs on NMDARs was also demonstrated in isolated neurones by specific receptor antagonists. After a control period to show constant responses to a mixture of kainate and NMDA (Fig. 4A1), we added 25 μM CPP (3-(2-carboxypiperazin-4-yl)propanephosphonic acid) to block the NMDAR-mediated current (Fig. 4A2). We take the peak of the difference between the control current and the current in the presence of CPP as INMDA, the current that flows through NMDAR channels while AMPAR channels are activated (Fig. 4B1–2). Following wash-out of CPP the response to kainate + NMDA recovered (Fig. 4A3). Next, 5 μM NBQX was applied to block AMPAR-mediated current (Fig. 4A4). In the presence of NBQX, 25 μM CPP was re-introduced to again block the NMDAR-mediated current (Fig. 4A5). We take the peak of the difference between the current in the presence of NBQX and the (almost zero) current during the last pulse in the presence of NBQX + CPP as INMDA* (Fig. 4B4–5), the current that flows through NMDAR channels when AMPAR channels are blocked. In six cells the average amplitude of INMDA* (1.89 ± 0.05 nA) was significantly larger than that of INMDA (1.12 ± 0.12 nA, P = 0.002; Student's paired t test, n = 6). To better compare current through NMDAR channels despite differences in cell surface area, we calculated fINMDA and fINMDA*, the fractions of current through NMDAR channels out of the total current (see Fig. 4 legend). As shown in Fig. 4C, the mean fINMDA* (0.59 ± 0.03) was significantly larger than the mean fINMDA (0.36 ± 0.04, P < 0.001, n = 6). Thus, NMDAR conductance is lower when AMPAR channels are open than when they cannot open, suggesting once again that open AMPAR channels inhibit NMDAR channels.
Figure 4. The amplitude of NMDAR current in isolated CA1 pyramidal cells is larger when AMPARs are blocked.
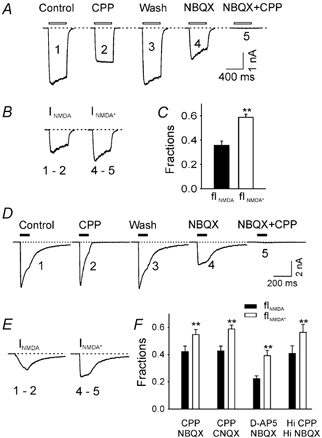
A, currents (VH = −60 mV) from an isolated CA1 hippocampal cell induced by a brief pulse (400 ms) of a mixture of kainate (200 μM) and NMDA (50 μM, open bars): 1, under control conditions; 2, in the presence of 25 μM CPP; 3, after wash-out of CPP; 4, in the presence of 5 μM NBQX; 5, in the presence of NBQX + CPP. B, CPP-sensitive current in the control condition (INMDA = I1 – I2) and in the presence of NBQX (INMDA* = I4 – I5). C, fraction of total current carried by NMDAR channels in the absence (fINMDA = (I1 – I2) / I1) and presence of NBQX (fINMDA* = (I4 – I5) / I3). fINMDA* is significantly larger than fINMDA. D, currents induced by a brief pulse of l-glutamate (0.5 mm for 100 ms, filled bars) from a single isolated hippocampal neurone during: 1, control conditions; 2, in the presence of 50 μM CPP; 3, wash; 4, in the presence of 10 μM NBQX; 5, in the presence of NBQX + CPP. E, CPP-sensitive current in the control condition (INMDA = I1 – I2) and in the presence of NBQX (INMDA* = I4 – I5). F, fraction of total current carried by NMDAR channels in the absence (fINMDA) and presence of NBQX (fINMDA*). fINMDA* was reliably larger than fINMDA regardless of which antagonist pair was used. The smaller values for fINMDA and fINMDA* observed with the NMDAR antagonist D-AP5 (20 μM) are due to incomplete block of NMDAR current at this D-AP5 concentration. The concentrations of the antagonists used were: CPP (25-50 μM), NBQX (10 μM), CNQX (10 μM), D-AP5 (20 μM). A near-saturating concentration of l-glutamate (3 mm) and higher concentrations of CPP (Hi CPP, 100 μM) and NBQX (Hi NBQX, 20 μM) were used for the last pair of bars.
Since l-glutamate is the native neurotransmitter and a full agonist for both AMPAR and NMDAR, it is important to determine whether there is an inhibitory effect from AMPAR to NMDAR when using l-glutamate as an agonist. Current responses to 100 ms pulses of 0.5 mm l-glutamate (applied every 30 s) were recorded in control conditions, during the application of the NMDAR antagonist CPP, during the application of the AMPAR antagonist NBQX, or during the application of both antagonists. In control conditions, the glutamate-induced current peaked rapidly (10-90 % rise time = 4.0 ± 0.2 ms, n = 43), desensitized during the glutamate pulse and deactivated after the end of the pulse (Fig. 4D1). Responses to glutamate pulses in each phase of the experiment are shown, including during the presence of CPP alone (Fig. 4D2), NBQX alone (Fig. 4D4) and NBQX + CPP (Fig. 4D5), as well as in control conditions (Fig. 4D1) and after CPP wash-out (Fig. 4D3). Consistent with the data obtained with antagonists added in the presence of subtype-selective agonists, INMDA* (2.0 ± 0.2 nA) was significantly larger than INMDA (1.5 ± 0.2 nA, P < 0.001, n = 8) and the mean fINMDA* (0.55 ± 0.04) was significantly larger than the mean fINMDA (0.42 ± 0.04, P < 0.001, n = 8). This inhibitory effect is not peculiar to NBQX and CPP; very similar results, shown in Fig. 4F, were obtained using CNQX and CPP, NBQX and 20 μM D-AP5 (D(-)-2-amino-5-phosphonopentanoic acid). We have also observed similar inhibitory actions with a near-saturating concentration of l-glutamate (3 mm)-induced current and higher concentrations of NBQX (20 μM) and CPP (100 μM, Fig. 4F). The lower normalized values of fINMDA and fINMDA* when D-AP5 was used as an NMDAR antagonist are due to the incomplete block of NMDARs at the tested concentration of D-AP5. We also tested the possibility that CNQX or NBQX acts directly on NMDARs to enhance INMDA. NBQX did not affect NMDA-induced current (97 ± 2 % of controls, n = 4) and CNQX caused a small but significant decrease of NMDA-induced current (85 ± 2 % of controls, P = 0.003; Student's paired t test, n = 8), indicating there is no facilitation of NBQX or CNQX on NMDARs.
We have tested the inhibitory action during the activation of AMPARs. To determine whether the inhibitory signal only exists during the open state of the AMPAR channel or is sustained for some time after the deactivation of AMPARs, a series of NMDA pulses (50 μM for 200 ms) were applied to a dissociated hippocampal neurone during control conditions and shortly after a prepulse of kainate (200 μM for 500 ms, Fig. 5A). As illustrated in Fig. 5A, there is no obvious reduction in the NMDA-induced current after the kainate prepulse, suggesting that the inhibitory signal mediated by the activation of AMPARs was rapidly diminished after the deactivation of AMPARs. By the time of the first testing point, 14 ms after kainate application, an estimated 72 % recovery of NMDA-induced current was observed (Fig. 5B). A complete recovery was seen 200 ms after the kainate pulse. It appears that the kainate-induced inhibitory signal through AMPARs is closely associated with the open state of the AMPAR.
Figure 5. The recovery time course of NMDA current after activation of AMPARs.
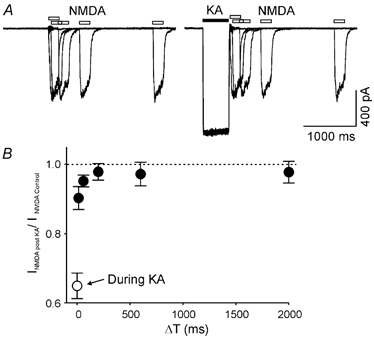
A, two sets of superimposed current recordings obtained from the same isolated neurones in response to a brief pulse of NMDA (100 μM, open bars) in control conditions (left panel) and at variable times after a prepulse of kainate (200 μM, filled bar, right panel). B, summarized data show the time course of the recovery process of NMDA-induced current shortly after kainate-induced current (•, n = 10). ○, inhibition of NMDA-induced current during the kainate-induced current.
Having established in several ways that AMPAR channel activation inhibits NMDAR channel conductance in conditions where good space clamp is expected, some possible mechanisms for the inhibition were investigated. In one experiment we asked if changing the direction and magnitude of univalent cation fluxes by varying the holding potential from −60 to +60 mV would affect the inhibition. As seen in Fig. 6A and B, INMDA was consistently less than INMDA* at all holding potentials, indicating both that the inhibition of NMDAR channels by AMPAR channels is voltage-independent and that it is unlikely to be mediated by fluxes of Na+ and K+. The reversal potential of nearly 0 mV for INMDA was unchanged.
Figure 6. The inhibition of NMDAR current by AMPARs is independent of voltage, the direction of monovalent cation flow and the availability of extracellular monovalent cations.
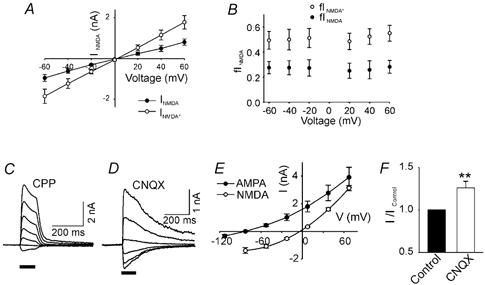
A, NMDAR current in the absence (INMDA, •) and presence of CNQX (10 μM, INMDA*, ○) plotted against voltage clamp holding potential. B, fINMDA* is larger than fINMDA at all holding potentials (-60≈+60 mV, P < 0.01, two-way ANOVA, n = 4). D-AP5 (20 μM) was used to isolate NMDAR current. C and D, current responses to glutamate pulses at different holding potentials (70, 40, 10, −20, −50, −80, −110 mV) in Na+- and K+-free, 10 mm Ca2+ extracellular medium. C, the AMPAR component of the glutamate-induced current was measured after blocking the NMDAR component with 25 μM CPP. D, the NMDAR component of the glutamate-induced response was measured after blocking the AMPAR component with 10 μM CNQX. E, I-V curves for the AMPAR and NMDAR current components under conditions described for C, and D. Note the very different reversal potentials for the two channel subtypes. F, after voltage clamping at the AMPAR channel reversal potential (VH −77 mV), when the average AMPAR channel current is zero, the NMDAR current increased significantly in the presence of 10 μM CNQX (n = 7). A later application of CPP abolished the glutamate-induced current in the presence of CNQX (data not shown).
To examine further the role of Na+ and K+ flow through AMPAR channels, these ions were removed from the extracellular solution leaving only Ca2+ as an extracellular charge carrier. In these circumstances, the reversal potential for AMPAR channels in the presence of CPP was −77.0 ± 3.0 mV (n = 7), whereas the reversal potential for NMDAR channels in the presence of CNQX was −3.8 ± 1.1 mV (n = 7; Fig. 6C-E). We then voltage clamped to the AMPAR channel reversal potential and found that CNQX still caused an increase in INMDA: the average increase was 26.1 ± 7.7 % (P = 0.008; Student's paired t test, n = 7, Fig. 6F).
We examined several other possible mechanisms for the inhibitory effect of AMPAR channels on NMDAR conductance. First, it is known that increases in intracellular calcium concentration, local to the synapse, may lead to calcium-calmodulin-dependent inactivation of NMDAR channels (Mayer & Westbrook, 1985; MacDermott et al. 1986; Rosenmund et al. 1996). We routinely used 11 mm EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid) in the recording pipette solution to minimize the possible contributions of calcium-calmodulin-dependent inactivation of NMDARs and in separate experiments we used a 10 mm intracellular concentration of the faster chelator BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid) instead of EGTA. These manipulations eliminate, or reduce significantly, the calcium-calmodulin-dependent inactivation of NMDARs (Rosenmund et al. 1996; Lu et al. 2000) but failed to alter the CNQX-induced increase of fINMDA (Fig. 7), reinforcing the conclusion that the inhibition is not mediated via intracellular calcium. Second, we used cyclothiazide to reduce or eliminate the rapid desensitization of AMPARs during application of glutamate. This desensitization was abolished in the presence of cyclothiazide (data not shown, see also Patneau et al. 1993), but fINMDA (0.10 ± 0.01) was still reliably lower than the fINMDA* (0.25 ± 0.04 nA, P = 0.008; Student's paired t test, n = 8,). The decrease in fractions of both the fINMDA and fINMDA* are presumably due to the increased AMPAR conductance caused by cyclothiazide and the incomplete block of NMDA current by D-AP5 (as shown in Fig. 4F). This result suggests that the AMPAR-based inhibition is not mediated through the desensitized state of AMPARs. Third, we asked if the metabotropic effects of activating AMPAR (Wang et al. 1997) might be involved in the inhibition of NMDAR channels. We determined the role of GTP-binding protein (G-protein) signals induced by AMPAR in producing differences between fINMDA and fINMDA* by including either 400 μM GTP-γ-S or 2 mm GDP-β-S in the recording pipette to activate or inhibit G-protein activities, respectively. Neither of these G-protein modulators significantly altered the difference between fINMDA* and fINMDA (Fig. 7). Fourth, AMPAR activation is able to signal through the Src family non-receptor protein tyrosine kinase Lyn (Hayashi et al. 1999). To test the roles of protein tyrosine kinases we included in the pipette solution a specific inhibitor – Lavendustin A (1 μM) – and this manipulation failed to change the difference between fINMDA* and fINMDA (Fig. 5). Finally, several protein kinases, including protein kinase C (PKC), are able to modulate NMDAR function (for a review see MacDonald et al. 2001). To test this we used a selective PKC inhibitor chelerythrine (10 μM) and a non-selective protein kinase inhibitor H7 (100 μM) separately in individual pipette solutions. Again, none of these treatments altered the ability of AMPARs to inhibit NMDARs (Fig. 7). The mechanisms underlying the cross-inhibition between AMPARs and NMDARs remain to be elucidated.
Figure 7. The role of intracellular calcium, G-proteins and protein kinases in AMPAR channel-induced inhibition of NMDAR current.
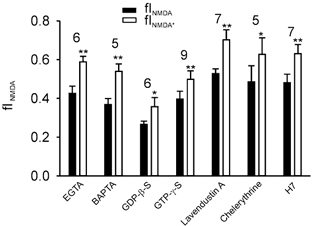
In each case, the fraction of current through the NMDAR channels was calculated as described in Fig. 3. In all cases, AMPARs were blocked with 10 μM CNQX and NMDARs with 50 μM CPP. The fINMDA was significantly lower than fINMDA* in the presence of divalent ion chelators (11 mm EGTA and 10 mm BAPTA), in the presence of either an agonist (400 μM GTP-γ-S) or an antagonist (2 mm GDP-β-S) of G-protein activity, in the presence of the protein tyrosine kinase inhibitor Lavendustin A (1 μM), protein kinase C inhibitor chelerythrine (10 μM) and also a general protein kinase inhibitor H7 (100 μM). The number of cells in each group of data is indicated at the top of the bars (*P < 0.05 and **P < 0.01; Student's paired t test).
Finally, we used hippocampal slices to ask if the inhibition of NMDAR channels can be seen with presynaptic release of glutamate instead of bath application. After blocking both GABAA and GABAB receptor-mediated inhibition, the whole cell patch method was used to voltage clamp CA1 pyramidal cells to +60 mV. Excitatory postsynaptic currents (EPSCs) induced by bursts of 10 stimuli at 100 Hz were recorded. At +60 mV, the magnesium block of NMDAR channels is relieved and variations in the contributions of voltage-gated sodium and calcium channels are eliminated. In parallel with results from isolated cells, the fraction of the EPSC through NMDAR channels was higher in the presence than in the absence of CNQX (Fig. 8). Thus, the inhibition of NMDAR conductance by activating AMPAR channels occurs in relatively intact hippocampal preparations, as well as in isolated cells.
Figure 8. The inhibitory effect of AMPAR activation on NMDAR channel current occurs at intact synapses in hippocampal slices.
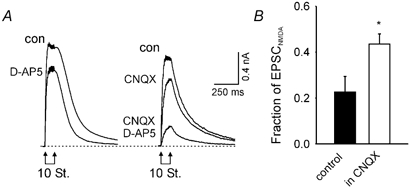
A, composite EPSCs from CA1 pyramidal cells during whole cell voltage clamp at +60 mV holding potential. The responses are evoked by 10 stimuli of the Schaffer-collateral pathway at 100 Hz. Responses before (con) and after the addition of 20 μM D-AP5 are shown (left panel). Responses before the addition of any drug (con), after the addition of 10 μM CNQX and after the further addition of 20 μM D-AP5 (right panel). B, fraction of the peak EPSC blocked by D-AP5. This fraction was significantly greater in the presence (n = 5) than in the absence of CNQX (n = 4, P = 0.04; Student's t test).
Discussion
Using rapid applications of drugs onto isolated CA1 pyramidal cells, we have found that activation of postsynaptic AMPAR channels causes inhibition of current through NMDAR channels. This inhibition can be demonstrated with an agonist of AMPAR channels, which results in a decrease of NMDAR conductance, or with an antagonist of AMPA-class glutamate channels, which results in an increase in NMDAR conductance; the effect is seen whether currents through the glutamate receptor subtypes are induced by specific agonists or by l-glutamate itself. We argue that the inhibitory action from AMPARs to the NMDARs is non-ionotropic based on the following evidence. First, a wide range of holding potentials (-60 mV to +60 mV) caused a drastic change in the electro-driving force for any cation flow through AMPA channels, yet we failed to see any significant differences for the inhibitory actions on NMDA current in this range (Fig. 6B). Secondly, alteration of the chemical gradient for conducting cations by removal of extracellular Na+ and K+ failed to eliminate the inhibitory action of AMPARs on NMDA current, indicating it is highly unlikely to be mediated through Na+ and K+. Finally, the use of high-affinity Ca2+ chelators, EGTA and BAPTA, in the recording pipette reduced the free Ca2+ level to a minimum and eliminated calcium-calmodulin-dependent inactivation of NMDARs (Rosenmund et al. 1995; Lu et al. 2000). Under these conditions the inhibitory action from AMPARs to NMDARs was not affected.
In other experiments we accumulated evidence that the inhibition of NMDAR current by activation of AMPAR channels is not an electrical artifact due to imperfect space clamping. First, the space clamping problems are not expected at the relatively low currents used in isolated neurones, especially when Cs+-based pipette solution is used. The magnitude of the inhibition was unchanged with wide variations of current magnitude or direction caused by voltage clamping over the range −60 to +60 mV. Second, the inhibition was seen when there was no net current through AMPAR channels by voltage clamping to the AMPAR reversal potential. Finally, the cross-inhibition exists in nucleated patches where not only the current amplitude was much smaller, but also the geometric shape and size of the nucleated patch make it very unlikely to have voltage-clamp escape. All the evidence provides a strong argument that the effect is not due to a shunting of NMDAR current by AMPAR channel activity.
A detailed inspection of the time course of the inhibitory signals generated from AMPARs indicates that the inhibition is closely associated with the open state of the AMPAR channels. Similar to other ligand-gated ion channels, the ligand-bound AMPARs can exist in at least three different states: closed, desensitized and open. The ligand-bound and closed state (for example in the presence of non-competitive antagonist, GYKI53655) of the AMPARs failed to demonstrate any inhibitory action on NMDA-induced current. Depending on the type and concentration of agonist used, the apparent rate and degree of desensitization varies dramatically for AMPAR-mediated current in neuronal preparations. In the present study kainate-induced current had minimum apparent desensitization or a constant amplitude in dissociated hippocampal neurones. Note that the inhibitory signal, i.e. the difference between INMDA and INMDA* (Fig. 4B), was also nearly constant. However, in the case of l-glutamate-induced current there was a prominent desensitization during the course of l-glutamate superfusion (Fig. 4D1). The difference between INMDA and INMDA* (Fig. 4E) clearly showed a gradual decrease of the inhibitory signal. In other words, with the increase in AMPAR desensitization, the inhibitory signal decreased, indicating that the desensitized state of AMPARs was also unable to generate the inhibitory signal to the NMDARs. Consistent with this conclusion, blocking AMPAR desensitization with cyclothiazide failed to remove the inhibitory signal. The open state of AMPARs appears to be the only state that generates the inhibitory signal. The inhibitory signal decayed with a time constant of a few milliseconds after removal of kainate (Fig. 5B). Such a short-lived signal is unlikely to be mediated through a diffusible molecule and/or a phosphorylation process. The identity of the inhibition is yet to be resolved.
Recent studies revealed cross-talk between different types of neurotransmitter receptors, including dopamine and adenosine receptors (Gines et al. 2000), dopamine and GABAA receptors (Liu et al. 2000), nicotinic acetylcholine receptors and ATP receptors (Nakazawa, 1994; Khakh et al. 2000) and GABAA receptors and ATP receptors (Sokolova et al. 2001). Some of these interactions are between G-protein-coupled receptors and ligand-gated ion channels (Gines et al. 2000; Liu et al. 2000), and others are between two types of ligand-gated ion channels (Nakazawa, 1994; Khakh et al. 2000; Sokolova et al. 2001). The mechanisms of the cross-talk between receptors could be via direct protein-protein interaction (Gines et al. 2000; Liu et al. 2000), or through influx/efflux of ions, such as Cl− and Ca2+ (Sokolova et al. 2001), or a receptor state-dependent mechanism (Khakh et al. 2000). Our findings provide the first evidence for cross-talk between two different ionotropic glutamate receptors. We eliminated the roles of cation fluxes in mediating the cross-talk. It is also unlikely to be mediated through G-proteins and protein kinases, at least not via lavendustin A, chelerythrine and H7-sensitive kinases. Other signalling molecules may exist directly or indirectly associated with subunits of AMPARs and NMDARs. So far an increasing number of proteins are found to be linked to the subunits of AMPARs and NMDARs (Hsui et al. 2000; Scannevin & Huganir 2000). However, there is no evidence for a direct protein-protein interaction between any subunits of AMPARs and NMDARs (Hsui et al. 2000; Scannevin & Huganir 2000). It would be interesting to determine in future studies the subunits required and the mechanisms for the cross-talk between AMPARs and NMDARs in the hippocampus.
The AMPAR-based inhibition of NMDAR conductance has important functional implications. It may play a neuroprotective role when both types of receptors are activated at the same time, for example during bursting activities and ischaemia when excessive neurotransmitter is released. Finally, inhibition of NMDAR channel activity by AMPAR channels may affect the ‘learning rule’ that describes how synaptic strength varies with the history of activation of the presynaptic and postsynaptic elements. A signalling pathway from AMPAR channels to NMDAR channels may modify the storage of information as synaptic strength.
Acknowledgments
We thank Drs Graham Collingridge, John MacDonald, Mike Salter, Yutian Wang, Weiyang Lu and John Georgiou for helpful discussions of this work. The work was supported by the Canadian Institute of Health Research and ALS society of Canada (J.C.R.), NIH grants NS20686 and NS37150, and an MRC (UK) Overseas Initiative Grant (R.U.M.). D.B. is a Michael Smith fellow of the Canadian Institute of Health Research.
References
- Bai D, Pennefather PS, MacDonald JF, Orser BA. The general anesthetic propofol slows deactivation and desensitization of GABAA receptors. Journal of Neuroscience. 1999;19:10 635–10 646. doi: 10.1523/JNEUROSCI.19-24-10635.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annual Review Neuroscience. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- Bleakman D, Ballyk BA, Schoepp DD, Palmer AJ, Bath CP, Sharpe EF, Woolley ML, Bufton HR, Kamboj RK, Tarnawa I, Lodge D. Activity of 2,3-benzodiazepines at native rat and recombinant human glutamate receptors in vitro: stereospecificity and selectivity profiles. Neuropharmacology. 1996;35:1689–1702. doi: 10.1016/s0028-3908(96)00156-6. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Fleck MW, Henze DA, Barrionuevo G, Palmer AM. Aspartate and glutamate mediate excitatory synaptic transmission in area CA1 of the hippocampus. Journal of Neuroscience. 1993;13:3944–3955. doi: 10.1523/JNEUROSCI.13-09-03944.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gines S, Hillion J, Torvinen M, Le Crom S, Casado V, Canela EI, Rondin S, Lew JY, Watson S, Zoli M, Agnati LF, Verniera P, Lluis C, Ferre S, Fuxe K, Franco R. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proceedings of the National Academy of Sciences of the USA. 2000;97:8606–8611. doi: 10.1073/pnas.150241097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Umemori H, Mishina M, Yamamoto T. The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature. 1999;397:72–76. doi: 10.1038/16269. [DOI] [PubMed] [Google Scholar]
- Hume RI, Dingledine R, Heinemann SF. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nature Neuroscience. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Zhou X, Sydes J, Galligan JJ, Lester HA. State-dependent cross-inhibition between transmitter-gated cation channels. Nature. 2000;406:405–410. doi: 10.1038/35019066. [DOI] [PubMed] [Google Scholar]
- Liu F, Wan Q, Pristupa ZB, Yu XM, Wang YT, Niznik HB. Direct protein-protein coupling enables cross-talk between dopamine D5 and gamma-aminobutyric acid A receptors. Nature. 2000;403:274–280. doi: 10.1038/35002014. [DOI] [PubMed] [Google Scholar]
- Lu WY, Jackson MF, Bai D, Orser BA, MacDonald JF. In CA1 pyramidal neurones of the hippocampus protein kinase C regulates calcium-dependent inactivation of NMDA receptors. Journal of Neuroscience. 2000;20:4452–4461. doi: 10.1523/JNEUROSCI.20-12-04452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Orser BA, MacDonald JF. Multiple sites of action of neomycin, Mg2+ and spermine on the NMDA receptors of rat hippocampal CA1 pyramidal neurones. Journal of Physiology. 1998;512:29–46. doi: 10.1111/j.1469-7793.1998.029bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Kotecha SA, Lu WY, Jackson MF. Convergence of PKC-dependent kinase signal cascades on NMDA receptors. Current Drug Targets. 2001;2:299–312. doi: 10.2174/1389450013348452. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Mody I, Salter MW. Regulation of N-methyl-d-aspartate receptors revealed by intracellular dialysis of murine neurones in culture. Journal of Physiology. 1989;414:17–34. doi: 10.1113/jphysiol.1989.sp017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation – a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Nakazawa K. ATP-activated current and its interaction with acetylcholine-activated current in rat sympathetic neurons. Journal of Neuroscience. 1994;14:740–750. doi: 10.1523/JNEUROSCI.14-02-00740.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Vyklicky LJ, Mayer ML. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. Journal of Neuroscience. 1993;13:3496–3509. doi: 10.1523/JNEUROSCI.13-08-03496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier JC, Hesson DP, Jones KA, Costa AM. Substituted 1,2-dihydrophthalazines: potent, selective, and noncompetitive inhibitors of the AMPA receptor. Journal of Medicinal Chemistry. 1996;39:343–346. doi: 10.1021/jm950740w. [DOI] [PubMed] [Google Scholar]
- Racca C, Stephenson FA, Streit P, Roberts JD, Somogyi P. NMDA receptor content of synapses in stratum radiatum of the hippocampal CA1 area. Journal of Neuroscience. 2000;20:2512–2522. doi: 10.1523/JNEUROSCI.20-07-02512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, FeltZ A, Westbrook GL. Calcium-dependent inactivation of synaptic NMDA receptors in hippocampal neurons. Journal of Neurophysiology. 1995;73:427–430. doi: 10.1152/jn.1995.73.1.427. [DOI] [PubMed] [Google Scholar]
- Satake S, Saitow F, Yamada J, Konishi S. Synaptic activation of AMPA receptors inhibits GABA release from cerebellar interneurons. Nature Neuroscience. 2000;3:551–558. doi: 10.1038/75718. [DOI] [PubMed] [Google Scholar]
- Sather W, Dieudonne S, MacDonald JF, Ascher P. Activation and desensitization of N-methyl-d-aspartate receptors in nucleated outside-out patches from mouse neurones. Journal of Physiology. 1992;450:643–672. doi: 10.1113/jphysiol.1992.sp019148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nature Review Neuroscience. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Sokolova E, Nistri A, Giniatullin R. Negative cross talk between anionic GABAA and cationic P2X ionotropic receptors of rat dorsal root ganglion neurons. Journal of Neuroscience. 2001;21:4958–4968. doi: 10.1523/JNEUROSCI.21-14-04958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B. Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. Journal of Physiology. 1995;482:325–352. doi: 10.1113/jphysiol.1995.sp020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi Y, RamireZ-leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nature Neuroscience. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nature Neuroscience. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Wang Y, Small DL, Stanimirovic DB, Morley P, Durkin JP. AMPA receptor-mediated regulation of a Gi-protein in cortical neurons. Nature. 1997;389:502–504. doi: 10.1038/39062. [DOI] [PubMed] [Google Scholar]
- Yu XM, Salter MW. Gain control of NMDA-receptor currents by intracellular sodium. Nature. 1998;396:469–474. doi: 10.1038/24877. [DOI] [PubMed] [Google Scholar]