

Abstract
In vivo and in vitro electrophysiological experiments were performed on the rat dorsal vagal complex (DVC, i.e. nucleus of the tractus solitarius, NTS, and dorsal motor nucleus of the vagus, DMV) to examine the effects of corticotropin releasing hormone (CRF) on the central components of the vago-vagal reflex control of gastric function. When applied to gastrointestinal projecting DMV neurones, CRF (10-300 nm) induced a concentration-dependent membrane depolarization, an increase in action potential firing rate and decrease in amplitude of the action potential afterhyperpolarization (P < 0.05). Pretreatment with the non-selective CRF antagonist, astressin (0.5-1 μM) or the selective CRF2 receptor antagonist, astressin 2B (500 nm) attenuated the CRF-induced increase in firing rate but did not alter basal discharge rate. CRF (30-300 nm) increased the amplitude of excitatory postsynaptic currents (EPSCs) evoked by stimulation of the NTS (P < 0.05). An alteration in the paired pulse ratio indicated the EPSC's increase occurred due to actions at presynaptic sites. In the in vivo anaesthetized rat preparation, bilateral microinjections (20 fmol in 20 nl for each site) of CRF in the DVC decreased gastric motility in rats pretreated with the muscarinic agonist, bethanecol (P < 0.05). The effects of CRF were abolished by systemic administration of the NOS inhibitor, l-NAME, or by bilateral vagotomy. We concluded that CRF had both a direct and an indirect excitatory effect on DMV neurones via activation of CRF2 receptors and the decrease in gastric motility observed following microinjection of CRF in the DVC is due to the activation of an inhibitory non-adrenergic non-cholinergic input to the gastrointestinal tract.
Sensory information from the gastrointestinal (GI) tract is received and integrated by neurones in the nucleus of the tractus solitarius (NTS). The output of these NTS neurones then influences the discharges of the dorsal vagal motoneurones of the dorsal motor nucleus of the vagus (DMV) (Altschuler et al. 1989, 1991; Rogers et al. 1999). The NTS utilizes, in the main, a tonic inhibitory GABAergic input to the DMV as well as an excitatory glutamatergic input (Rogers et al. 1995; Sykes et al. 1997). The DMV then provides efferent outflow to the GI tract through the subdiaphragmatic vagal branches (Norgren & Smith, 1988; Berthoud et al. 1991). The DMV preganglionic motoneurones have been shown to control both tonic excitatory cholinergic as well as inhibitory non-adrenergic, non-cholinergic (NANC) postganglionic neurones in the gut (Forster & Southam, 1993; Barnes et al. 1994; Yu, 1994; Panico et al. 1995; Takahashi & Owyang, 1995; Esplugues et al. 1996; Lawrence, 1997; Beltran et al. 1999; Krowicki et al. 1999; Zheng et al. 1999; Guo et al. 2001; Quintana et al. 2001).
The DMV also receives input from higher CNS centres, including the paraventricular nucleus of the hypothalamus (PVN) (Rogers et al. 1980), and Barrington's nucleus (Valentino et al. 1995). Corticotropin releasing factor (CRF) is one of a number of neuronal modulators released from neurones with soma in the PVN and Barrington's nucleus (Cummings et al. 1983; De Souza & Kuhar, 1986; Valentino et al. 1995). Nerve fibres immunoreactive for CRF and CRF binding sites are observed in both the NTS and the DMV (De Souza et al. 1985; Skofitsch et al. 1985; De Souza & Kuhar, 1986; De Souza, 1987; Sakanaka et al. 1987; Herbert & Saper, 1990; Bittencourt et al. 1999).
Various stressors such as abdominal surgery, restraint and cold have been shown to induce dramatic effects on the GI function in laboratory animals (Gue et al. 1987; Coskun et al. 1997; Martinez et al. 1997). These effects can be mimicked by injection of CRF in the cisterna magna, lateral ventricles, DVC or PVN and induce a decrease in gastric acid secretion, gastric emptying, small bowel transit time and an increased large bowel transit time (Tache et al. 1983, 1987; Garrick et al. 1988; Heymann-Monnikes et al. 1991; Monnikes et al. 1992; Smedh et al. 1995; Coskun et al. 1997; Martinez et al. 1997, 1998). Furthermore, the stress-related and the CRF-induced GI effects are blocked by pretreatment with CRF antagonists (Lenz et al. 1988; Coskun et al. 1997; Martinez et al. 1997).
Recent in vivo studies conducted by Tache's group using systemic administration of CRF and its analogues have suggested that the gastric inhibitory effects of CRF might be mediated by activation of CRF2 receptors (Martinez et al. 1998). Despite the tremendous volume of descriptive work, which supports the involvement of CRF in stress-induced alterations in GI function, little is known about the cellular mechanisms responsible for these actions of CRF in the DVC.
Based on previous studies showing that in different neuronal populations the prevalent postsynaptic effect of CRF is excitatory (Eberly et al. 1983; Siggins et al. 1985; Yamashita et al. 1991; Hille, 1992; Curtis et al. 1997; Page & Abercrombie, 1999; Haug & Storm, 2000), and that the NTS contains a more dense CRF-immunoreactivity (IR) and urocortin-IR fibre innervation than the DMV (Herbert & Saper, 1990; Bittencourt et al. 1999), we performed a series of experiments to test the hypotheses that: (1) the effects of CRF on identified gastric-projecting DMV neurones were mediated by both a direct membrane depolarization and by an increase in excitatory synaptic transmission between the NTS and the DMV, and (2) the inhibition of gastric motility induced by microinjection of CRF in the DVC was mediated by activation of the NANC pathway rather than withdrawal of cholinergic tone.
Methods
Animal care and experimental procedures were performed with the approval of the Animal Care and Utilization Committees of the University of Michigan (in vitro studies) and Ohio State University (in vitro studies).
In vitro brain slice studies
Slice preparation
Sprague-Dawley rats were purchased from Charles River Laboratories, Inc. (Wilmington, MA, USA). GI-projecting DMV neurones were labelled as described previously (Browning et al. 1999). Briefly, 11-day-old rat pups of either sex were anaesthetized deeply (indicated by abolition of the foot pinch withdrawal reflex) by inhalation of a 6 % solution of halothane in air (400-600 ml min−1) before an abdominal laparotomy was performed. During surgery, anaesthesia was maintained by placing the head of the rat in a custom-made anaesthetic chamber through which the halothane-air mixture was perfused. Crystals of the retrograde tracer DiI (1,1′-dioctadecyl-3,3,3''3′-tetramethylindocarbocyanine perchlorate (DiIC18(3); DiI; Molecular Probes, Eugene, OR, USA) were applied to the serosal surface of the gastric fundus, corpus or antrum/pylorus areas or duodenum and the application site was embedded in a fast-hardening epoxy resin that was allowed to dry for several minutes before the entire surgical area was washed with warm saline (Browning et al. 1999). The wound was closed with 5/0 suture and the animal allowed to recover for 10–15 days under the monitoring of veterinary staff.
The brainstems were removed as described previously (Travagli et al. 1991; Browning et al. 1999). Briefly, the rats were placed in an anaesthetic chamber and anaesthetized with 6% halothane and killed by severing the major blood vessels in the chest. The brainstem was removed and placed in oxygenated Krebs solution at 4 °C (see Solution composition). The site of DiI labelling in the stomach (i.e. fundus, corpus or antrum/pylorus) or in the intestine was confirmed by visual inspection of the organ. We sliced the brainstems only from those animals in which the glue covering the site of DiI application was still in place. Using a vibratome, six to eight coronal sections (200 μm thick) containing the DVC were cut and stored in oxygenated Krebs solution at 30 °C for at least 1 h prior to use. A single slice was transferred to a custom-made perfusion chamber (volume 500 μl) and kept in place using a nylon mesh. The chamber was maintained at 35 ± 1 °C by perfusion with warmed, oxygenated Krebs solution at a rate of 2.5-3.0 ml min−1.
Prior to electrophysiological recording, DiI-labelled GI-projecting DMV neurones were identified using a Nikon E600-FS microscope equipped with epifluorescent filters suitable for visualizing DiI. We exposed the DVC to fluorescent light using TRITC filters to identify the fluorescent neurone(s) and, once the identity of a labelled neurone was confirmed, we switched the illumination to brightfield. Whole-cell recordings were made under brightfield illumination.
Electrophysiological recording
Whole-cell recordings were made with patch pipettes (3-8 MΩ resistance) filled with a potassium gluconate solution using an Axoclamp-2B single electrode voltage clamp amplifier (Axon Instruments, Union City, CA, USA). Recordings were made only from neurones unequivocally labelled with DiI. Data were sampled every 100 μs and filtered at 2 kHz, digitized via a Digidata 1200C interface (Axon Instruments) acquired, stored and analysed on an IBM PC utilizing pCLAMP 8 software (Axon Instruments). Recordings were accepted only if the series resistance was < 15 MΩ. In addition, the action potential evoked following injection of depolarizing current had to have an amplitude of at least 60 mV and the membrane potential had to return to the baseline value following the action potential afterhyperpolarization.
Electrical stimulation
Bipolar tungsten electrodes were used to electrically stimulate the NTS. Paired stimuli (0.1-1.0 ms, 10–500 μA; 15–100 ms interval) were applied every 20 s to evoke submaximal excitatory (EPSCs) postsynaptic currents (Browning & Travagli, 2001). A minimum of six control EPSCs were obtained and averaged prior to each drug application. Drugs were applied to the bath via a series of manually operated valves.
In vivo rat studies
Animal preparation
Experiments were performed on rats (n = 15) weighing 250–400 g. Animals were anaesthetized with an intraperitoneal injection of thiobutabarbital (Inactin; 100 mg kg−1). An adequate depth of anaesthesia was assessed by the absence of the foot pinch withdrawal reflex. Body temperature was monitored by a rectal thermometer and maintained at 37 ± 1 °C with a heating pad.
Surgery
Rats were intubated to maintain an open airway. A jugular cannula was placed for the administration of intravenous drugs. In animals that underwent cervical vagotomy, loose 5/0 silk ligatures were tied around the vagus nerves and the trailing ends passed through a 3 cm length of PE 240 tubing. Later, the ligatures were pulled out through the tube to cut the nerve.
An abdominal laparotomy was performed and a strain gauge (RB Products, Madison, WI, USA) was sewn onto the surface of the gastric corpus as described in previous reports (Hermann & Rogers, 1995). The laparotomy was closed with the strain gauge leads exiting the abdomen. Strain gauge gastric motility data were digitized and stored using a DataPac2000 PC-based waveform analysis system (Run Technologies, Laguna Hills, CA, USA). Motility data were saved for analysis at a later time.
The animals were then positioned in a stereotaxic apparatus (David Kopf, Tujunga, CA, USA). A partial dorsal craniotomy was performed to expose the fourth ventricle. The cerebellum was retracted slightly while using a 26-gauge needle to cut the subarachnoid covering. Calamus scriptorius (CS) was viewed from the dorsal aspect and used as a point of reference (see below). A 1 h recovery period preceded experimental manipulations during which strain gauge activity was monitored continuously, digitized and stored.
Experimental procedure
Pipettes (tip diameter of between 10–30 μm) were filled with either CRF in phosphate-buffered saline (PBS), or PBS alone, and were placed in a hydraulic microdrive carrier. The pipette was directed toward the CS to establish a zero reference for subsequent placement of the pipette in the DVC. The pipette was first positioned at a point 0.3 mm anterior and 0.3 mm left from CS on the brainstem surface. The pipette was then advanced to a point 0.4 mm ventral to the brainstem surface. This location corresponds to the DVC, an area well known to contain the cell bodies of the main brainstem areas involved in the gastric vago-vagal reflex control circuitry (McCann & Rogers, 1992; Rogers et al. 1999). Baseline gastric motility was increased by systemic (i.v.) administration of the muscarinic agonist, bethanecol (50 μg kg−1 bolus followed by continuous i.v. infusion with 20 μg kg−1 h−1 for 20 min). Two minutes after i.v. administration of bethanecol, CRF (20 nl × 10−6m CRF = 20 fmol; n = 5) or PBS (20 nl; n = 4) were micropressure injected into the DVC under direct microscopic control (Hermann & Rogers, 1995). The procedure was then repeated on the right side. The bilateral DVC injection procedure took less than 1 min to complete.
In three additional cases, the nitric oxide synthase inhibitor, NG-nitro-l-arginine methyl ester (l-NAME; 10 mg kg−1 bolus i.v. injection) (Takahashi & Owyang, 1995, 1998) was injected 20 min prior to bethanecol administration. CRF was injected into the DVC as described. In three more cases, rats received bilateral cervical vagotomy 20 min prior to bethanecol administration. Although these animals developed apneustic breathing after vagal section, all survived without auxiliary ventilation.
Histological verification
At the end of the experiment all rats were killed with an i.v. dose of lidocaine (0.3 ml × 2 %). Brains were removed and fixed in a mixture of 4 % paraformaldehyde and 20 % sucrose in PBS for at least 48 h. The brains were cut into 40 μm thick coronal sections and stained with Nuclear Fast Red (Vector Labs; Berlingame, CA, USA). Location of nuclear groups was studied in relation to microinjection sites using the atlas of Paxinos & Watson (1986).
Data analysis
In vitro experiments
Electrophysiological data were analysed using Clampex (pCLAMP 8 software package, Axon Instruments). To assess the effects of drugs, each neurone served as its own control (i.e. the results obtained after administration of a drug were compared with those before administration using Student's paired t test).
Data were obtained from spontaneously active DMV neurones or from DMV neurones hyperpolarized to −65 mV by current injection. When tested on spontaneously active DMV neurones, the CRF-induced increase in firing rate was assessed as the number of action potentials counted during the 20 s preceding the administration of the drug and during the 20 s of maximal firing rate increase following drug superfusion. When tested on silent neurones, the response to CRF was assessed as a change in membrane potential. The amplitude of the evoked excitatory postsynaptic currents (EPSCs) was obtained from the average of three to six EPSCs in control and following superfusion with CRF or its antagonists.
All concentration-response curves were constructed only from neurones in which at least three concentrations (at 5–15 min intervals and always given in random order) were tested. DMV neurones were classified as CRF responders if a 1 min-long perfusion with 100 nm CRF resulted in either a minimum 15 % change in the frequency of action potential firing, a variation of at least 1.25 mV in membrane potential, or at least 10 % change in the EPSC amplitude all of which returned to baseline values upon washout of CRF.
Results are expressed as means ± s.e.m. Significance was set at P < 0.05.
In vivo experiments
Motility data were analysed using DataPac2000. Motility records were divided into three epochs; 2 min before bethanecol (basal period), 2 min after i.v. bethanecol (maximum motility) and 2 min after completion of the DVC injection. The area under the motility curves was determined for each 2 min epoch and expressed in volts per second. Data from all three epochs were subjected to an analysis of variance followed by Tukey's multiple comparison post hoc test.
Results are expressed as means ± s.e.m. Significance was set at P < 0.05.
Solution composition
Intracellular electrode solution was composed of (mm): 128 potassium gluconate, 10 KCl, 0.3 CaCl2, 1 MgCl2, 1 Hepes, 1 EGTA, 2 ATP, 0.25 GTP, adjusted to pH 7.35 with KOH. Krebs solution was composed of (mm): 126 NaCl, 25 NaHCO3, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4 and 11 dextrose, maintained at pH 7.4 by bubbling with 95 % O2-5 % CO2. Phosphate-buffered saline (PBS) was composed of (mm): 124 NaCl, 26 NaHCO3, 2 KH2PO4 adjusted to 304 mosmol kg−1 and pH 7.4. A histological marker (Pontamine Blue, 5 mg ml−1) for locating injection sites was added to PBS.
Drugs and chemicals
All drugs were purchased from Sigma Chemical Co., St Louis, MO, USA except for astressin 2B which was a generous gift from Professor J. Rivier (Salk Institute, San Diego, CA, USA).
Results
In vitro studies
Whole-cell patch clamp studies were conducted on 203 neurones; 172 identified gastric-projecting (36 fundus-, 60 corpus- and 76 antrum/pylorus-projecting neurones) and 31 duodenal-projecting neurones. Results did not differ among the various projection groups and were thus pooled.
Effects of CRF on DMV neurones
Sixty-four percent of neurones (i.e. 103 of 160) responded to 100 nm CRF with either a change in discharge rate or membrane depolarization at or above the minimum criteria. Sixty cells (49 gastric- and 11 intestinal-projecting neurones) showed an increase in discharge rate from 0.9 ± 0.07 Hz in control to 1.7 ± 0.12 Hz in 100 nm CRF (P < 0.05), two cells showed a decrease in discharge rate (one cell decreased from 0.8 to 0 Hz in CRF and the second cell decreased from 2.0 to 1.28 Hz in CRF; both cells recovered upon washout, this type of response was not investigated further). Additionally, CRF 100 nm depolarized 41 cells (35 gastric- and 6 intestinal-projecting neurones) by 4.0 ± 0.23 mV (P < 0.05), while no cells were hyperpolarized and 57 cells were unresponsive. Concentration-response curves were constructed from cells in which at least three concentrations of CRF (10-300 nm) were tested (Fig. 1).
Figure 1. CRF induced a concentration-dependent increase in discharge rate and membrane depolarization.
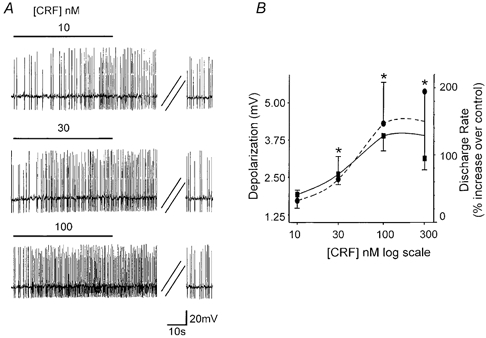
A, representative traces from a gastric-projecting DMV neurone illustrating the concentration-dependent increase in discharge rate induced by superfusion of nanomolar concentrations of CRF. Neurones were spontaneously active and were monitored for stability for a minimum of 2 min before the superfusion of CRF. The horizontal line above the traces indicates perfusion of CRF for a time sufficient to reach the response plateau. In the depicted example the firing rate was 0.95 Hz in control, 1.35, 1.7 and 2.45 Hz in 10, 30 and 100 nm CRF, respectively. The increase in discharge rate did not desensitize even at high concentrations. The parallel oblique lines indicate a 3.5 min period. A recovery period of at least 5 min was allowed between successive applications, during which the firing rate returned to baseline values. B, concentration-response curve for the CRF-induced increase in discharge rate (•; n = 7) and membrane depolarization (○; n = 8). Each neurone was tested with at least three different concentrations of CRF. *P < 0.05 vs. baseline.
The response to CRF did not show tachyphylaxis. In fact, two 1 min superfusions of 100 nm CRF within 5 min gave similar results. In detail, the first superfusion of CRF depolarized the membrane by 3.2 ± 0.55 mV (P < 0.05 vs. control; n = 7) or increased the frequency of action potential firing from 0.97 ± 0.09 to 2.1 ± 0.3 Hz (i.e. 122 ± 32.7 %; P < 0.05; n = 6) while the second superfusion of CRF depolarized the membrane by 3.2 ± 0.49 mV or increased the firing rate by 156 ± 63.7 % (P < 0.05 vs. control; P > 0.05 vs. first application; Fig. 2).
Figure 2. The CRF-induced depolarization of the DMV membrane does not show tachyphylaxis.
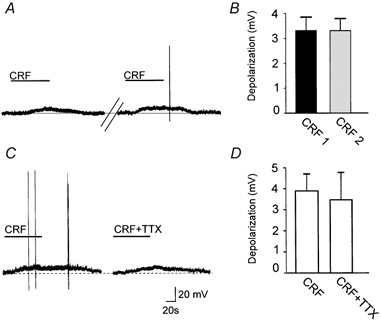
A, representative current-clamp traces compare the amplitude of the CRF-induced depolarization when the superfusions of CRF (100 nm) were initiated 5 min apart. Holding potential, −65 mV. B, summary comparing the amplitudes of the subsequent CRF applications to individual neurones. The plot compares the amplitude of membrane depolarization (n = 7; P > 0.05). C, representative current-clamp traces showing that the CRF (100 nm)-induced depolarization is unaffected by 10 min pretreatment with TTX. Holding potential, −65 mV. D, summary comparing the mean depolarization amplitude of CRF and CRF + TTX (n = 5; P > 0.05).
To ascertain whether the CRF-induced depolarization was due to a direct effect of CRF on the DMV cell, we compared the amplitude of the CRF-induced membrane depolarization in the absence and presence of the synaptic transmission blocker, tetrodotoxin (TTX; 0.3 μM). In ten neurones, CRF induced a 3.3 ± 0.44 mV depolarization that recovered to baseline upon washout. Following 10 min of perfusion with TTX, reapplication of CRF in the presence of TTX induced a 3.1 ± 0.63 mV depolarization (i.e. 92 ± 14 % of control, P > 0.05 vs. CRF alone; Fig. 2).
In five cells, CRF increased the discharge rate from 1.1 ± 0.23 to 2.5 ± 0.67 Hz (i.e. 182 ± 67 % of control; P < 0.05). Following washout of CRF and 10 min pretreatment with the non-selective CRF antagonist, astressin (0.5 – 1 μM), the baseline firing rate did not change, but the CRF-induced increase in action potential firing rate was attenuated from 182 ± 67 % in control to 14 ± 9 % in astressin (P < 0.05 vs. control; Fig. 3). In a further four cells, CRF increased the discharge rate from 1.1 ± 0.28 to 1.9 ± 0.37 Hz (i.e. 75 ± 13 % of control; P < 0.05). Following washout of CRF and 10 min superfusion with the selective CRF2 antagonist (Chen et al. 2001) astressin 2B (500 nm), the baseline firing rate did not change, but the CRF-induced increase in discharge rate was reduced from 75 ± 13 % in control to 16 ± 16 % in astressin 2B (P < 0.05 vs. control; Fig. 3B).
Figure 3. The CRF-induced increase in discharge rate was attenuated by the non-selective CRF antagonist, astressin, and the selective CRF2 antagonist, astressin 2B.
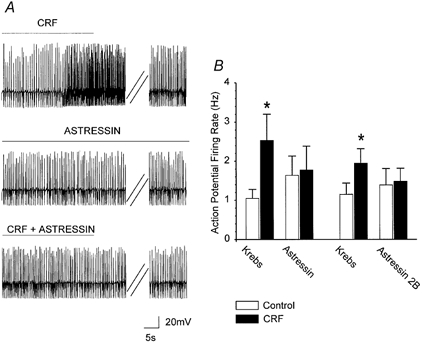
A, upper trace, representative current-clamp trace from a gastric-projecting DMV neurone illustrating the CRF-induced increase in discharge rate (from 2.1 to 3.9 Hz in control and following perfusion with 100 nm CRF, respectively). Middle trace, after a 5 min washout of CRF, the CRF antagonist, astressin (1 μM), was perfused for at least 7 min. Astressin had no effect on the basal firing rate (1.8 Hz before and during astressin perfusion). Lower trace, in the presence of astressin (1 μM), perfusion with CRF did not increase significantly the firing rate (from 1.8 to 1.9 Hz in astressin and in astressin + CRF, respectively). The astressin-mediated blockade of the CRF response was reversible (data not shown). Double parallel oblique lines indicate a 3.5 min period. B, summary showing the effects of astressin and astressin 2B on the excitatory effect of CRF. Bar graph shows the mean basal discharge rate and the CRF effect on the discharge rate before and in the presence of astressin (n = 5) or astressin 2B (n = 4) (*P < 0.05 vs. control).
The effect of CRF on the shape of the action potential was tested in six CRF-responsive neurones. The cells were hyperpolarized to −60 mV before being injected with a 10 ms-long pulse of DC sufficient to evoke a single action potential at its offset. The amplitude of the afterhyperpolarization (AHP) in control conditions was 15.3 ± 0.72 mV. Following CRF (100 nm) perfusion, current was injected to return the membrane potential to baseline values and a 10 ms-long pulse of DC was injected. In the presence of CRF, the amplitude of the action potential AHP was reduced to 14 ± 0.7 mV (P < 0.05 vs. control; Fig. 4).
Figure 4. CRF decreased the amplitude of the afterhyperpolarization (AHP).
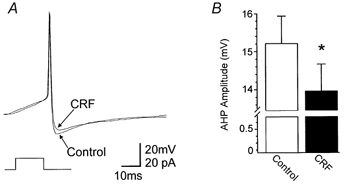
To evoke single action potentials, neurones were maintained at −60 mV by constant current injection before passing a short (10 ms) depolarizing current pulse of sufficient intensity to evoke an action potential at the offset of the pulse. A, current-clamp trace shows a single action potential before and after superfusion with 100 nm CRF. B, summary of data illustrating the CRF-induced decrease in AHP relative to the control AHP (n = 6). *P < 0.05.
The DMV membrane input resistance (calculated as the average of 20–40 electrotonic potentials evoked by injection of DC (50 ms on-950 ms off) sufficient to hyperpolarize the membrane by 5–7 mV) was measured in 15 neurones. Superfusion with CRF (100 nm) induced a 3.3 ± 0.4 mV depolarization; negative DC was then injected to restore the membrane potential to the value before CRF application. The input resistance was 461 ± 46 MΩ in control and 449 ± 57 MΩ in presence of CRF (P > 0.05).
In seven other neurones, the input resistance was measured in the presence of TTX. In these neurones, superfusion with CRF (100 nm) induced a 2.75 ± 0.32 mV depolarization; negative DC was then injected to restore the membrane potential to the value before CRF application. In four of the seven neurones, the input resistance in presence of CRF increased from 301 ± 84 to 337 ± 93 MΩ (P < 0.05) while in three other neurones no variations in the input resistance were observed.
Effects of CRF on EPSCs
EPSCs were evoked in 43 neurones by electrical stimulation of the NTS. In 16 of 43 (37 %) neurones, CRF (100 nm) increased the evoked EPSC amplitude from 218 ± 18.3 to 266 ± 21.7 pA (i.e. an increase of 22 ± 2.2 %; P < 0.05; Fig. 5). The paired pulse ratio is the ratio of the amplitude of the second evoked EPSC to the first evoked EPSC (C2/C1) and it is used as an indication of the site of drug action; a change in the ratio is taken as indication of a presynaptic site of action (Travagli & Williams, 1996; Browning & Travagli, 2001). In the aforementioned 16 cells, the paired pulse ratio was 0.56 ± 0.05 in control and 0.47 ± 0.06 in CRF thus indicating that CRF acts at presynaptic sites (P < 0.05 vs. control; Fig. 5). Concentration-response curves were constructed from cells in which all three concentrations of CRF (30-300 nm) were tested (n = 5; Fig. 5). In detail, 30 nm CRF increased the mean EPSC amplitude from 241 ± 29.3 to 250 ± 29.1 pA (i.e. 4 ± 5.2 % over control), while 100 nm CRF increased the mean EPSC amplitude from 260 ± 40.0 to 305 ± 42.8 pA (i.e. 18 ± 2.6 % over control) and 300 nm CRF increased the mean EPSC amplitude from 222 ± 17.8 to 283 ± 38.4 pA (i.e. 27 ± 13.7 % over control).
Figure 5. Activation of presynaptic CRF receptors increases the amplitude of evoked EPSCs.
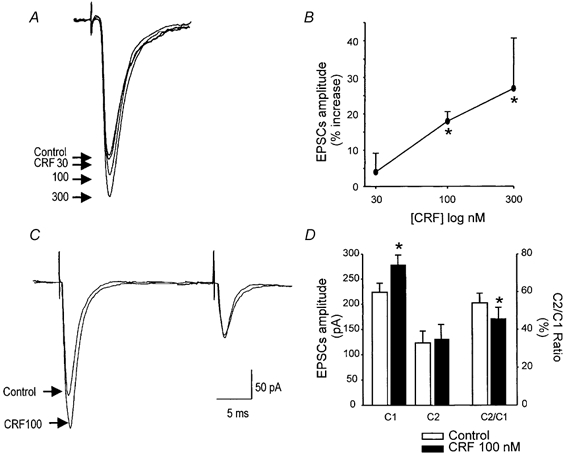
EPSCs were evoked by stimulation of the NTS. Traces are the average of three to six EPSCs each. Holding potential, −60 mV. CRF (30-300 nm) increased the amplitude of the EPSCs in a concentration-dependent manner (A, representative traces; B, summary graphic). Pairs of EPSCs were evoked 30–100 ms apart. CRF significantly increased the amplitude of the first evoked EPSC (C1) leaving unaffected the amplitude of the second EPSC (C2) thus altering the paired pulse ratio (C2/C1) (C, representative traces; D, summary bar graph). *P < 0.05 vs. control.
Figure 6. Microinjection sites for CRF.
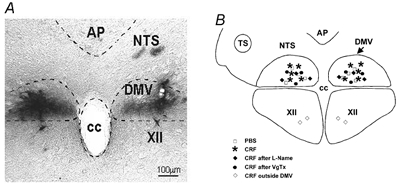
A, representative micrograph showing the location of the microinjection sites of CRF (dark precipitate). B, schematic diagram representing the brainstem areas in which the microinjections of CRF were performed. AP, area postrema; NTS, nucleus of the tractus solitarius; DMV, dorsal motor nucleus of the vagus; CC, central canal; XII, nucleus of the hypoglossus.
In vivo studies
Effects of CRF on gastric motility
Vagally mediated GI functions are controlled by two separate pathways: a cholinergic-excitatory muscarinic pathway and a NANC inhibitory pathway (Abrahamsson, 1986; Boeckxstaens et al. 1991; Desai et al. 1991; Lefebvre et al. 1992; Meulemans et al. 1995; Takahashi & Owyang, 1995; Hozer-Petsche & Moser, 1996; Mashimo et al. 1996; Kim et al. 1999). A vagally mediated decrease in gastric motility could then be obtained either by withdrawal of cholinergic tone or by activation of NANC pathways.
Experiments were performed on 15 anaesthetized rats in which baseline gastric motility was increased by systemic administration of the muscarinic agonist, bethanecol (50 μg kg−1 bolus followed by continuous i.v. bethanecol infusion 20 μg kg−1 h−1 for 20 min). Two minutes after administration of bethanecol either CRF (n = 5) or PBS (n = 4) was delivered bilaterally to the DVC. If, despite the supramaximal cholinergic stimulation via bethanecol, CRF reduced gastric motility, we could then infer that the effects of CRF are not mediated by withdrawal of cholinergic tone to the stomach but rather by activation of NANC pathways.
In the basal state, gastric motility was 11 ± 3.2 V s, following intravenous injection of bethanecol gastric motility significantly increased within 30 s of perfusion to reach a plateau value of 343 ± 31.6 V s (n = 15; P < 0.05; Fig. 7).
Figure 7. The CRF-induced decrease in gastric motility is abolished by the nitric oxide synthase inhibitor, l-NAME and vagotomy.
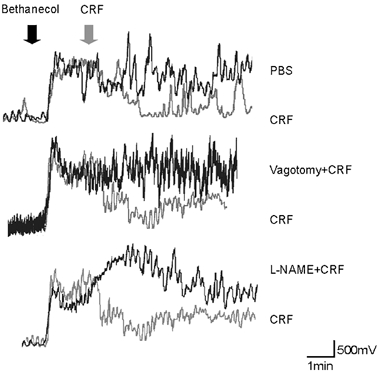
A, representative raw motility traces showing that the bethanecol-induced increase in gastric motility was decreased by the microinjection of CRF in the DVC. B, vagotomy eliminated the CRF-induced decrease in gastric motility. C, intravenous l-NAME also eliminated the CRF-induced decrease in gastric motility.
In four rats basal motility was 11 ± 9 V s, following bethanecol infusion, motility increased to 417 ± 34 V s and following bilateral microinjection of PBS in the DVC motility was unchanged (394 ± 87.5 V s; n = 4; P > 0.05). Compared with the effects of 20 nl of PBS injected into the DVC, bilateral microinjection of CRF in the DVC produced a very large reduction in gastric motility (i.e. from 17 ± 8.2 V s in control, to 373 ± 62.5 V s following bethanecol to 63 ± 34.3 V s in CRF; n = 5; P < 0.05; Fig. 7 and Fig. 8). The measures of gastric motility following CRF injections into the DVC were not statistically different from the basal condition (Fig. 8). In two animals, we injected CRF outside the borders of the DMV; no alteration in gastric motility or tone was observed (data not shown). Intravenous administration of l-NAME or bilateral vagotomy antagonized the inhibition of gastric motility induced by CRF microinjections in the DVC (Fig. 7 and Fig. 8). In either cases (l-NAME and vagotomy), gastric motility remained at levels not significantly different from the maximal levels observed after bethanecol, but were significantly greater than the levels observed after CRF alone. In detail, bethanecol infusion in the presence of l-NAME increased the motility from 6 ± 4.4 to 250 ± 64.4 V s, bilateral microinjection of CRF in the DVC did not significantly affect motility (i.e. 328.7 ± 107 V s; n = 3; P > 0.05). Similarly, bethanecol infusion following vagotomy increased the motility from 6 ± 3.2 to 286 ± 71 V s, bilateral microinjection of CRF in the DVC did not significantly affect motility (i.e. 280 ± 80.3 V s; n = 3; P > 0.05). The analysis of variance showed the treatment effect across all groups to be statistically significant; P < 0.05).
Figure 8. Summary of the CRF-induced inhibition of the bethanecol-induced increase in gastric motility.
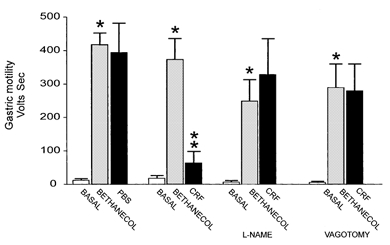
Two minutes after i.v. bethanecol, either CRF or PBS (control) was bilaterally microinjected in the DVC. From left to right: the first group of columns compares the bethanecol-stimulated gastric motility to control and following the microinjection of PBS in the DVC; the second group of columns compares the bethanecol-stimulated gastric motility to control and following the microinjection of CRF in the DVC; the third group of columns compares the effect of the NOS inhibitor, l-NAME, on the bethanecol-stimulated gastric motility vs. control and following the microinjection of CRF in the DVC; the fourth group of columns compares the effect of bilateral vagotomy on the bethanecol stimulated gastric motility vs. control and following the microinjection of CRF in the DVC. *P < 0.05 vs. control, **P < 0.05 vs. bethanecol.
Discussion
In this study we have shown that (1) in the in vitro rat brainstem slice preparation, perfusion with CRF induced a concentration-dependent excitation of vagal neurones. The CRF-mediated excitation was due to a direct depolarization of the DMV membrane as well as to an increase of the excitatory neurotransmission between the NTS and DMV neurones; and (2) in the in vivo anaesthetized rat preparation, bilateral microinjection of CRF in the DVC produced a vagally mediated inhibition of gastric motility via activation of inhibitory NANC neurones. Our data led us to conclude that the gastric inhibitory actions of CRF in the DVC result from the activation of CRF2 receptors, which increase activity in NANC inhibitory pathways. The following experimental evidence supports our conclusions.
CRF excites GI-projecting DMV neurones
Superfusion with CRF induced a concentration-dependent excitation of identified gastric- and duodenal-projecting DMV neurones via two separate mechanisms. The first mechanism was a direct depolarization of DMV neurones, most likely determined by a reduction in the amplitude of the AHP. The CRF-induced depolarization was mediated by activation of CRF2 receptors because both the non-selective CRF antagonist, astressin, as well as the selective CRF2 antagonist, astressin 2B, significantly attenuated the CRF-induced excitation of the DMV membrane. The second mechanism was via an increase in the amplitude of the EPSCs evoked by electrical stimulation of the NTS.
Our data on the excitatory effects of CRF are in line with current literature reporting that in the majority of the CNS neurones CRF has a depolarizing effect, most likely due to a decrease in the amplitude of the AHP (Eberly et al. 1983; Siggins et al. 1985; Yamashita et al. 1991; Hille, 1992; Fox & Gruol, 1993; Curtis et al. 1997; Page & Abercrombie, 1999; Haug & Storm, 2000).
Our pharmacological data confirmed and extended the suggestion put forward by Tache's group which suggested that the centrally mediated CRF inhibition of gastric emptying was likely mediated via the CRF2 receptor (Martinez et al. 1998).
CRF in the DVC significantly inhibited gastric motility
Vagally mediated GI functions are controlled by two separate pathways: a cholinergic-muscarinic pathway, whose activation induces an increase of GI functions, and a NANC pathway, whose activation induces a decrease in GI functions mainly via release of nitric oxide (NO) on to gastric smooth muscle (Abrahamsson, 1986; Boeckxstaens et al. 1991; Desai et al. 1991; Lefebvre et al. 1992; Meulemans et al. 1995; Takahashi & Owyang, 1995; Hozer-Petsche & Moser, 1996; Mashimo et al. 1996; Kim et al. 1999). A vagally mediated decrease in gastric motility could then be obtained either by withdrawal of the excitatory cholinergic tone or by activation of inhibitory NANC pathways.
Results from our in vitro study showed that the effects of CRF within the DVC were exclusively excitatory while our in vivo studies, as well as the studies already available in literature (Tache et al. 1983, 1987; Garrick et al. 1988; Heymann-Monnikes et al. 1991; Monnikes et al. 1992; Smedh et al. 1995; Coskun et al. 1997; Martinez et al. 1997, 1998), show that the effects of CRF microinjection in the DVC were exclusively gastroinhibitory. The in vitro and in vivo results obtained in the present study would then suggest that the gastroinhibitory effects of CRF were due to activation of the NANC pathway, possibly, via release of NO in the gastric smooth muscle. We conducted the following experiments to test the hypothesis that NANC pathways, but not cholinergic pathways, were involved in the gastric relaxation induced by CRF. Our experimental design was based on the premise that, to study the inhibitory effects of CRF on gastric motility, the cholinergic activity of the stomach would have to be substantially increased. If CRF were to antagonize the muscarinic-cholinergic activity of the stomach we would have not been able to ascertain it with the use of systemic injection of the muscarinic antagonist atropine, because gastric motility would be decreased to a level such that it would not be possible to detect a further decrease in motility carried by the activation of the inhibitory NANC pathway. To circumvent this problem, we increased gastric motility by systemic injection of the muscarinic agonist, bethanecol at a supramaximal dose. If the microinjection of CRF in the DVC failed to decrease gastric motility in the bethanecol-treated rats, then we would surmise that the actions of CRF were mediated via muscarinic-cholinergic receptors. If instead, CRF were to decrease gastric motility in the bethanecol-treated rats, this would suggest that the CRF-induced decrease in gastric motility was mediated by a mechanism other than the cholinergic-muscarinic pathway, perhaps the NANC pathway. Indeed, our in vivo results showed that despite a supramaximal activation of muscarinic cholinergic receptors, microinjections of CRF in the DVC still induced a decrease in gastric motility, thus arguing in favour of an NANC pathway, but against the withdrawal of a cholinergic pathway as the gastric inhibitory mechanism.
Much of the vagal NANC inhibitory effect on the stomach is mediated by the release of nitric oxide onto gastric smooth muscle (Abrahamsson, 1986; Boeckxstaens et al. 1991; Desai et al. 1991; Lefebvre et al. 1992; Meulemans et al. 1995; Takahashi & Owyang, 1997; Kim et al. 1999). Therefore, we tested the hypothesis that the nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester (l-NAME) would block the effects of CRF in the DVC despite the bethanecol-induced increase in gastric motility.
Indeed, bilateral microinjections of CRF in the DVC significantly inhibited gastric motility in rats pretreated with bethanecol, and the effects of CRF on GI motility were abolished by systemic injection of l-NAME (or bilateral vagotomy), thus suggesting that excitation of a vagal pathway that uses NO as neurotransmitter mediated the effects of CRF. Our results are in contrast to a recent report that, by using extracellular recordings from isolated ‘efferent’ vagal fibres, suggested that the gastric relaxation induced by intracisternal administration of CRF was determined by a decrease in vagal firing (Kosoyan et al. 1999). This discrepancy may be due to the fact that, in his work, Kosoyan recorded from intact vagal preparations, which include fibres projecting to other GI regions (Berthoud et al. 1991), making his analysis difficult to interpret.
Physiological significance
Numerous studies in rats have shown that injection of CRF in the cisterna magna, lateral ventricles, DVC and PVN induce a decrease in gastric acid secretion, gastric emptying, small bowel transit and an increased large bowel transit (Tache et al. 1983, 1987; Garrick et al. 1988; Heymann-Monnikes et al. 1991; Monnikes et al. 1992; Smedh et al. 1995; Coskun et al. 1997; Martinez et al. 1997, 1998). Such varied GI effects are similar to those observed in response to stress. Indeed, the effects that are observed in response to stress are mimicked by the i.c. injection of CRF and both the stress-related and the CRF-induced GI-effects are blocked by the various CRF antagonists such as α-helical CRF9-41, d-Phen CRF12-41 and astressin (Lenz et al. 1988; Coskun et al. 1997; Martinez et al. 1997). In the absence of exogenous CRF or stress, the CRF antagonists do not affect gastric activity, suggesting that CRF does not modulate basal digestive activity (Lenz et al. 1988; Smedh et al. 1995; Martinez et al. 1997, 1998). Taken together, the evidence suggests that, in the time of stress, CRF acts as a central neurotransmitter in the DMV to control gastric function.
In summary, CRF acts within the DVC to decrease gastric motility and intragastric pressure. The effect of CRF in the DVC is mediated via activation of NANC inhibitory pathways to the GI tract and requires an intact vagus nerve. Evidence suggests that, within the DVC, CRF has a dual role. In the NTS, CRF increases the probability of release of an excitatory neurotransmitter, most probably glutamate, however, the predominant role of CRF in the DVC is to directly excite DMV neurones by depolarization and a reduction in the amplitude of the AHP.
Acknowledgments
The in vivo portion of this work was performed while Drs Rogers and Hermann were at the Department of Neuroscience, Ohio State University, Columbus, OH, USA. This work was supported by NIH grants DK56373 and DK55530.
References
- Abrahamsson H. Non-adrenergic non-cholinergic nervous control of gastrointestinal motility patterns. Archives internationals de pharmacodynamie et de therapie. 1986;280:50–61. [PubMed] [Google Scholar]
- Altschuler SM, Bao X, Bieger D, Hopkins DA, Miselis RR. Viscerotopic representation of the upper alimentary tract in the rat: sensory ganglia and nuclei of the solitary and spinal trigeminal tracts. Journal of Comparative Neurology. 1989;283:248–268. doi: 10.1002/cne.902830207. [DOI] [PubMed] [Google Scholar]
- Altschuler SM, Ferenci DA, Lynn RB, Miselis RR. Representation of the cecum in the lateral dorsal motor nucleus of the vagus nerve and commissural subnucleus of the nucleus tractus solitarii in rat. Journal of Comparative Neurology. 1991;304:261–274. doi: 10.1002/cne.903040209. [DOI] [PubMed] [Google Scholar]
- Barnes KL, McWueeney AJ, Barrett WR, Knowles WD. Morphology and projections of neurobiotin-labeled nucleus tractus solitarii neurons recorded in vitro. Brain Research Bulletin. 1994;4:339–348. doi: 10.1016/0361-9230(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Beltran B, Barrachina MD, MendeZ A, Quintero E, Esplugues JV. Synthesis of nitric oxide in the dorsal motor nucleus of the vagus mediates the inhibition of gastric acid secretion by central bombesin. British Journal of Pharmacology. 1999;127:1603–1610. doi: 10.1038/sj.bjp.0702717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR, Carlson NR, Powley TL. Topography of efferent vagal innervation of the rat gastrointestinal tract. American Journal of Physiology. 1991;260:R200–207. doi: 10.1152/ajpregu.1991.260.1.R200. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC, Vaughan J, Arias C, Rissman RA, Vale WW, Sawchenko PE. Urocortin expression in rat brain: evidence against a pervasive relationship of urocortin-containing projections with targets bearing type 2 CRF receptors. Journal of Comparative Neurology. 1999;415:285–312. [PubMed] [Google Scholar]
- Boeckxstaens GE, Pelckmans PA, Bogers J, Bult H, De Man JG, Oosterbosch L, Herman AG, Van Maercke YM. Release of nitric oxide upon stimulation of noadrenergic noncholinergic nerves in the rat gastric fundus. Journal of Pharmacology and Experimental Therapeutics. 1991;256:441–447. [PubMed] [Google Scholar]
- Browning KN, Renehan WE, Travagli RA. Electrophysiological and morphological heterogeneity of rat dorsal vagal neurones which project to specific areas of the gastrointestinal tract. Journal of Physiology. 1999;517:521–532. doi: 10.1111/j.1469-7793.1999.0521t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. The peptide TRH uncovers the presence of presynaptic 5-HT1A receptors via activation of a second messenger pathway in the rat dorsal vagal complex. Journal of Physiology. 2001;531:425–435. doi: 10.1111/j.1469-7793.2001.0425i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Million M, Adelson DW, MartineZ V, Rivier J, Tache Y. Intracisternal urocortin inhibits vagally stimulated gastric motility in rats: role of CRF2. British Journal of Pharmacology. 2002;136:237–247. doi: 10.1038/sj.bjp.0704713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun T, Bozkurt A, Alican I, Ozkutlu U, Kurtel H, Yegen BC. Pathways mediating CRF-induced inhibition of gastric emptying in rats. Regulatory Peptides. 1997;69:113–120. doi: 10.1016/s0167-0115(96)02066-6. [DOI] [PubMed] [Google Scholar]
- Cummings S, Elde R, Ells J, Lindall A. Corticotropin-releasing factor immunoreactivity is widely distributed within the central nervous system of the rat: an immunohistochemical study. Journal of Neuroscience. 1983;3:1355–1368. doi: 10.1523/JNEUROSCI.03-07-01355.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AL, Lechner SM, Pavcovich LA, Valentino RJ. Activation of the locus coeruleus noradrenergic system by intracoerulear microinfusion of corticotropin-releasing factor: effects on discharge rate, cortical norepinephrine levels and cortical electroencephalographic activity. Journal of Pharmacology and Experimental Therapeutics. 1997;281:163–172. [PubMed] [Google Scholar]
- De Souza EB. Corticotropin-releasing factor receptors in the rat central nervous system: characterization and regional distribution. Journal of Neuroscience. 1987;7:88–100. doi: 10.1523/JNEUROSCI.07-01-00088.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza EB, Insel TR, Perrin MH, Rivier J, Vale WW, Kuhar MJ. Corticotropin-releasing factor receptors are widely distributed within the rat central nervous system: an autoradiographic study. Journal of Neuroscience. 1985;5:3189–3203. doi: 10.1523/JNEUROSCI.05-12-03189.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza EB, Kuhar MJ. Corticotropin-releasing factor receptors in the pituitary gland and central nervous system: methods and overview. Methods in Enzymology. 1986;124:560–590. doi: 10.1016/0076-6879(86)24040-9. [DOI] [PubMed] [Google Scholar]
- Desai KM, Sessa WC, Vane JR. Involvement of nitric oxide in the reflex relaxation of the stomach to accommodate food or fluid. Nature. 1991;351:477–479. doi: 10.1038/351477a0. [DOI] [PubMed] [Google Scholar]
- Eberly LB, Dudley CA, Moss RL. Iontophoretic mapping of corticotropin-releasing factor (CRF) sensitive neurons in the rat forebrain. Peptides. 1983;4:837–841. doi: 10.1016/0196-9781(83)90077-3. [DOI] [PubMed] [Google Scholar]
- Esplugues JV, Barrachina MD, Beltran B, Calatayud S, Whittle BJR, Moncada S. Inhibition of gastric acid secretion by stress: a protective reflex mediated by cerebral nitric oxide. Proceedings of the National Academy of Sciences of the USA. 1996;93:14839–14844. doi: 10.1073/pnas.93.25.14839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster ER, Southam E. The intrinsic and vagal extrinsic innervation of the rat stomach contains nitric oxide synthase. NeuroReport. 1993;4:275–278. doi: 10.1097/00001756-199303000-00012. [DOI] [PubMed] [Google Scholar]
- Fox EA, Gruol DL. Corticotropin releasing factor suppresses the afterhyperpolarization in cerebellar purkinje neurons. Neuroscience Letters. 1993;149:103–107. doi: 10.1016/0304-3940(93)90358-r. [DOI] [PubMed] [Google Scholar]
- Garrick T, Veiseh A, Sierra A, Weiner H, Tache Y. Corticotropin-releasing factor acts centrally to suppress stimulated gastric contractility. Regulatory Peptides. 1988;21:173–181. doi: 10.1016/0167-0115(88)90101-2. [DOI] [PubMed] [Google Scholar]
- Gue M, Fioramonti J, Frexinos J, Alvinerie M, Bueno L. Influence of acoustic stress by noise on gastrointestinal motility in dogs. Digestive Diseases and Sciences. 1987;32:1411–1417. doi: 10.1007/BF01296668. [DOI] [PubMed] [Google Scholar]
- Guo JJ, Browning KN, Rogers RC, Travagli RA. Catecholaminergic neurons in rat dorsal motor nucleus of vagus project selectively to gastric corpus. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2001;280:G361–367. doi: 10.1152/ajpgi.2001.280.3.G361. [DOI] [PubMed] [Google Scholar]
- Haug T, Storm JF. Protein kinasse A mediates the modulation of the slow Ca2+-dependent K+ current, IsAHP, by the neuropeptides CRF, VIP, and CGRP in hippocampal pyramidal neurons. Journal of Neurophysiology. 2000;83:2071–2079. doi: 10.1152/jn.2000.83.4.2071. [DOI] [PubMed] [Google Scholar]
- Herbert H, Saper CB. Cholecystokinin-, galanin-, and corticotropin-releasing factor-like immunoreactive projections from the nucleus of the solitary tract to the parabrachial nucleus in the rat. Journal of Comparative Neurology. 1990;293:581–598. doi: 10.1002/cne.902930405. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC. Tumor necrosis factor-alpha in the dorsal vagal complex suppresses gastric motility. Neuroimmunomodulation. 1995;2:74–81. doi: 10.1159/000096874. [DOI] [PubMed] [Google Scholar]
- Heymann-Monnikes I, Tache Y, Trauner M, Weiner H, Garrick T. CRF microinjected into the dorsal vagal complex inhibits TRH analog- and kainic acid-stimulated gastric contractility in rats. Brain Research. 1991;554:139–144. doi: 10.1016/0006-8993(91)90181-t. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland, MA, USA: Sinauer Associates Inc.; 1992. [Google Scholar]
- Hozer-Petsche U, Moser RL. Participation of nitric oxide in the relaxation of the rat gastric corpus. Naunyn-Schmiedeberg's Archives of Pharmacology. 1996;354:348–354. doi: 10.1007/BF00171067. [DOI] [PubMed] [Google Scholar]
- Kim CD, Goyal RK, Mashimo H. Neuronal NOS provides nitrergic inhibitory neurotransmitter in moouse lower esophageal sphincter. American Journal of Physiology. 1999;277:G280–284. doi: 10.1152/ajpgi.1999.277.2.G280. [DOI] [PubMed] [Google Scholar]
- Kosoyan HP, Wei JY, Tache Y. Intracisternal sauvagine is more potent than corticotropin-releasing factor to decrease gastric vagal efferent activity in rats. Peptides. 1999;20:851–858. doi: 10.1016/s0196-9781(99)00072-8. [DOI] [PubMed] [Google Scholar]
- Krowicki ZK, Sivarao DV, Abrahams TP, Hornby PJ. Excitation of dorsal motor vagal neurons evokes non-nicotinic receptor-mediated gastric relaxation. Journal of the Autonomic Nervous System. 1999;77:83–89. [PubMed] [Google Scholar]
- Lawrence AJ. Nitric oxide as a modulator of medullary pathways. Clinical and Experimental Pharmacology and Physiology. 1997;24:760–763. doi: 10.1111/j.1440-1681.1997.tb02128.x. [DOI] [PubMed] [Google Scholar]
- Lefebvre RA, Hasrat J, Gobert A. Influence of NG-nitro-l-arginine methyl ester on vagally induced gastric relaxation in the anaesthetized rat. British Journal of Pharmacology. 1992;105:315–320. doi: 10.1111/j.1476-5381.1992.tb14252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz HJ, Raedler A, Greten H, Vale WW, Rivier JE. Stress-induced gastrointestinal secretory and motor responses in rats are mediated by endogenous corticotropin-releasing factor. Gastroenterology. 1988;95:1510–1517. doi: 10.1016/s0016-5085(88)80070-2. [DOI] [PubMed] [Google Scholar]
- McCann MJ, Rogers RC. Impact of antral mechanoreceptor activation on the vago-vagal reflex in the rat: functional zonation of responses. Journal of Physiology. 1992;453:401–411. doi: 10.1113/jphysiol.1992.sp019235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez V, Barquist E, Rivier J, Tache Y. Central CRF inhibits gastric emptying of a nutrient solid meal in rats: the role of CRF2 receptors. American Journal of Physiology. 1998;274:G965–970. doi: 10.1152/ajpgi.1998.274.5.G965. [DOI] [PubMed] [Google Scholar]
- Martinez V, Rivier J, Wang L, Tache Y. Central injecton of a new corticotropin-releasing factor (CRF). antagonist, astressin, blocks CRF- and stress-related alterations of gastric and colonic motor function. Journal of Pharmacology and Experimental Therapeutics. 1997;280:754–760. [PubMed] [Google Scholar]
- Mashimo H, He XD, Huang PL, Fishman MC, Goyal RK. Neuronal constitutive nitric oxide synthase is involved in murine enteric inhibitory neurotransmission. Journal of Clinical Investigation. 1996;98:8–13. doi: 10.1172/JCI118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulemans AL, Eelen JG, Schuurkes JA. NO mediates gastric relaxation after brief vagal stimulation in anesthetized dogs. American Journal of Physiology. 1995;269:G255–261. doi: 10.1152/ajpgi.1995.269.2.G255. [DOI] [PubMed] [Google Scholar]
- Monnikes H, Schmidt BG, Raybould HE, Tache Y. CRF in the paraventricular nucleus mediates gastric and colonic motor response to restraint stress. American Journal of Physiology. 1992;262:G137–143. doi: 10.1152/ajpgi.1992.262.1.G137. [DOI] [PubMed] [Google Scholar]
- Norgren R, Smith GP. Central distribution of subdiaphragmatic vagal branches in the rat. Journal of Comparative Neurology. 1988;273:207–223. doi: 10.1002/cne.902730206. [DOI] [PubMed] [Google Scholar]
- Page ME, Abercrombie ED. Discrete local application of corticotropin-releasing factor increases locus coeruleus discharge and extracellular norepinephrine in rat hippocampus. Synapse. 1999;33:304–313. doi: 10.1002/(SICI)1098-2396(19990915)33:4<304::AID-SYN7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Panico WH, Cavuto NJ, Kallimanis G, Nguyen C, Armstrong DM, Benjamin SB, Gillis RA, Travagli RA. Functional evidence for the presence of nitric oxide synthase in the dorsal motor nucleus of the vagus. Gastroenterology. 1995;109:1484–1491. doi: 10.1016/0016-5085(95)90634-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2. San Diego: Academic Press, Inc.; 1986. [DOI] [PubMed] [Google Scholar]
- Quintana E, Garcia-Zaragoza E, MartineZ-Cuesta MA, Calatayud S, Esplugues JV, Barrachina MD. A cerebral nitrergic pathway modulates endotoxin-induced changes in gastric motility. British Journal of Pharmacology. 2001;134:325–332. doi: 10.1038/sj.bjp.0704258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE, Travagli RA. Brainstem pathways responsible for oesophageal control of gastric motility and tone in the rat. Journal of Physiology. 1999;514:369–383. doi: 10.1111/j.1469-7793.1999.369ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, Kita H, Butcher LL, Novin D. Afferent projections to the dorsal motor nucleus of the vagus. Brain Research Bulletin. 1980;5:365–373. doi: 10.1016/s0361-9230(80)80006-2. [DOI] [PubMed] [Google Scholar]
- Rogers RC, McTigue DM, Hermann GE. Vagovagal reflex control of digestion: afferent modulation by neural and ‘endoneurocrine’ factors. American Journal of Physiology. 1995;268:G1–10. doi: 10.1152/ajpgi.1995.268.1.G1. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Shibasaki T, Lederis K. Corticotropin releasing factor-like immunoreactivity in the rat brain as revealed by a modifyed cobalt-glucose oxidase-diaminobenzidine method. Journal of Comparative Neurology. 1987;260:256–298. doi: 10.1002/cne.902600209. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Gruol D, Aldenhoff JB, Pittman Q. Electrophysiological actions of corticotropin-releasing factor in the central nervous system. Federation Proceedings. 1985;44:237–242. [PubMed] [Google Scholar]
- Skofitsch G, Insel TR, JacobowitZ DM. Binding sites for corticotropin releasing factor in sensory areas of the rat hindbrain and spinal cord. Brain Research Bulletin. 1985;15:519–522. doi: 10.1016/0361-9230(85)90043-7. [DOI] [PubMed] [Google Scholar]
- Smedh U, Uvnas-moberg K, Grill HJ, Kaplan JM. Fourth ventricle injection of corticotropin-releasing factor and gastric emptying of glucose during gastric fill. American Journal of Physiology. 1995;269:G1000–1003. doi: 10.1152/ajpgi.1995.269.6.G1000. [DOI] [PubMed] [Google Scholar]
- Sykes RM, Spyer KM, Izzo PN. Demonstration of glutamate immunoreactivity in vagal sensory afferents in the nucleus tractus solitarius of the rat. Brain Research. 1997;762:1–11. doi: 10.1016/s0006-8993(97)00368-5. [DOI] [PubMed] [Google Scholar]
- Tache Y, Goto Y, Gunion MW. Inhibition of gastric acid secretion rats by intracerebral injection of corticotropin-releasing factor. Science. 1983;222:935–937. doi: 10.1126/science.6415815. [DOI] [PubMed] [Google Scholar]
- Tache Y, Maeda-Hagiwara M, Turkelson CM. Central nervous system action of corticotropin-releasing factor to inhibit gastric emptying in rats. American Journal of Physiology. 1987;253:G241–245. doi: 10.1152/ajpgi.1987.253.2.G241. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Owyang C. Vagal control of nitric oxide and vasoactive intestinal polypeptide release in the regulation of gastric relaxation in rat. Journal of Physiology. 1995;484:481–492. doi: 10.1113/jphysiol.1995.sp020680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Owyang C. Characterization of vagal pathways mediating gastric accomodation reflex in rats. Journal of Physiology. 1997;504:479–488. doi: 10.1111/j.1469-7793.1997.479be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Owyang C. Regional differences in the nitrergic innervation between the proximal and the distal colon in rats. Gastroenterology. 1998;115:1504–1512. doi: 10.1016/s0016-5085(98)70029-0. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA, Rossiter CD, Vicini S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. American Journal of Physiology. 1991;260:G531–536. doi: 10.1152/ajpgi.1991.260.3.G531. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Williams JT. Endogenous monoamines inhibit glutamate transmission in the spinal trigeminal nucleus of the guinea-pig. Journal of Physiology. 1996;491:177–185. doi: 10.1113/jphysiol.1996.sp021205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino RJ, Pavcovich LA, Hirata H. Evidence for corticotropin-releasing hormone projections from Barrington's nucleus to the periaqueductal gray and dorsal motor nucleus of the vagus in the rat. Journal of Comparative Neurology. 1995;363:402–422. doi: 10.1002/cne.903630306. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Kasai M, Inenaga K. Effects of corticotropin-releasing factor on neurons in the hypothalamic paraventricular nucleus in vitro. Brain Research Bulletin. 1991;27:321–325. doi: 10.1016/0361-9230(91)90119-5. [DOI] [PubMed] [Google Scholar]
- Yu W-H A. Nitric oxide synthase in motor neurons after axotomy. Journal of Histochemistry and Cytochemistry. 1994;42:451–457. doi: 10.1177/42.4.7510317. [DOI] [PubMed] [Google Scholar]
- Zheng ZL, Rogers RC, Travagli RA. Selective gastric projections of nitric oxide synthase-containing vagal brainstem neurons. Neuroscience. 1999;90:685–694. doi: 10.1016/s0306-4522(98)00586-7. [DOI] [PubMed] [Google Scholar]