

Abstract
Paired pulse depression (PPD) is a common form of short-term synaptic plasticity. The aim of this study was to characterise PPD at the level of a single inhibitory bouton. Low-density collicular cultures were loaded with the Ca2+ indicator Oregon Green-1, active boutons were stained with RH414, and action potentials were blocked with TTX. Evoked IPSCs (eIPSCs) and presynaptic Ca2+ transients were recorded in response to direct presynaptic depolarisation of an individual bouton. The single bouton eIPSCs had a low failure rate (< 0.1), large average quantal content (3-6) and slow decay (τ1 = 15 ms, τ2 = 81 ms). The PPD of eIPSCs had two distinct components: PPDfast and PPDslow (τ = 86 ms and 2 s). PPDslow showed no dependence on extracellular Ca2+ concentration, or on the first eIPSC's failure rate or amplitude. Most probably, it reflects a release-independent inhibition of exocytosis. PPDfast was only observed in normal or elevated Ca2+. It decreased with the failure rate and increased with the amplitude of the first eIPSC. It coincided with paired pulse depression of the presynaptic Ca2+ transients (τ = 120 ms). The decay of the latter was accelerated by EGTA, which also reduced PPDfast. Therefore, a suppressive effect of residual presynaptic Ca2+ on subsequent Ca2+ influx is considered the most likely cause of PPDfast. PPDfast may also have a postsynaptic component, because exposure to a low-affinity GABAA receptor antagonist (TPMPA; 300 μM) counteracted PPDfast, and asynchronous IPSC amplitudes were depressed for a short interval following an eIPSC. Thus, at these synapses, PPD is produced by at least two release-independent presynaptic mechanisms and one release-dependent postsynaptic mechanism.
In many systems, a single synaptic activation will depress a subsequent synaptic response. This form of synaptic plasticity lasts from tens of milliseconds to several seconds, and it plays an important role in neural coding (O'Donovan & Rinzel, 1997). Short-term synaptic depression acts as low-pass filter (Fortune & Rose, 2001), it can serve as a gain control mechanism during and after burst activation (Varela et al. 1999), and it may contribute to the generation of synchronous network activity (Tsodyks et al. 2000). Long-term changes of synaptic transmission are often associated with a modification of short-term synaptic plasticity (Abbott & Nelson, 2000). Whether a connection is prone to depression or facilitation may depend on its developmental state (Pouzat & Hestrin, 1997; Jüttner et al. 2001), or the contacted target cell (Rozov et al. 2001). Usually, connections with higher average release probability will be more likely to display depression when activated at short intervals (Thomson, 2000).
The mechanisms underlying the various forms of short-term plasticity are not yet clear. Traditionally, short-term depression is attributed to decreased transmitter release from the presynaptic terminal (O'Donovan & Rinzel, 1997). Earlier studies stressed the significance of release-dependent depletion of a vesicle pool (Elmquist & Quastel, 1965; Mennerick & Zorumski, 1995; Stevens & Tsujimoto, 1995; Debanne et al. 1996; Stevens & Wesseling, 1998). However, it has become increasingly evident that the probabilities with which a given vesicle is released and then recovered during a certain time interval are both highly non-uniform. Recent detailed studies in the calyx of Held suggested that a subpool of rapidly replenished, but reluctantly released vesicles maintain synaptic function when a more slowly recovering but readily released vesicle population is exhausted (Sakaba & Neher, 2001). An increase in presynaptic bulk Ca2+ can increase the fraction of vesicles available for immediate release (Dittman & Regehr, 1998; Stevens & Wesseling, 1998; Wang & Kaczmarek, 1998; Sakaba & Neher, 2001). Therefore, the kinetics of paired pulse depression (PPD) will depend on both previous release and the residual Ca2+ concentration in the terminal.
Other studies revealed that a major component of short-term depression is release independent (Betz, 1970; Hsu et al. 1996; Bellingham & Walmsley, 1999; Thomson & Bannister, 1999; Wu & Borst, 1999; Kraushaar & Jonas, 2000; Waldeck et al. 2000). In this case depression can even occur in the absence of exocytosis. Release-independent depression may result from decreased presynaptic Ca2+ influx due to failures in action potential invasion (Hatt & Smith, 1976; Streit et al. 1992; Brody & Yue, 2000), reduction or shortening of presynaptic action potentials (Brody & Yue, 2000; Geiger & Jonas, 2000; He et al. 2002), inactivation of presynaptic Ca2+ channels (Patil et al. 1998) or post-pulse hyperpolarisation due to Ca2+-activated K+ and/or Cl− conductances (Marrion & Tavalin, 1998; Oliver et al. 2000).
Finally, short-term depression has been attributed to postsynaptic receptor desensitisation (Trussell et al. 1993; Mennerick & Zorumski, 1996). This mechanism of depression requires enhanced transmitter release (Oleskevich et al. 2000).
Most previous studies on short-term depression in the central nervous system (for reviews see Thomson, 2000; Schneggenburger et al. 2002; Zucker & Regehr, 2002) were based on the analysis of composite unitary synaptic responses. This approach is hampered by the lack of information on the propagation/invasion of the presynaptic action potentials and the size of presynaptic Ca2+ signals induced by individual stimuli. Furthermore, in a vast majority of central connections postsynaptic currents are generated by an unknown number of heterogeneous release sites which most probably operate in an intermittent fashion. This makes it technically difficult to determine the relative strength and the site of expression of a modulatory mechanism.
Recent advances in the visualisation of small synapses offered a chance to circumvent some of these problems by activating single terminals in culture by application of a short depolarising pulse in the area of contact (Kirischuk et al. 1999b). The presynaptic Ca2+ transients could be recorded in parallel with the postsynaptic response, and deliberately be varied by changing the stimulus intensity (Kirischuk et al. 1999a). By applying this approach to GABAergic terminals in cultures from the neonatal rat superior colliculus we intended to obtain answers to the following questions. (1) Do release-dependent and release-independent mechanisms of PPD coexist in the same synaptic contact? (2) What role does the residual Ca2+ elevation play in the induction of PPD? (3) Is there a postsynaptic contribution to PPD at inhibitory synapses?
A preliminary account of this work has already appeared (Kirischuk & Grantyn, 2001).
Methods
Cultures
Cell cultures were prepared as described previously (Perouansky & Grantyn, 1989). Neonatal rats (embryonic day 21) were anaesthetised with halothane before decapitation. Superior colliculi were removed and dissociated. The neurones were grown at low density (about 5000 cells cm−2) on laminin-coated glass coverslips. Experiments were performed on cultures between 10 and 36 days in vitro. All experiments were carried out according to the guidelines laid down by the Landesamt für Arbeitsschutz, Gesundheitsschutz und technische Sicherheit Berlin (T0406/98).
Imaging
A detailed description of the method is given elsewhere (Kirischuk et al. 1999b). Briefly, cultures were incubated in standard extracellular solution (mm: 140 NaCl, 3 KCl, 1 MgCl2, 5 CaCl2, 20 Hepes, 30 glucose, pH set to 7.4 with NaOH) supplemented with either Oregon Green 488 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetate acetoxymethyl ester (OGB-1-AM; 5 μM, 15–20 min, at 36 °C) or Magnesium Green (MG; 5 μM, 30 min, at 36 °C) and then kept for an additional 20 min in standard saline to ensure deesterification. Next, synaptic vesicles were stained with a fluorescent marker. FM1-43 (10 μM) was used in early experiments, but was replaced in later studies by RH414. RH414 was loaded in two steps. Cultures were incubated first in solution containing high potassium (50 mm) and RH414 (50 μM) for 15–20 s, and were then switched to extracellular solution containing RH414 (50 μM) for a further 30 s, before being washed twice in extracellular solution alone. The coverslip with the stained cultures formed the bottom of a recording chamber on the stage of an inverted microscope (Axiovert 100, Zeiss, Jena, Germany). A ×100 phase contrast oil immersion objective with a numerical aperture of 1.3 (Zeiss) was used in all experiments. The excitation wavelengths were controlled by a fast monochromator system, and fluorescence signals were recorded using a CCD camera (TILL Photonics, München, Germany). All measurements were performed using 4 × 4 binning (1 pixel = 0.4 μm × 0.4 μm). The acquisition rate for [Ca2+] measurement was one image per 10 ms (OGB-1) or 20 ms (MG). The probes were excited at 490 nm. The excitation and emission light was separated using a 510 nm dichroic mirror. The emitted light was filtered at 550 ± 30 nm for calcium-sensitive probes and at 600 nm for RH414. Phase contrast and RH414 images were captured at the beginning of each experiment. The RH414 image was converted to binary format using a threshold set to half-maximal intensity above the background. The binary image was used as a mask (by multiplication) to define the region of interest for subsequent OGB-1 or MG images. Thus, the presynaptic region of interest was defined as the area of vesicle accumulation. The background fluorescence originating from glial cells was determined from a region in the immediate vicinity of the stimulated bouton and subtracted. To decrease contamination of the presynaptic signal by fluorescence from the underlying dendrite, all measurements were performed after at least 15 min dialysis of the postsynaptic neurone. Fluorescence signals were expressed as the relative change from the pre-stimulus level (ΔF/F0).
EGTA-AM loading
A 100 mm stock solution of EGTA-AM in dimethyl sulfoxide (DMSO) was aliquoted and frozen. Immediately before use, aliquots were diluted in extracellular solution to the final concentration, and OGB-1-AM was added. The cells were then incubated for 20 min at 36 °C in standard saline containing OGB-1-AM and EGTA-AM.
Patch clamp recordings
Whole-cell patch clamp recordings were performed using glass pipettes containing (mm): 100 potassium gluconate, 50 KCl, 5 NaCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 20 Hepes, and pH was set to 7.2 with KOH. The holding potential was −70 mV, and ECl was −20 mV. Signals were acquired at 10 kHz using an EPC-7 patch clamp amplifier and TIDA 3.7 acquisition software (HEKA Electronics, Lambrecht, Pfalz, Germany). Series resistance (10-12 MΩ) was compensated up to 70 %.
Superfusion
All experiments were performed at room temperature (23-25 °C). A superfusion system with a flow rate of 0.5 ml min−1 was used. dl-2-amino-5-phosphonopentanoic acid (APV; 50 μM) and 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μM) were added to prevent glutamatergic synaptic transmission. Tetrodotoxin (TTX; 1 μM) was added to prevent action potential generation, unless otherwise stated. In experiments using lower (< 5 mm) Ca2+ solutions, Ca2+ was replaced with an equimolar concentration of Mg2+ to keep the total concentration of divalent ions constant. OGB-1, MG, RH414 and EGTA-AM were obtained from Molecular Probes; CGP55845A was a kind gift from Novartis (Basel, Switzerland); all other chemicals were from Sigma-Aldrich (Deisenhofen, Germany).
Selection of synaptic boutons
All experiments were carried out on well-isolated GABAergic axo-dendritic boutons, preferably located on the side of a first order dendrite (Fig. 1A and Fig. 2A and B). The largest diameter of the bouton was on average 1.06 μm (s.d. = 0.41, n = 50). The following precautions ensured that only a single bouton was activated. Experiments were confined to terminal boutons, or boutons formed when an axon crossed a dendrite at close to 90 deg. The selected bouton was always > 2 μm away from its closest RH414-labelled neighbour. A small (≈1 μm) displacement of the stimulation pipette reversibly abolished both the Ca2+ transient and evoked IPSC (eIPSC). A Ca2+ response was detected only in the stimulated boutons.
Figure 1. Response of a single visualised inhibitory bouton to a presynaptic action potential.
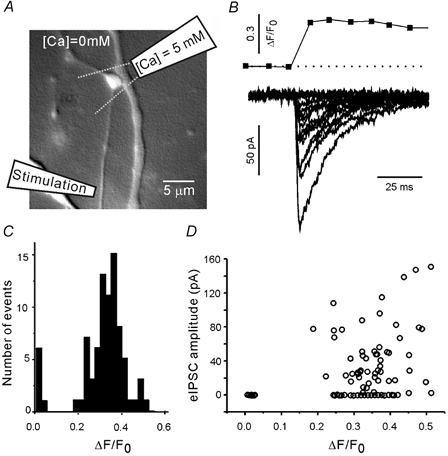
A, recording configuration. The extracellular solution contained no added Ca2+. A glass pipette was used to apply 5 mm Ca2+ selectively to the bouton of interest. Dashed lines indicate the approximate dimensions of the Ca2+ solution plume. The axon was focally stimulated via a second patch pipette placed 50–100 μm from the axon terminal. B, an action potential-evoked presynaptic Ca2+ transient and several eIPSC traces recorded from the same terminal. C, amplitude histogram of the presynaptic Ca2+ responses. D, the evoked IPSC (eIPSC) amplitude was weakly correlated with the amplitude of Ca2+ transients, and exhibited failures and fluctuations at all levels of presynaptic Ca2+ transients.
Figure 2. Response of a single bouton to focal stimulation in the presence of TTX.
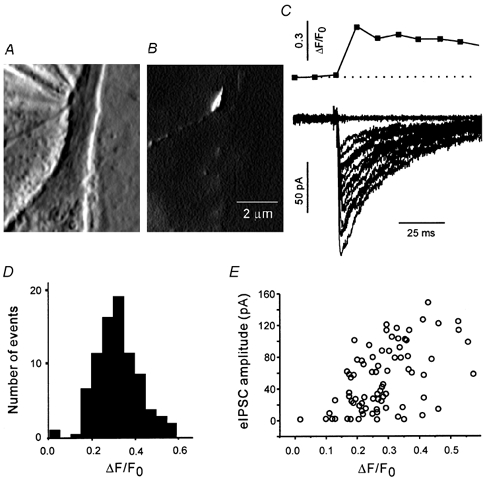
Phase contrast (A) and fluorescence (B) images of a single terminal. The phase contrast image shows a stimulation pipette in close proximity to a well-isolated bouton. The fluorescence image shows that the bouton was labelled with RH414, and no other active boutons were present in the field of view. C, a presynaptic Ca2+ transient and several eIPSCs evoked by focal stimulation of the bouton shown in A and B. D, amplitude histogram of the Ca2+ responses. E, the eIPSC amplitude was correlated with the amplitude of Ca2+ transients and fluctuated at all levels of presynaptic Ca2+ transients.
Selection of the stimulus pulse
Preliminary experiments were performed to select a depolarising focal stimulus pulse that approximated the presynaptic effects of an action potential. First, action potential-evoked responses were recorded (Fig. 1). TTX was omitted from the extracellular solution, and the culture was maintained in Ca2+-free solution (2.5 mm MgCl2, no added CaCl2). A single visualised bouton was locally superfused with extracellular solution containing 5 mm Ca2+ and 1 mm Mg2+. A glass stimulation pipette (10-15 MΩ) was filled with extracellular solution and placed on an axon 50–100 μm from the terminal (Fig. 1A). Electrical stimulation of the axon (1 ms, 1–2 μA) induced fluctuating presynaptic and postsynaptic responses. Ca2+ transients were only observed in the perfused terminal. Figure 1B represents a typical presynaptic Ca2+ transient and a set of eIPSCs from the synapse shown in Fig. 1A. The peak amplitudes of individual Ca2+ responses (ΔF/F0) were distributed normally (Fig. 1C). The eIPSC amplitude was weakly correlated with Ca2+ response amplitude (Fig. 1D). The eIPSCs exhibited transmission failures and amplitude fluctuations at all measured levels of the presynaptic Ca2+ transient. The average amplitude of action potential-mediated Ca2+ responses was 0.4 ± 0.02 (3 boutons, at least 30 trials per bouton). The eIPSC amplitudes ranged from 0 to 250 pA (3 boutons).
Next, synaptic responses were evoked by focal stimulation of a bouton in the absence of action potential generation. The Ca2+ imaging procedure bleached the indicator in the first bouton, and it could not be reused. Therefore, a fresh bouton was selected in a new visual field. The bath solution was replaced with standard extracellular solution containing TTX (Fig. 2). The stimulating pipette was placed at a distance of approximately 1 μm from an RH414-labelled terminal (Fig. 2A and B), and an isolated stimulator delivered short depolarising pulses. Figure 2C shows the response of the bouton in Fig. 2A and B to a depolarising pulse (2 ms and 1.5 μA). The Ca2+ transient amplitudes were distributed normally (Fig. 2D), and eIPSC amplitude increased as a function of ΔF/F0 (Fig. 2E). A range of stimulus pulse widths and amplitudes were explored. The amplitude of Ca2+ responses was averaged over several boutons and compared with the average amplitude of action potential-mediated responses. Pulses of 2 ms and 1.5-2 μA induced Ca2+ elevation (0.35 ± 0.04, n = 15) similar to the action potential-mediated Ca2+ increase in the absence of TTX. Therefore a 2 ms, 2 μA pulse was selected as the standard single-terminal stimulus for all subsequent experiments. Only boutons with a failure rate of < 0.2 were used. On average, the failure rate was 0.1 (range 0.02-0.18, n = 16).
Validity of the single bouton approach
To the best of our knowledge, PPD has not previously been analysed in single GABAergic boutons. It is therefore necessary to clarify some methodological aspects of this work. In the present study, the term ‘single bouton’ refers to a single axonal swelling of about 1 μm in diameter which contained a single fluorescence spot when stained with a vesicle marker (FM1-43 or RH414). A GABAergic bouton can possess several release sites, although generally not more than two (Warton et al. 1990; Sur et al. 1995). Nonetheless, it is reasonable to assume that all release sites in a terminal experience similar conditions during the depolarisation by an electrical pulse, and transmitter released from any site can access all postsynaptic receptors under that terminal (Clements et al. 1992).
The interpretation of some of our results is strongly dependent on the assumption that only one single presynaptic bouton is activated at a time. Ca2+ imaging provides a sensitive test for selective activation, because postsynaptic responses are not elicited when presynaptic Ca2+ transients are undetectable (Kirischuk et al. 1999a). Moreover, the threshold at which a Ca2+ transient can be resolved with the high-affinity Ca2+ indicator OGB-1 is lower than the threshold at which transmitter release occurs. Therefore, we can state that the eIPSCs described here always reflected the activity of a single presynaptic bouton.
Modelling
The simulation of paired eIPSC amplitude fluctuations was performed using a small custom program (available upon request) written using AxoGraph 4.6 (Axon Instruments, USA). The model assumed that at a given terminal there was a fixed number of docked vesicles, all with the same release probability. A random number generator determined the number of vesicles released at each stimulus. Vesicle depletion was simulated by making any vesicle released at the first stimulus unavailable for release at the second paired stimulus.
Statistics
All results are presented as means ± s.e.m., unless otherwise stated. The error bars in all figures indicate ± s.e.m. All comparisons between means were tested for significance using Student's unpaired t test, unless otherwise stated.
Results
Single GABAergic boutons display paired pulse depression
An RH414-labelled terminal that was well separated from neighbouring terminals (Fig. 1A and Fig. 2A and B) was selected according to the criteria described in Methods. A saline-filled glass pipette was placed approximately 1 μm from the terminal, and an isolated stimulator delivered short depolarising current pulses (2 ms, 2 μA). The focal stimulation of a single bouton produced an inhibitory postsynaptic response with a double exponential decay. The average decay time constants were 15 ± 3 and 81 ± 7 ms (n = 12). The GABAergic nature of the synaptic response was identified by its sensitivity to bicuculline methiodide (BMI, 1–10 μM; data not shown).
Paired pulse activation of single GABAergic boutons always induced depression (Fig. 3A). At short inter-stimulus intervals, the second response was superimposed on the decay of the first, so the amplitude of eIPSC2 was measured after subtracting an average eIPSC scaled to match eIPSC1. The period between one pair of stimuli and the next pair was 5 s, which was sufficient for full recovery of the first eIPSC. The dependency of the paired pulse ratio (eIPSC2/eIPSC1) on the inter-stimulus interval was determined in 26 boutons stimulated in elevated extracellular Ca2+ after loading OGB-1. The recovery from depression exhibited two kinetically distinct components, with time constants of 86 ms and 2.0 s (Fig. 3B). These two components will be referred to as PPDfast and PPDslow. At the shortest interval tested (20 ms) the combined depression of the eIPSC amplitude was 73 %. The mechanism(s) responsible for PPDslow reduced the amplitude by 50 %, while PPDfast contributed 23 % to the depression.
Figure 3. Paired pulse depression of eIPSCs at a single inhibitory terminal in elevated (5 mm) extracellular Ca2+.
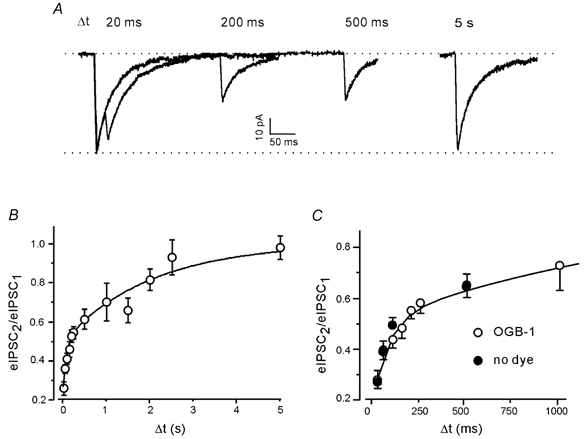
A, eIPSCs at several different inter-stimulus intervals (Δt). Each trace is the average of 25 responses. B, PPD as a function of inter-stimulus interval. Pooled data from 26 boutons after loading OGB-1. The continuous line represents a double exponential fit. C, PPD of eIPSCs was not changed by the presence of Ca2+-sensitive dye.
It is well known that the paired pulse behaviour of a terminal is influenced by the presynaptic Ca2+ buffers (see for instance Ohana & Sakmann, 1998; Rozov et al. 2001; Sakaba & Neher, 2001). In the present study, we used OGB-1, a high-affinity Ca2+ indicator (KD ≈200 nm), to monitor the presynaptic Ca2+ levels in the stimulated bouton. The use of a membrane-permeant form of a Ca2+ indicator precludes the precise estimation of its concentration in the cell. However, its approximate value was obtained by comparing the bouton fluorescence with the fluorescence produced on glass by droplets of artificial intracellular solution containing OGB-1 at various concentrations. We estimate that the OGB-1 concentration in the terminal was 30 μM or less.
To test whether PPD was affected by the presence of this Ca2+ probe, six boutons were tested in the absence of OGB-1. Figure 3C illustrates that the dye had no significant effect on the magnitude or the time course of PPD. For example, at an inter-stimulus interval of 20 ms the PPD ratio was 0.24 ± 0.01 in the absence of OGB-1, and 0.26 ± 0.03 in its presence (P > 0.8, Student's t test). The failure rate was also not affected. It was 0.08 ± 0.04 and 0.09 ± 0.03 in the absence and presence of the indicator, respectively. We therefore assumed that the Ca2+ buffering strength of OGB-1 was low in comparison with the strength of endogenous Ca2+ buffers in these terminals. All the following experiments were carried out in the presence of OGB-1.
GABAB receptors do not contribute to PPD
At some central inhibitory synapses, PPD is mediated by presynaptic GABAB autoreceptors. Activation of GABAB receptors suppresses voltage-activated Ca2+ currents (Pfrieger et al. 1994) and enhances presynaptic voltage-gated K+ channel activity (Scholz & Miller, 1991) via a G protein-mediated mechanism. To test whether GABAB receptors contribute to PPD in this preparation, we applied a competitive antagonist, CGP55845A (CGP) (Davies et al. 1993). CGP (5 μM) had no effect on PPD. For example, at an inter-stimulus interval of 100 ms, the PPD ratio was 0.41 ± 0.03 in control (n = 17) vs. 0.44 ± 0.05 in CGP (n = 8). The recovery from depression in the presence of CGP was fitted by a double exponential function with time constants of 92 ms and 1.9 s, which were similar to control values (86 ms and 2 s). Thus, activation of GABAB receptors does not contribute to PPD at GABAergic synapses in these collicular cultures.
A release-independent presynaptic mechanism accounts for PPDslow
To examine the possible role of residual Ca2+ in paired pulse responses, EGTA-AM was loaded into collicular cultures. EGTA binds Ca2+ with relatively slow kinetics. Therefore, moderate concentrations of this chelator should not interfere with the brief, localised, high-concentration Ca2+ transients necessary for transmitter release (Adler et al. 1991; Llinás et al. 1992), but will speed up the clearance of residual Ca2+(Atluri & Regehr, 1996). Indeed, incubation in 5 μM EGTA-AM for 20 min accelerated the decay of the presynaptic Ca2+ transient more than 10-fold, from 427 ± 30 ms (n = 16) in control to 35 ± 3 ms (n = 5) in EGTA, without reducing the peak amplitude (Fig. 4A and B). Since a membrane-permeant form of EGTA was used in these experiments, the concentration of the chelator in the presynaptic terminals was unknown. However, EGTA increased the failure rate of eIPSCs from 0.08 ± 0.02 (n = 16) in control to 0.11 ± 0.02 (n = 14), which suggests that EGTA-AM-loading produced a higher presynaptic buffer concentration than loading of OGB-1-AM. This result probably reflects the faster loading rate of EGTA-AM. To minimise the effect of the EGTA-induced increase in the failure rate of the first eIPSC, we limited the following analysis to boutons with failure rates of less than 0.1 (11 out of 14). This subpopulation had a mean failure rate similar to that of untreated terminals. The mean amplitudes of the first eIPSCs (excluding failures) were also similar, i.e. 131 ± 84 pA (mean ± s.d., n = 16) in control and 125 ± 91 pA (mean ± s.d., n = 11) in EGTA. The experiments showed that PPDslow was unaffected by the accelerated clearance of the residual presynaptic Ca2+ (Fig. 4C).
Figure 4. Presynaptic residual Ca2+ does not determine PPDslow.
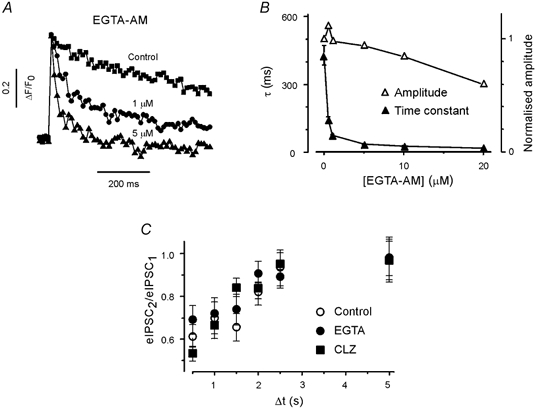
A, typical presynaptic Ca2+ transients under control conditions and after pre-incubation with 1 and 5 μM EGTA-AM in addition to OGB-1. Extracellular Ca2+ concentration: 5 mm. B, effect of EGTA-AM on the peak amplitude (▵) and decay time constants (τ, ▴) of presynaptic Ca2+ transients. C, paired pulse ratios of eIPSCs at inter-stimulus intervals > 500 ms were not affected by EGTA and CLZ.
It is unlikely that postsynaptic receptor desensitisation plays a role in PPDslow, because experiments in hippocampal neurones have shown that GABAA receptors recover from desensitisation within a few hundred milliseconds (Jones & Westbrook, 1995). Nevertheless, we examined the significance of postsynaptic receptor activity by applying the benzodiazepine clonazepam (CLZ). CLZ enhances the affinity of the GABAA receptor and prolongs GABAA receptor occupancy (for review see Costa, 1998). In the presence of CLZ, the eIPSC decay was fitted with a double exponential (τ = 15 and 160 ms). The second time constant was significantly larger in CLZ (160 ± 15 ms, n = 6) than in controls (81 ± 7 ms, n = 12, P < 0.01, data not shown), but the paired pulse ratio at long inter-stimulus intervals (≥ 1 s) was not changed by the presence of CLZ (at 1 s, 0.67 ± 0.04, n = 8 vs. 0.7 ± 0.09, n = 16, in control). Thus, unavailability of postsynaptic GABAA receptors is not the basis of PPDslow (Fig. 4C).
This leaves two potential PPD mechanisms with slow recovery kinetics: vesicle depletion and release-independent modification of vesicle release probability. If PPDslow was caused by depletion of the releasable pool, then the PPD ratio should change with the failure rate. We therefore examined the paired pulse responses at 1 s intervals in three different extracellular Ca2+ concentrations (Fig. 5A). The failure rates and the mean eIPSC amplitudes were, respectively, 0.08 ± 0.02 and 131 ± 24 pA in elevated Ca2+ (5 mm, n = 16), 0.29 ± 0.05 and 57 ± 13 pA in normal Ca2+ (2 mm, n = 9), and 0.53 ± 0.03 and 32 ± 8 pA in low Ca2+ (1 mm, n = 5). A correlation of the paired pulse ratio with the failure rate was not observed (Fig. 5B).
Figure 5. PPDslow is not dependent on the release probability.
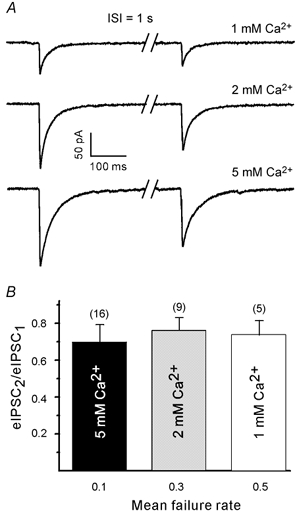
A, eIPSC pairs at an interval of 1 s in various extracellular Ca2+ concentrations. Each trace is the average of 20 responses. Traces were obtained from different cells. B, paired pulse ratio at 1 s was not dependent on the response probability/extracellular Ca2+ concentration. Numbers in parentheses indicate the number of boutons tested. ISI, inter-stimulus interval.
Next, we investigated the relationship between the amplitudes of the first and second eIPSC in a pair. If vesicle depletion plays a role in PPD, there will be fewer vesicles remaining for immediate release by the second stimulus if the first eIPSC in a pair is larger than average. Therefore, the second eIPSC will be smaller than average. We modelled eIPSC amplitude fluctuations in a paired pulse paradigm to determine whether the predicted correlation could be detected reliably with only a small sample of eIPSC amplitude measurements. The simulation parameters were set as follows: 40 paired eIPSCs, eight vesicles in the readily releasable pool before the first stimulus of each pair, release probability was 0.4, the probability that a released vesicle re-docked between the paired stimuli was 0.1, each vesicle produced an average quantal response of 30 pA, and the quantal amplitude exhibited intrinsic variability with a coefficient of variation (CV) of 0.3. The model predicts PPD of about 50 %, which is close to the average level of PPDslow at an inter-stimulus interval of 1 s. Ten data sets were simulated. Despite the small sample size and the large scatter in the amplitudes, the slope of the regression line was negative in all 10 cases. The correlation was statistically significant in five of the 10 cases. So, if vesicle depletion is responsible for PPDslow, then negative correlation is expected in the majority of data sets, and it should be statistically significant in at least some recordings.
Nine boutons were tested for negative correlation between the first and second eIPSC at an inter-stimulus interval of 1 s. The slope of the regression line was negative in only 3/9 boutons, and there was no significant correlation in any of the data sets. To increase the sensitivity of the search for a correlation, data from all cells were pooled. For each cell, the first and second eIPSC amplitudes were divided by the mean amplitude of the first eIPSC. The normalised data were pooled and a regression line was fitted (Fig. 6A). No correlation was found between the paired eIPSCs.
Figure 6. PPDslow is independent of previous release.
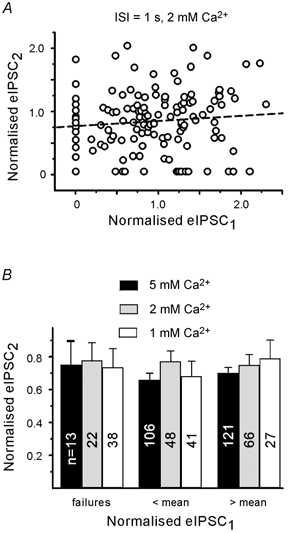
A, plot of second against first eIPSC for individual pairs at 1 s inter-stimulus interval. Amplitudes were normalised to the mean amplitude of the first eIPSC in the entire data set (9 boutons). Dashed line represents linear regression. There was no significant correlation between eIPSC peak amplitudes (P > 0.4). B, binning of second eIPSCs according to the amplitude of first eIPSC in each pair. Records in three different extracellular Ca2+ concentrations. Depression was observed even after a failure of the first response. Note that the amplitude of the second eIPSC is not related to the amplitude of the first eIPSC. Numbers on columns indicate eIPSC number. Pooled data from different boutons.
We then divided the normalised first eIPSC into three groups (failures, responses smaller than average and responses larger than average) and compared the second eIPSCs at different extracellular Ca2+ concentrations (Fig. 6B). The amount of depression at 1 s was again unrelated to the amplitude of the first eIPSC. Even after failure of the first response there was a reduction of the second eIPSC. The average paired pulse ratio was 0.7 ± 0.09 (n = 16), 0.76 ± 0.04 (n = 9) and 0.74 ± 0.05 (n = 5) for elevated, normal and low extracellular Ca2+, respectively.
Having eliminated both a postsynaptic mechanism and a release-dependent presynaptic mechanism, the most plausible cause of PPDslow is a release-independent down-regulation of vesicle release probability.
A release-independent, but Ca2+-dependent presynaptic mechanism may underlie PPDfast
In contrast to PPDslow, PPDfast displayed a strong dependency on the extracellular Ca2+ concentration. At an inter-stimulus interval of 20 ms, PPDfast depressed the second eIPSC by 23 % in elevated extracellular Ca2+ (5 mm, n = 26), but only by 13 % in normal extracellular Ca2+ (2 mm, n = 9). The duration of PPDfast was also less in 2 mm Ca2+. In low extracellular Ca2+ (1 mm, n = 5) PPDfast was almost undetectable (Fig. 7A). We then tested for a dependence of the paired pulse ratio on the failure rate of the first eIPSC (Fig. 7B). The depression at an inter-stimulus interval of 20 ms increased significantly when the failure rate decreased. Figure 7C shows that at 50 ms the depression of the second eIPSC in normal and elevated Ca2+ was significantly lower after a failure, even if compared with a weaker than average first eIPSC. This contrasts with PPD at 1 s, which was independent of the failure rate
Figure 7. PPDfast depends on previous release and is stronger under high release probability conditions.
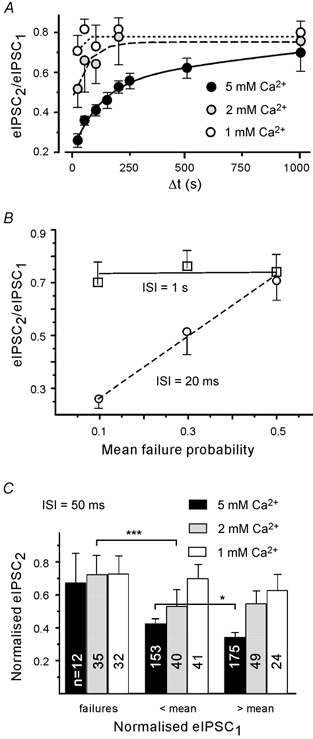
A, dependency of PPDfast on extracellular Ca2+ concentration. Lines represent single exponential fits. B, differential dependency of PPDfast and PPDslow on the failure rate. C, dependence of PPD on the amplitude of the first eIPSC. Binning of second eIPSCs according to the amplitude of first eIPSC in each pair. Records shown for three different extracellular Ca2+ concentrations. In this and subsequent figures, the asterisks refer to significance levels. * P < 0.05, *** P < 0.001. Numbers on columns indicate eIPSC number. Pooled data from different boutons.
Next we examined the relationship between PPDfast and the amplitude of the first eIPSC (Fig. 7C). As in Fig. 6B, normalised second eIPSCs were binned in three groups based on the amplitude of the first eIPSC, which was categorised either as a failure, a response smaller than average or a response larger than average. In contrast to the results at 1 s, the paired pulse ratio in elevated extracellular Ca2+ was sensitive to the amplitude of the first eIPSC. The PPD ratio was significantly different after smaller first eIPSCs (0.45 ± 0.02) vs. larger first eIPSCs (0.37 ± 0.02, P < 0.05).
It is noteworthy that paired pulse facilitation (PPF) was never detected. Even when the mean amplitude of first eIPSCs (excluding failures) approached the mean amplitude of miniature IPSCs (mIPSCs) (in 1 mm extracellular Ca2+), a larger than control second response was not observed. However, we cannot exclude that a superimposed synaptic facilitation influences the paired pulse response at intervals shorter than 20 ms.
To further investigate the dependency of PPDfast on previous Ca2+ influx, we applied the following protocol (Fig. 8A). At an inter-stimulus interval of 100 ms, boutons were alternatively stimulated by two different stimulus pairs. One pair consisted of a weak pulse (1 μA) followed by a standard pulse. The other pair consisted of two standard pulses (2 μA). The weaker pulse induced a smaller presynaptic Ca2+ transient and a smaller first eIPSC than the standard pulse (Kirischuk et al. 1999a). The eIPSC1 amplitude evoked by the weak pulse was 0.72 ± 0.05 (n = 5) of the amplitude after the standard pulse. The eIPSC1 failure rate was 0.21 ± 0.03 after a weak pulse vs. 0.08 ± 0.03 (n = 5) after a standard pulse.
Figure 8. Presynaptic residual Ca2+ contributes to PPDfast.
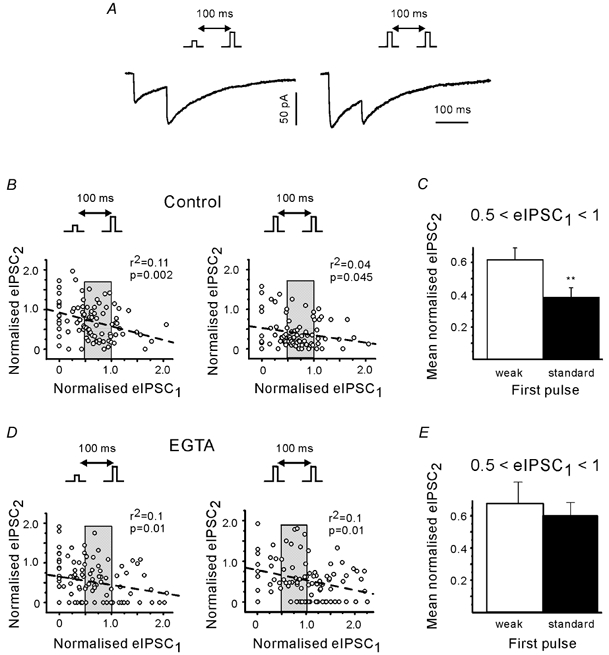
A, eIPSCs induced by a pair of standard stimuli (right) or by a standard pulse preceded by a weak pulse (left). Insets illustrate the stimulation protocols. Each trace represents the average of 20 responses. Both traces from the same cell. Recordings in 5 mm Ca2+. B, plot of the second eIPSC amplitude against the amplitude of the first eIPSC in individual pairs. Amplitudes were normalised to the mean amplitude of the first eIPSC (5 boutons). Normalisation is based on responses to the standard pulses. Pooled data. Dashed lines represent the results of linear regression. C, only data from trials with a narrow range of first eIPSCs. Amplitudes of the first eIPSCs were between 0.5 and 1 (shaded range in B). ** P < 0.01. D and E, same as B and C, but in the presence of EGTA. Note stronger PPD at short interval after stronger first pulse in controls, but not in EGTA.
The plots of Fig. 8B suggest that the negative correlation between second and first eIPSCs at intervals of 100 ms was more pronounced in trials with a weak first pulse. However, taking advantage of the wide range of eIPSC fluctuations we were able to select cases where the eIPSC1 amplitudes induced by the weak and standard pulses were similar both in their mean and standard deviation amplitude (shaded range in Fig. 8B and D). If a release-independent but Ca2+-dependent mechanism contributes to PPDfast, the PPD ratio should vary with the strength of the first pulses (because they cause different Ca2+ influx) even though the amplitudes of the first eIPSCs (the amount of released transmitter) were similar. In all five boutons tested in this way, the mean amplitude of the second eIPSCs was indeed larger after the weaker first pulse. The difference was statistically significant in 4/5 boutons (P < 0.05). When the data sets from the five boutons were pooled the difference in the depression after the weak and standard pulse was different at P < 0.01 (Fig. 8C). The second eIPSCs were depressed to 0.61 ± 0.07 (n = 38) and 0.38 ± 0.06 (n = 37) after the weak and standard pulse, respectively. Similar results were obtained for a larger bin width (normalised first eIPSCs between 0.5 and 1.5). To further clarify the origin of PPDfast, we repeated the same experimental protocol after loading of EGTA-AM (Fig. 8D). Figure 8E shows that the presence of EGTA removed the difference in the depression of the second eIPSCs after the weak and standard pulse (pooled data from 5 boutons).
One explanation for these results is that the residual Ca2+ after the first stimulus depresses the Ca2+ influx produced by the second stimulus. We therefore examined the Ca2+ transients obtained in individual paired pulse trials (Fig. 9A). Experiments in 10 boutons showed that at short inter-stimulus intervals the second Ca2+ responses were indeed depressed (ΔF2/ΔF1 = 0.44 ± 0.06, at an interval of 20 ms). The Ca2+ responses recovered from PPD with a time constant of 180 ms (Fig. 9B). However, the observed depression of the second presynaptic Ca2+ transient could reflect the saturation of the high-affinity Ca2+-sensitive dye. If this were the case, we should expect a negative correlation between the amplitudes of the first and second Ca2+ response, but there was no correlation (results not shown). To provide further evidence that the depression of the second Ca2+ transient did not result from OGB-1 saturation, additional experiments (6 boutons) were performed in cultures loaded with the low-affinity Ca2+ indicator MG. Again, the amplitudes of the second Ca2+ transients were smaller (ΔF2/ΔF1 = 0.52 ± 0.12, at an interval of 20 ms), and recovered with a time constant of 120 ms (Fig. 9B). Thus, the paired pulse depression of the presynaptic Ca2+ transient was not due to saturation of OGB-1.
Figure 9. Residual presynaptic Ca2+ inhibits the Ca2+ influx.
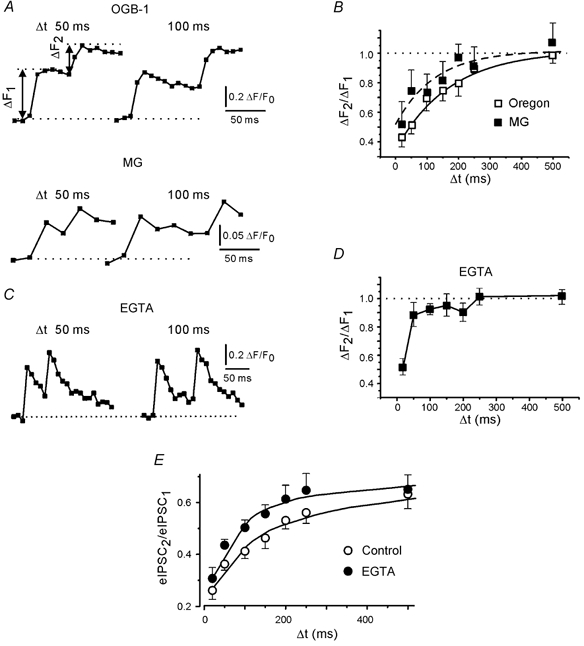
A, examples of presynaptic Ca2+ transients in response to standard stimulus pairs at intervals of 50 and 100 ms. Records based on loading of either OGB-1 or MG. MG trace: average of five responses. B, depression of presynaptic Ca2+ transients as a function of stimulus interval. Points are averages of 100 (OGB-1) and 60 (MG) trials. Ten (OGB-1) and 6 (MG) boutons were tested. The curves represent exponential fits. C, examples of paired presynaptic Ca2+ transients recorded with OGB-1 in the presence of EGTA. D, depression of presynaptic Ca2+ transients as a function of stimulus interval in the presence of EGTA (results from 7 boutons). E, PPDfast in the presence and absence of EGTA. Continuous lines represent exponential fits.
EGTA accelerates the clearance of residual Ca2+, so at short intervals it should reduce the depression of the second Ca2+ response. This was the case (Fig. 9C and D). We also examined the effect of EGTA on the paired pulse behaviour of postsynaptic currents and found that EGTA reduced PPDfast (Fig. 9E). At an inter-stimulus interval of 50 ms, the PPD ratio was 0.44 ± 0.05 in EGTA (n = 7), but 0.36 ± 0.02 in control (n = 16; difference significant at P < 0.01). The magnitude and the time constant of PPDfast decreased from 23 % and 86 ms, respectively, in the controls to 15 % and 61 ms in EGTA. We therefore conclude that in this preparation the decrease of the presynaptic Ca2+ influx by residual Ca2+ represents an additional release-independent mechanism that may cause IPSC depression at short intervals.
A release-dependent postsynaptic mechanism may also contribute to PPDfast
The above results do not rule out the possibility that a component of the depression at short intervals increases with the amount of the released transmitter. For example, the inverse relationship between the first and the second eIPSCs at an interval of 100 ms after a weak first pulse (left graph in Fig. 8B) could be due to a transient unavailability of postsynaptic transmitter receptors, especially in trials with multiquantal release. It is known that a brief, high concentration pulse of GABA can cause both saturation and desensitisation of GABAA receptors, making them unresponsive to a second pulse of transmitter delivered a few milliseconds later (Jones & Westbrook, 1995). As GABA diffuses away, the number of unbound receptors increases but, due to receptor desensitisation, full recovery of responsiveness takes > 100 ms. This mechanism has the correct time course to contribute to PPDfast.
If PPDfast results, in part, from decreased receptor availability, then CLZ should prolong and augment PPDfast. This was the case. At a 50 ms inter-stimulus interval, CLZ reduced the PPD ratio to 0.26 ± 0.04 (n = 10), compared with a control value of 0.36 ± 0.02 (n = 17, P < 0.05, not shown). To further characterise the postsynaptic component of PPDfast, paired pulse experiments were carried out in the presence of competitive GABA antagonists. The rationale for using GABAA receptor antagonists of low affinity with fast kinetics and of high affinity with slow kinetics is similar to that of previous experiments with AMPA or NMDA receptor blockers (Liu et al. 1999; Chen et al. 2002). Briefly, in the extreme case of complete saturation, all GABAA receptors would bind GABA on the first pulse and no receptors would be available for a second pulse at a sufficiently short interval. This would result in a failure to evoke a second eIPSC. When an antagonist blocks a subset of GABAA receptors, GABA activates the available receptors eliciting a first eIPSC that is smaller than the control. However, if the dissociation rate of the antagonist is shorter than or comparable with the time of GABA being present in the synaptic cleft (Clements et al. 1992), some receptors become available for a second GABA pulse. As these receptors were protected by the antagonist they did not enter a desensitised state and can now generate a second eIPSC. Therefore, in the presence of the antagonist the paired pulse ratio will increase. In contrast, if a high-affinity antagonist with a slow off-rate blocks the same fraction of GABAA receptors, the antagonist will remain bound after the first stimulus, leaving no receptors available for the second stimulus. The paired pulse ratio will remain nearly unchanged by the presence of the antagonist. The same reasoning applies when the receptor saturation is not complete and predicts that if GABAA receptor saturation is playing a role, a high-affinity/slow off-rate antagonist will have little effect on PPDfast, while a low-affinity antagonist/fast off-rate antagonist should decrease PPDfast.
To perform this test we used 1,2,5,6-tetrahydropyridin-4-yl-methylphosphinic acid (TPMPA; 300 μM) and bicuculline methiodide (BMI; 1 μM). TPMPA is a low-affinity, rapidly equilibrating antagonist at the GABAA receptor (koff ≈0.6 ms), while BMI is a high-affinity, slowly equilibrating GABA antagonist (koff ≈20 ms) (Jones et al. 2001). The concentrations of TPMPA and BMI were chosen to be close to their reported IC50 values for GABA-induced Cl− currents (Ragozzino et al. 1996; Jonas et al. 1998). The results from six experiments with TPMPA and four experiments with BMI were in agreement with these predictions. In the case of TPMPA (Fig. 10A and B), but not BMI (Fig. 10B), the second eIPSC was less inhibited than the first eIPSC. Consequently, the PPD ratio was increased by TPMPA, but not by BMI. These results support the idea that a component of PPDfast may have a postsynaptic origin.
Figure 10. Postsynaptic contribution to PPDfast.
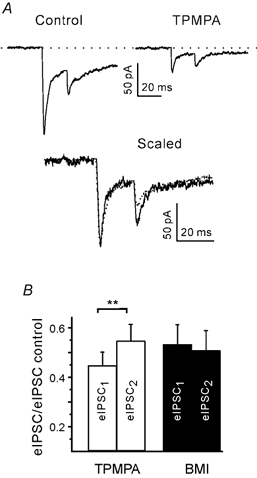
A, eIPSCs at an interval of 20 ms in the presence of 300 μM TPMPA. Top: average traces from 20 trials. Bottom: superposition of average traces after normalisation to the peak of the first eIPSC. Both traces from same bouton. B, summary of results for TPMPA and similar experiments with 1 μM BMI. At an interval of 20 ms TPMPA, but not BMI, blocked the second eIPSC less than the first eIPSC. ** P < 0.01.
Short-term synaptic depression due to postsynaptic receptor saturation or desensitisation may influence the summation of so-called asynchronous IPSCs (aIPSCs), which under some conditions follow the stimulus-locked eIPSC (Goda & Stevens, 1994). We examined how the amplitudes of aIPSCs depend on the time of their occurrence after the stimulus inducing the eIPSC (Fig. 11A). The low rate of occurrence of aIPSCs limited the analysis to few boutons. A reasonable number of aIPSCs (> 20) were obtained in five boutons tested in elevated (5 mm) Ca2+. Individual aIPSCs were normalised to the mean amplitude of mIPSCs and pooled for the graphs of Fig. 11B and C. Larger than average eIPSCs were selected for this test. We found that aIPSCs occurring within less than 100 ms after the stimulus were significantly suppressed. Their normalised amplitude was 0.63 ± 0.04 (51 events), whereas the amplitude of aIPSCs occurring between 100 and 300 ms after the pulse was 0.99 ± 0.03 (86 events). The time constant of recovery from aIPSC suppression was 50 ms (Fig. 11C). Although mIPSC frequencies were low (0.5 ± 0.3 Hz, n = 16) in the low-density cultures, we have to consider a possible contribution of mIPSCs stemming from other contacts. However, provided that mIPSCs occur randomly, their summation with the eIPSCs should not vary with the time interval. It is also possible that the time-dependent depression of aIPSCs is due to a local depolarisation or shunt in the dendrite. However, neither the data sets from individual boutons nor the pooled data (Fig. 11D) showed a negative correlation between eIPSC and aIPSC amplitudes, which argues against local depolarisation or shunt as a cause of aIPSC reduction at short intervals. Note that in all these experiments eIPSCs were substantially larger than aIPSCs, which means that eIPSCs were, most probably, induced by multiquantal release. Thus, the results from aIPSC recordings are in line with the results from the earlier experiments which suggested that, under conditions of high release efficacy, PPDfast may also have a postsynaptic component.
Figure 11. Functional limits of single bouton transmission.
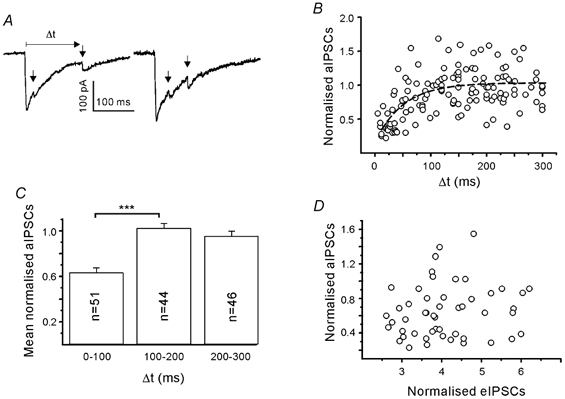
A, time-dependent superposition of asynchronous IPSCs (aIPSCs; arrow) and eIPSCs. Experiment in 5 mm Ca2+. B, aIPSCs were normalised to the mean miniature IPSC (mIPSC) amplitude and plotted against the interval after the pulse. Dashed line represents exponential fit. C, dependence of aIPSC depression on the interval after the stimulus used to induce eIPSCs. Same data as in B. One hundred millisecond binning of intervals after the pulse. There was suppression of aIPSCs at short intervals (*** P < 0.001). D, scatter diagram to show that aIPSCs recorded within 100 ms after the stimulus were unrelated to the amplitude of eIPSCs. Pooled data from 5 boutons. All IPSC amplitudes were normalised to the mean amplitude of mIPSCs.
Discussion
In this study, focal stimulation of single visualised GABAergic boutons in culture provided detailed insight into the mechanisms of short-term synaptic plasticity. The experiments demonstrated two kinetically distinct components in the PPD of eIPSCs. The relative contribution of the fast and slow components can vary with the strength of Ca2+ influx or presynaptic Ca2+ buffering.
Concerning the origin of PPDslow, our experiments are in agreement with previous results at another inhibitory connection which suggested that PPDslow reflects a presynaptic depression and is mainly caused by a release-independent down-regulation of exocytosis (Kraushaar & Jonas, 2000). Whatever the underlying process, it is insensitive to changes in extracellular Ca2+ concentration and residual presynaptic Ca2+.
PPDfast depends much more on the activation conditions than PPDslow. Our data suggest that PPDfast is caused by two processes with similar decay time constants: a presynaptic release-independent depression which probably reflects an inhibition of Ca2+ influx by residual Ca2+, and a postsynaptic release-dependent depression which is likely to result from the unavailability of postsynaptic receptors.
While inhibitory synapses in the neonatal collicular slice display strong PPD, the prevailing response in the more mature superior colliculus is PPF (Jüttner et al. 2001). It is therefore not excluded that in the present preparation a process of synaptic facilitation exists, but is either masked by the superimposed depression or is undetectable due to its very rapid decay (Gupta et al. 2000). In any case, in collicular cultures a net PPF was not observed even under conditions of low extracellular Ca2+ concentration. We therefore focus the following discussion on the possible mechanisms of synaptic depression.
Presynaptic release-independent mechanisms of PPD
The mechanism(s) underlying PPDslow
Experiments on specialised synapses with a large number of active zones have shown that depletion of a vesicle pool can cause short-term synaptic depression (Liley & North, 1954). Later this result was extended to CNS connections with multiple (Mennerick & Zorumski, 1995; Debanne et al. 1996; Jensen et al. 1999) or single (Stevens & Tsujimoto, 1995) small terminals. However, a constant vesicle release or recovery rate could not adequately describe the experimentally determined time course of release. Wu & Borst (1999) therefore suggested that the average probability of vesicle release is not constant but decreases with consecutive stimuli. Recent detailed studies in the calyx of Held introduced a further modification of the vesicle depletion hypothesis which takes into account that the probability of vesicle release and recovery is not the same from vesicle to vesicle (see Schneggenburger et al. 2002, for a review). It is still unclear whether this modified vesicle depletion hypothesis can be extended to GABAergic synapses, but it seems quite possible that vesicles are also heterogeneous in small GABAergic terminals. After complete depletion of the fast releasing vesicle pool by a very strong first pulse in high Ca2+ the second response may be generated by a different type of release process, because the IPSCs following the second pulse are very variable in latency and amplitude, reminiscent of asynchronous IPSCs (N. Veselovsky, S. Kirischuk & R. Grantyn, unpublished result).
The remaining possibility is that one or several components of the release machinery are modified by the near-membrane, high Ca2+ microdomain (Chapman et al. 1995) or a direct effect of membrane voltage (Parnas et al. 2000). The suggestion that a ubiquitous presynaptic protein is involved in PPDslow seems convincing because release-independent PPDslow has been observed at many different synapses, including the neuromuscular junction (Betz, 1970), the squid giant synapse (Hsu et al. 1996), a goldfish axo-axonic connection (Waldeck et al. 2000), cerebellar and cortical glutamatergic contacts (Dittman & Regehr, 1998; Thomson & Bannister, 1999), the calyx of Held (Bellingham & Walmsley, 1999; Wu & Borst, 1999), and the hippocampal inhibitory interneurone to principal neurone synapse (Kraushaar & Jonas, 2000). However, more experiments are necessary to fully understand this important mechanism.
The mechanism(s) underlying PPDfast
In contrast to PPDslow, PPDfast was dependent on extracellular Ca2+ concentration, and it was reduced by loading EGTA-AM. There are three candidate presynaptic mechanisms which could satisfy both the time course and the residual Ca2+ sensitivity of PPDfast: (a) Ca2+-dependent replenishment of a vesicle pool, (b) desensitisation of a Ca2+-sensor, and (c) decreased Ca2+ influx.
The time constant of PPDfast roughly coincided with the time course of Ca2+-dependent replenishment of a releasable vesicle pool at glutamatergic synapses (Dittman & Regehr, 1998; Wang & Kaczmarek, 1998; Sakaba & Neher, 2001). However, the effect produced by introducing the slow Ca2+ buffer EGTA was the opposite of what would be expected if this mechanism governs PPD in our preparation (reduction instead of increase in PPD).
Our results are consistent with the hypothesis that the presynaptic component of PPDfast is partially due to a Ca2+-induced and/or voltage-driven modification of a component of the exocytotic molecular apparatus. This possibility has been repeatedly discussed since Chapman et al. (1995) showed that Ca2+ increases the affinity between synaptotagmin and syntaxin 1. In addition, an adaptation of the Ca2+ sensor was demonstrated by photolysis of caged Ca2+ (Hsu et al. 1996).
Our experiments provide evidence for a use-dependent decrease in Ca2+ influx. There was a marked paired pulse depression of the presynaptic Ca2+ transient which was reduced by loading EGTA. We also took advantage of the fact that the presynaptic Ca2+ transients displayed fluctuations, such that in individual trials equally sized eIPSCs could be produced by different presynaptic Ca2+ transients (Kirischuk et al. 1999a). This allowed us to compare the second eIPSC amplitudes after weaker and stronger Ca2+ influx. This experiment showed that PPDfast was enhanced after larger presynaptic Ca2+ influx. Accordingly, PPDfast was reduced by loading EGTA. These results underline the significance of the time course of residual Ca2+. The faster decline of the presynaptic Ca2+ transient after introducing an exogenous buffer reduced but did not abolish PPDfast.
The Ca2+ influx through voltage-activated Ca2+ channels can be reduced by a variety of mechanisms, including reduction or shortening of presynaptic action potentials (Brody & Yue, 2000; Geiger & Jonas, 2000; He et al. 2002), depletion of Ca2+ in the extracellular space (Borst & Sakmann, 1999; King et al. 2001), increased Ca2+-activated K+ and/or Cl− conductances (Marrion & Tavalin, 1998; Oliver et al. 2000), or Ca2+-dependent inactivation of voltage-gated Ca2+ channels (Patil et al. 1998). Variable action potential shape was not an issue in our experiments, because the boutons were activated by a standard electrical pulse in the presence of TTX. At well-separated small boutons in culture, Ca2+ depletion in the extracellular space is not a candidate for PPD on a time scale of 100 ms, because the replenishment of [Ca2+]o via diffusion is faster than 10 ms (Borst & Sakmann, 1999). At present, we favour the idea that the presynaptic component of PPDfast is largely due to a Ca2+-dependent inactivation of voltage-gated Ca2+ channels (Forsythe et al. 1998). The experiments showed that the second Ca2+ transient was significantly reduced. By applying Ca2+ imaging, we found that collicular inhibitory terminals possess a mixture of N-, P/Q- and R-type Ca2+ channels, with a predominance of N-type channels in culture (S. Kirischuk & R. Grantyn, unpublished observation). Both N- and P-type channels were reported to display considerable inactivation when activated repetitively by brief, action potential-like depolarisations (Patil et al. 1998).
The postsynaptic release-dependent mechanism of PPDfast
The EGTA-resistant negative correlation between the second and the first eIPSCs at short intervals (Fig. 8D) points to the existence of an additional component of PPDfast. We tentatively attributed this negative correlation to a postsynaptic mechanism. This was then confirmed by the experiments with CLZ and TPMPA and by the test for aIPSC summation. The postsynaptic component of PPDfast comes into play when release is strong. Multivesicular release and a high degree of postsynaptic receptor occupancy were previously reported for excitatory (Trussell et al. 1993; Oleskevich et al. 2000) and inhibitory synapses (Auger et al. 1998).
In the present study, a high release efficacy could deliberately be established by selecting stimulus parameters that evoked larger presynaptic Ca2+ transients (Kirischuk et al. 1999a). In elevated extracellular Ca2+/Mg2+, a majority of terminals generated maximal eIPSCs that were three to seven times larger than mIPSCs. In the experiments with delayed release, individual eIPSCs were almost always larger than aIPSCs. We therefore concluded that in 5 mm Ca2+ low-frequency activation typically induced multivesicular release. As a consequence, GABA will bind to a significant fraction of the available synaptic GABAA receptors and desensitise them (Jones & Westbrook, 1995) thereby reducing the number of receptors that are available for another release event, either stimulus-locked or asynchronous.
A distinct slow component in the IPSC decay could reflect the transition of the GABAA receptor-channel complex from a desensitised to an open state (Jones & Westbrook, 1995). The likelihood of observing a postsynaptic component of PPD will strongly depend on the properties of postsynaptic receptors. However, the subunit composition and the kinetic properties of synaptic GABAA receptors display a high degree of variability (Rudolph et al. 2001). This could explain why, despite a high probability of release, a postsynaptic locus of PPD was not observed in a hippocampal interneurone-principal cell synapse (Kraushaar & Jonas, 2000). The presence of IPSCs with slow decay, as in the present collicular preparation (with a total duration of up to 80 ms), makes it, of course, easier to detect a postsynaptic component of PPDfast.
In collicular slices, prolonged IPSC decay and robust PPD at physiological extracellular Ca2+/Mg2+ concentrations is a characteristic feature of immature inhibitory synapses (Jüttner et al. 2001). Moreover, at an early postnatal stage, individual neurones display a high variability in the duration of unitary IPSCs. Interestingly, the amount of PPD at short intervals (10-25 ms) is then negatively correlated with the duration of eIPSCs. These observations suggested that in the immature brain PPD may not represent, as often assumed (O'Donovan & Rinzel, 1997), a purely presynaptic phenomenon. When inhibitory terminals release GABA in a multiquantal fashion, short-term depression of aIPSCs can be caused by slow recovery of postsynaptic GABAA receptors, at least under low-frequency activation conditions.
It should be stressed that at another developmental stage and synapse type, and under different activation conditions, the relative contribution and functional importance of the depression mechanisms delineated here might be different. For example, a decrease in release probability should reduce the postsynaptic contribution to PPD. However, a short train of stimuli would elevate the presynaptic Ca2+ concentration to levels higher than were seen in this study, and would promote postsynaptic receptor saturation. Thus, our conclusions are not necessarily in conflict with previous studies reporting different results.
Possible physiological role of PPDfast in developing synapses
What could be the function of PPD in an immature synapse? Kraushaar & Jonas (2000) suggested that PPDslow enables tonic inhibition by conserving vesicles at the beginning of a long action potential train and releasing them later on. The release-independent, Ca2+-related presynaptic component of PPDfast may have a similar function: to conserve the readily releasable vesicle pool by preventing exocytosis at a time when the postsynaptic receptors are not available for signal transmission. At the immature inhibitory synapse a prolonged IPSC decay was matched by an equally slow decay of the residual presynaptic Ca2+, and this caused depression of subsequent presynaptic Ca2+ transients and IPSCs. It is tempting to speculate that during development of this system the increasing expression of GABAA receptors with faster deactivation kinetics will be paralleled by an increasing efficacy of presynaptic Ca2+ clearance. This would result in a reduction of PPDfast and may allow for a net facilitation of IPSCs at intervals < 100 ms, as seen in inhibitory synapses of the mature colliculus.
Acknowledgments
We would like to express our particular gratitude to Steve Redman and Bruce Walmsley (Canberra, Australia) and Erwin Neher and Ralf Schneggenburger (Göttingen, Germany) for helpful discussion of the experimental results. We also thank Uwe Heinemann for useful comments on an earlier version of the manuscript. The technical assistance of Mr Bernd Schacht and Mrs Karin Przezdziecki is highly appreciated. This study was supported by the DFG (Sonderforschungbereich 515, Project Grant B2 to R.G.).
References
- Abbott LF, Nelson SB. Synaptic plasticity: Taming the beast. Nature Neuroscience. 2000;3:1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. Journal of Neuroscience. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. Journal of Neuroscience. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger C, Kondo S, Marty A. Multivesicular release at single functional synaptic sites in cerebellar stellate and basket cells. Journal of Neuroscience. 1998;18:4532–4547. doi: 10.1523/JNEUROSCI.18-12-04532.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- BetZ WJ. Depression of transmitter release at the neuromuscular junction of the frog. Journal of Physiology. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. Journal of Physiology. 1999;521:123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. Journal of Neuroscience. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. Journal of Biological Chemistry. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- Chen C, BlitZ DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron. 2002;33:779–788. doi: 10.1016/s0896-6273(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RAJ, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Costa E. From GABAA receptor diversity emerges a unified vision of GABAergic inhibition. Annual Review of Pharmacology and Toxicology. 1998;38:321–350. doi: 10.1146/annurev.pharmtox.38.1.321. [DOI] [PubMed] [Google Scholar]
- Davies CH, PoAZZ MF, Collingridge GL. CGP 55845A: a potent antagonist of GABAB receptors in the CA1 region of rat hippocampus. Neuropharmacology. 1993;32:1071–1073. doi: 10.1016/0028-3908(93)90073-c. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, GÄHWILER BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. Journal of Physiology. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. Journal of Neuroscience. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist D, Quastel DMA. A quantitative study of end-plate potentials in isolated human muscle. Journal of Physiology. 1965;178:505–529. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Fortune ES, Rose GJ. Short-term synaptic plasticity as a temporal filter. Trends in Neurosciences. 2001;24:381–385. doi: 10.1016/s0166-2236(00)01835-x. [DOI] [PubMed] [Google Scholar]
- Geiger JRP, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proceedings of the National Academy of Sciences of the USA. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Wang Y, Markram H. Organizing principles for a diversity of GABAergic interneurons and synapses in the neocortex. Science. 2000;287:273–278. doi: 10.1126/science.287.5451.273. [DOI] [PubMed] [Google Scholar]
- Hatt H, Smith DO. Synaptic depression related to presynaptic axon conduction block. Journal of Physiology. 1976;259:367–393. doi: 10.1113/jphysiol.1976.sp011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zorumski CF, Mennerick S. Contribution of presynaptic Na+ channel inactivation to paired pulse synaptic depression in cultured hippocampal neurons. Journal of Neurophysiology. 2002;87:925–936. doi: 10.1152/jn.00225.2001. [DOI] [PubMed] [Google Scholar]
- Hsu SF, Augustine GJ, Jackson MB. Adaptation of Ca2+-triggered exocytosis in presynaptic terminals. Neuron. 1996;17:501–512. doi: 10.1016/s0896-6273(00)80182-8. [DOI] [PubMed] [Google Scholar]
- Jensen K, Lambert JDC, Jensen MS. Activity-dependent depression of GABAergic IPSCs in cultured hippocampal neurons. Journal of Neurophysiology. 1999;82:42–49. doi: 10.1152/jn.1999.82.1.42. [DOI] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Sandkühler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- Jones MV, Jonas P, Sahara Y, Westbrook GL. Microscopic kinetics and energetics distinguish GABA(A) receptor agonists from antagonists. Biophysical Journal. 2001;81:2660–2670. doi: 10.1016/S0006-3495(01)75909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- JÜTTNER R, Meier J, Grantyn R. Slow IPSC kinetics, low levels of α1 subunit expression and paired pulse depression are distinct properties of neonatal inhibitory GABAergic synaptic connections in the mouse superior colliculus. European Journal of Neuroscience. 2001;13:2088–2098. doi: 10.1046/j.0953-816x.2001.01587.x. [DOI] [PubMed] [Google Scholar]
- King RD, Wiest MC, Montague PR. Extracellular calcium depletion as a mechanism of short-term synaptic depression. Journal of Neurophysiology. 2001;85:1952–1959. doi: 10.1152/jn.2001.85.5.1952. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Grantyn R. Short-term plasticity of synaptic transmission at single inhibitory synaptic boutons in culture. Society for Neuroscience Abstracts. 2001;27:923. [Google Scholar]
- Kirischuk S, Veselovsky N, Grantyn R. Relationship between presynaptic calcium transients and postsynaptic currents at single gamma-aminobutyric acid (GABA)ergic boutons. Proceedings of the National Academy of Sciences of the USA. 1999a;96:7520–7525. doi: 10.1073/pnas.96.13.7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Veselovsky N, Grantyn R. Single-bouton-mediated synaptic transmission: postsynaptic conductance changes in their relationship with presynaptic calcium signals. Pflügers Archiv. 1999b;438:716–724. doi: 10.1007/s004249900075. [DOI] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. Journal of Neuroscience. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liley AW, North KAK. An electrical investigation of effects of repetitive stimulation on mammalian neuromuscular junction. Journal of Neurophysiology. 1954;16:509–527. doi: 10.1152/jn.1953.16.5.509. [DOI] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a presynaptic terminal. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Paired pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. Journal of Physiology. 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Postsynaptic modulation of NMDA synaptic currents in rat hippocampal microcultures by paired pulse stimulation. Journal of Physiology. 1996;490:405–407. doi: 10.1113/jphysiol.1996.sp021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MJ, Rinzel J. Synaptic depression: a dynamic regulator of synaptic communication with varied functional roles. Trends in Neurosciences. 1997;20:431–433. doi: 10.1016/s0166-2236(97)01124-7. [DOI] [PubMed] [Google Scholar]
- Ohana O, Sakmann B. Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. Journal of Physiology. 1998;513:135–148. doi: 10.1111/j.1469-7793.1998.135by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Clements J, Walmsley B. Release probability modulates short-term plasticity at a rat giant terminal. Journal of Physiology. 2000;524:513–523. doi: 10.1111/j.1469-7793.2000.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Klocker N, Schuck J, BaukrowitZ T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000;26:595–601. doi: 10.1016/s0896-6273(00)81197-6. [DOI] [PubMed] [Google Scholar]
- Parnas H, Segel L, Dudel J, Parnas I. Autoreceptors, membrane potential and the regulation of transmitter release. Trends in Neurosciences. 2000;23:60–68. doi: 10.1016/s0166-2236(99)01498-8. [DOI] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Perouansky M, Grantyn R. Separation of quisqualate- and kainate-selective glutamate-receptors in cultured neurons from the rat superior colliculus. Journal of Neuroscience. 1989;9:70–80. doi: 10.1523/JNEUROSCI.09-01-00070.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Gottmann K, Lux H-D. Kinetics of GABAB receptor-mediated inhibition of calcium currents and excitatory synaptic transmission in hippocampal neurons in vitro. Neuron. 1994;12:97–107. doi: 10.1016/0896-6273(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Pouzat C, Hestrin S. Developmental regulation of basket/stellate cell⇒Purkinje cell synapses in the cerebellum. Journal of Neuroscience. 1997;17:9104–9112. doi: 10.1523/JNEUROSCI.17-23-09104.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino D, Woodward RM, Murata Y, Eusebi F, Overman LE, Miledi R. Design and in vitro pharmacology of a selective gamma-aminobutyric acid C receptor antagonist. Molecular Pharmacology. 1996;50:1024–1030. [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. Journal of Physiology. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Mohler H. GABA(A) receptor subtypes: dissecting their pharmacological functions. Trends in Pharmacological Sciences. 2001;22:188–194. doi: 10.1016/s0165-6147(00)01646-1. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short term synaptic depression: lessons from a large synapse. Trends in Neurosciences. 2002;25:206–212. doi: 10.1016/s0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- ScholZ KP, Miller RJ. GABAB receptor-mediated inhibition of Ca2+ currents and synaptic transmission in cultured rat hippocampal neurones. Journal of Physiology. 1991;444:669–686. doi: 10.1113/jphysiol.1991.sp018900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proceedings of the National Academy of Sciences of the USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Streit J, LÜSCHER C, LÜSCHER H-R. Depression of postsynaptic potentials by high-frequency stimulation in embryonic motoneurons grown in spinal cord slice cultures. Journal of Neurophysiology. 1992;68:1793–1803. doi: 10.1152/jn.1992.68.5.1793. [DOI] [PubMed] [Google Scholar]
- Sur C, Triller A, Korn H. Morphology of the release site of inhibitory synapses on the soma and dendrite of an identified neuron. Journal of Comparative Neurology. 1995;351:247–260. doi: 10.1002/cne.903510205. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends in Neurosciences. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Bannister AP. Release-independent depression at pyramidal inputs onto specific cell targets: dual recordings in slices of rat cortex. Journal of Physiology. 1999;519:57–70. doi: 10.1111/j.1469-7793.1999.0057o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- Tsodyks M, Uziel A, Markram H. Synchrony generation in recurrent networks with frequency-dependent synapses. Journal of Neuroscience. 2000;20:RC50. doi: 10.1523/JNEUROSCI.20-01-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela JA, Song S, Turrigiano GG, Nelson SB. Differential depression at excitatory and inhibitory synapses in visual cortex. Journal of Neuroscience. 1999;19:4293–4304. doi: 10.1523/JNEUROSCI.19-11-04293.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired pulse depression at a central synapse. Journal of Neuroscience. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Warton SS, Perouansky M, Grantyn R. Development of GABAergic synaptic connections in vivo and in cultures from the rat superior colliculus. Developmental Brain Research. 1990;52:95–111. doi: 10.1016/0165-3806(90)90225-n. [DOI] [PubMed] [Google Scholar]
- Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annual Review of Physiology. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]