

Abstract
We measured the ortho-para conversion rate in solid hydrogen by using Raman scattering in a diamond-anvil cell, extending previous measurements by a factor of 60 in pressure. We confirm previous experiments that suggested a decrease in the conversion rate above about 0.5 GPa. We observe a distinct minimum at 3 GPa followed by a drastic increase in the conversion rate to our maximum pressure of 58 GPa. This pressure enhancement of conversion is not predicted by previous theoretical treatments and must be due to a new conversion pathway.
Keywords: high-pressure physics, Raman spectroscopy, solid hydrogen
Solid hydrogen is a highly compressible quantum molecular solid whose behavior changes drastically over experimentally accessible pressures. Recently, a great deal of attention has been paid to the orientational ordering behavior of solid hydrogen at very high pressures (1–6). Driving this interest is the interpretation of several high-pressure phase transitions as involving orientational ordering, as well as the realization that the insulator-to-metal transition in solid hydrogen depends critically on the orientational properties of high-pressure hydrogen. Crucial to understanding these phenomena is a detailed knowledge of the ortho-para state of the solid at high pressures.
The molecular wavefunction for any isolated homonuclear diatomic molecule must be symmetric or antisymmetric under nuclear exchange. For hydrogen the molecular wavefunction must be antisymmetric under proton exchange, which leads to two distinct types of hydrogen: ortho with total nuclear spin I = 1 and para with I = 0. If we restrict ourselves to ground electronic states, the total protonic wavefunction must be antisymmetric and can be written as a product of vibrational, spin, and rotational parts, ψmol = ψvibψspinψrot. ψvib is always symmetric, and ψspin is symmetric for the triplet state I = 1 and antisymmetric for the singlet state I = 0. Thus ψrot must be antisymmetric for ortho-hydrogen (I = 1) and symmetric for para-hydrogen (I = 0). For an isolated hydrogen molecule ψrot is given by spherical harmonics, YJmj, with the symmetry restrictions that J is odd for ortho-hydrogen and even for para-hydrogen. These species are very stable and do not intermix for isolated molecules. For solid hydrogen the rotational states are only slightly perturbed from the gaseous state, and ψrot is still accurately described by spherical harmonics.
The equilibrium ortho-hydrogen concentration is easily calculated by using the rotational energy of a diatomic simple rotator Erot = BJ(J + 1), where B = 59.3 cm−1 is the rotational constant for hydrogen (7), and assuming a Boltzmann distribution. The equilibrium concentration of ortho-hydrogen drops from 75% at room temperature, to 61% at 100 K, to 0.1% at 20 K (7). While ortho-para conversion is strictly forbidden for isolated molecules, conversion does occur in the solid. At ambient pressure ortho-para conversion in the solid is very slow, taking weeks for a sample to equilibrate (7, 8). Although there have been numerous experimental studies of the ortho-para conversion rate at pressures below 1 GPa (9–13), little is known about the conversion rate at higher pressures. Driessen et al. (9) found a maximum conversion rate at 0.5 GPa followed by a drop up to 0.7 GPa. We have confirmed these results, finding that K slows to a minimum at about 3 GPa followed by a dramatic increase that persists to at least 58 GPa. Preliminary results of this study at lower pressures were presented in ref. 14. The results are compared with recent NMR measurements to 12.8 GPa (15).
We loaded research-grade hydrogen at room temperature into a modified Mao-Bell diamond-anvil cell. The cell was placed in a liquid-helium dewar cryostat, and all pressure adjustments were made at sample temperatures between 90 and 120 K. Pressure was determined by using standard ruby fluorescence techniques corrected for low temperatures. Raman spectra were excited with an argon-ion laser at 514.5 nm and recorded with a Dilor triple-grating spectrograph and a liquid-nitrogen-cooled charge-coupled device (CCD) detector. The spectral resolution of the system was 3 cm−1.
Once we obtained a desired pressure, we held the sample temperature near 100 K for at least 24 h and sometimes as long as 72 h. This ensured that full para-ortho reconversion would take place and that our sample would be at equilibrium ortho concentration when we began cooling to liquid helium temperatures. We cooled from roughly 100 K to below 20 K in 7 to 10 min. The quick cooling time assured us that minimal conversion would take place during the cooling period and that our sample was still in the high-temperature equilibrium condition when we began collecting spectra. Once the sample temperature was below 20 K, we began measuring rotational Raman spectra at 2- to 5-min intervals for 3–24 h, depending on how fast a conversion rate we observed. Temperature was held between 6 and 10 K during conversion, and more than 3,000 spectra were collected from three separate samples.
We determined the ortho concentration, x, of our samples from the integrated intensities of the rotational Raman peaks,
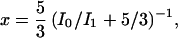 |
1 |
where x is the ortho concentration and I0 and I1 are the integrated intensities of the S0(0) (J = 2 ← 0) and the S0(1) (J = 3 ← 1) transitions, respectively. This expression is simplified compared to the corresponding equations given by Silvera (7) and Driessen et al. (9) because kBT ≪ 6B, 10B ≪ ℏωL. In many of our spectra we observed an optical phonon overlapping the rotational lines. The intrinsic Raman cross-section of the phonon is very small so that its intensity is primarily due to hybridization with the rotational transitions and the total Raman cross-section is conserved (16–18); thus we assigned the phonon intensity to the nearest rotational line. Neglect of the phonon intensity has a negligible effect on the calculated rates. In addition, at our highest pressures the S0(0) and S0(1) Raman peaks broaden and begin to overlap, causing some uncertainty in the assignment of spectral intensity. Although this could call into question some of the assumptions used in deriving Eq. 1, this possible problem is relevant only at the very highest pressures of our study (e.g., >40 GPa).
We confirmed that Eq. 1 gives reliable measurements of x at low temperatures by extrapolating our measurements of x during conversion back to the concentration at the beginning of the cool down. We found agreement between the initial x and our equilibration temperatures within the scatter of our data. Additionally, to verify that Eq. 1 extended to high temperatures (7, 9) holds at our high pressures, we analyzed the rotational spectra of many equilibrium experiments performed at liquid nitrogen and higher temperatures to extract x. We found for spectra between 0 and 58 GPa that ortho concentrations deviated from the equilibrium concentrations by less than 5%. For example, at 58 GPa and 105 K we measured x = 0.607 while the equilibrium concentration is x = 0.626. We are confident that our method of determining the ortho concentration is accurate over the pressure-temperature range relevant to our study.
Fig. 1 shows several spectra at 21.6 GPa. The decrease in the integrated intensity of the S0(1) peak with respect to the S0(0) peak with time is obvious. The phonon peak is also apparent and was included simply as a contribution to S0(0) in our analyses. Fig. 2 shows the ortho concentration, plotted as 1/x, as a function of time for several pressures calculated from Eq. 1 and our best fits as t → 0 as discussed below.
Figure 1.

Representative Raman spectra at 21.6 GPa. Each spectrum is labeled by the time elapsed from cool-down completion.
Figure 2.

Inverse ortho concentration as a function of time for a series of pressures. The solid lines are the results of the Monte Carlo simulation (see text). The dot-dashed lines are linear fits to the short-time data.
According to the theoretical description of ortho-para conversion in solid hydrogen as developed by Motizuki and Nagamiya (19), an ortho-para transition requires a large magnetic field gradient acting on a molecule to flip a proton spin. This field gradient is supplied by the rotational magnetic moment of neighboring l = 1 (ortho) molecules. Thus the rate of conversion is proportional to the average number of nearest ortho neighbors, M, and the ortho concentration,
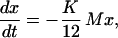 |
2 |
where K is the conversion rate. If the ortho distribution is random, then M = 12x [12 nearest neighbors in a hexagonal close-packed (hcp) lattice] and the rate equation is second order in x (bimolecular), dx/dt = −Kx2. The analytic solution to this rate equation is simple,
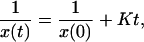 |
3 |
and is shown by the dot-dashed line in Fig. 3.
Figure 3.

Our data at 21.6 GPa along with several different fitting functions. The dot-dashed line is the fast-diffusion bimolecular result (Eq. 3), the dashed line is the no-diffusion mean-field result (Eq. 4), and the solid line is the average of four 8,000-molecule Monte Carlo simulations.
The bimolecular rate equation is only valid for rapid molecular diffusion where the ortho molecules maintain a random distribution. For slow molecular diffusion, ortho-hydrogen nearest neighbors will become depleted so that M < 12x and conversion slows compared to Eq. 3. Thus, in previous experiments when conversion was measured far below the melting temperature, short excursions to temperatures approaching melting were employed to keep the ortho distribution random (9). In our experiments this was not feasible because of the very high melting temperatures. We could extract the conversion rate from our data using Eq. 3 by restricting our analysis to the initial decay at very short times (see Fig. 2). We also decided to look for a way to fit our data to Eq. 2 directly, thereby using our entire data set. We consider two methods for solving Eq. 2, a mean-field rate equation, and a Monte Carlo simulation.
First, we consider the mean-field rate equation for no diffusion given by Schmidt (20, 21). In the mean-field approximation, Schmidt derives a no-diffusion form of Eq. 2,
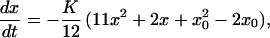 |
where x0 is the initial ortho concentration, having the closed-form solution,
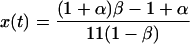 |
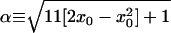 |
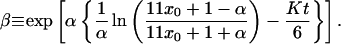 |
4 |
This is shown as the dashed line in Fig. 3.
Alternatively, Perrell and Haase (22) have presented a Monte Carlo algorithm for ortho-para conversion in hydrogen in one and two dimensions. We have extended their algorithm to a hcp lattice to model our data. Perrell and Haase’s algorithm for no diffusion is as follows. We set up the topology of nearest neighbors according to an hcp lattice, as appropriate for hydrogen in this pressure range (23) and randomly distributed an initial concentration x0 of ortho molecules on the lattice. Stepping through the lattice sites we determine if a site is ortho. If so, we count the number of ortho nearest neighbors M. The probability for an ortho molecule to convert to a para molecule in a single Monte Carlo step is KM/12 according to Eq. 2. We continue sampling lattice sites until equilibrium is achieved. The time unit for this simulation is the number of Monte Carlo steps per lattice site (MCS/LS), which we can easily scale with K for fitting to our data.
To check our algorithm, we ran several simulations using x0 = 0.65 and K = 0.25, using 150, 1,000, 4,096, and 8,000 lattice sites with periodic boundary conditions. There was no lattice size dependence on our results. We also ran simulations using the bimolecular conversion probability, P(x) = Kx2, for rapid diffusion and verified agreement with Eq. 3. As expected, for short times or large x the two simulations (no diffusion and rapid diffusion) showed identical results. Finally, we verified that the simulations for different values of K scaled linearly with time as expected.
Fig. 3 shows fits to our data with the three models discussed: the bimolecular rapid diffusion case (Eq. 3), Schmidt’s no-diffusion mean-field theory (Eq. 4), and an average of 4 Monte Carlo simulations with 8,000 lattice sites. We see that for x > 0.3 all three models are in good agreement, while for x < 0.3 the rapid diffusion case underestimates the experimental concentration (as expected for our temperature far below melting), the mean-field model overestimates the concentration [as expected for a mean-field model (22)], and the Monte Carlo simulation fits our data quite well. Thus we fit all of our data to the Monte Carlo simulation, scaling the time axis linearly to determine K and treating the cool-down time as an adjustable time-offset parameter. We found good agreement with our concentration vs. time data using this fitting method at all pressures as illustrated in Fig. 2.
Fig. 4 shows our experimental determinations of the rate constant K versus density. [Below 2.5 GPa we used the equation of state for para-hydrogen at 10 K (23); above 2.5 GPa we used the equation of state for hydrogen at room temperature (24).] At very low densities we find excellent agreement between our results and previous experiments. As the pressure increases, K decreases until it reaches a minimum at about 3 GPa, nearly a factor of two below the ambient pressure rate constant. At higher pressures the conversion rate increases rapidly to about 260%/h at 58 GPa.
Figure 4.

Our results for the ortho-para conversion rate as a function of density. The hollow circles are from refs. 9–13, the hollow squares are from Pravica and Silvera (15), the filled region is the theoretical result of Berlinsky (25), and the solid line is a guide to the eye.
According to Motizuki and Nagamiya (19) the rotational energy difference between a J = 1 ortho molecule and a J = 0 para molecule, ΔE0←1 = 2B = 171 K, released by ortho-para conversion must be absorbed by the lattice. At ambient pressure this energy conservation requires the creation two phonons and at moderate pressures one phonon. Berlinsky (25) has given a detailed theory of these processes, finding that the one-phonon conversion rate should initially increase with pressure before falling above about 0.5 GPa because of density of states factors and the rapid increase in phonon energy. This theory explains the apparent peak in conversion rate observed experimentally at around 0.5 GPa. Previous low-pressure determinations of the conversion rate as well as Berlinsky’s theory are plotted as the hollow circles and the solid curve in the inset of Fig. 4. Our data confirm that the conversion rate does fall above 1 GPa to well below the ambient pressure value.
Above 3 GPa our conversion rate increases very rapidly. A similar observation has been reported on the basis of a contemporaneous study of the conversion by NMR techniques, albeit to lower maximum pressures (15). There is generally excellent agreement except for the two highest pressure points of the above study (at 10.4 and 12.8 GPa), for which higher rates are reported. Our result in this low-pressure range is unaffected by possible scrambling of the rotational modes under pressure (breakdown in quantum number J), as the rotons remain well resolved and the pressure is well below the orientiational ordering transition (110 GPa); moreover, the reported rates are essentially independent of how the roton-phonon mixing is treated. Possible discrepancies could arise from temperature differences in the two studies. Notably, the theory developed by Berlinsky fails to explain the striking increase in conversion rate. To generate the very rapid conversion we observe, another pathway for energy conservation is needed. One possibility is that rotational excitations (rotons) rather than lattice vibrations carry away the excess energy. As the density of the solid increases, so too does the crystal field in which the molecules reside. The associated broadening of the J = 2 roton band may be sufficient to lower its energy enough to allow it to carry away the rotational energy released by an ortho-para transition. In fact, such a softening of rotons has been predicted for the face-centered cubic (fcc) and hcp structures of molecular hydrogen and deuterium (26). Thus, it is likely that phonon-roton, roton-roton, and single roton processes will become important channels for high-pressure ortho-para conversion energy conservation. Developing a rigorous theory for these conversion channels is a theoretical challenge that would greatly aid our understanding of dense solid hydrogen.
Acknowledgments
We thank V. V. Struzhkin for experimental assistance, P. Gillet for help in the preparation of the manuscript, and the National Science Foundation and the National Aeronautics and Space Administration for financial support.
References
- 1.Goncharov A F, Eggert J H, Mazin I I, Hemley R J, Mao H K. Phys Rev B. 1996;54:R15590–R15593. doi: 10.1103/physrevb.54.r15590. [DOI] [PubMed] [Google Scholar]
- 2.Hemley R J, Mao H K. J Non-Crystalline Solids. 1996;205–207:282–289. [Google Scholar]
- 3.Mazin I I, Hemley R J, Goncharov A F, Hanfland M, Mao H K. Phys Rev Lett. 1997;78:1066–1969. [Google Scholar]
- 4.Goncharov A F, Mazin I I, Eggert J H, Hemley R J, Mao H K. Phys Rev Lett. 1995;75:2514–2517. doi: 10.1103/PhysRevLett.75.2514. [DOI] [PubMed] [Google Scholar]
- 5.Cui L, Chen N H, Silvera I F. Phys Rev Lett. 1995;74:4011–4014. doi: 10.1103/PhysRevLett.74.4011. [DOI] [PubMed] [Google Scholar]
- 6.Natoli V, Martin R M, Ceperley D. Phys Rev Lett. 1995;74:1601–1604. doi: 10.1103/PhysRevLett.74.1601. [DOI] [PubMed] [Google Scholar]
- 7.Silvera I F. Rev Mod Phys. 1980;52:393–452. [Google Scholar]
- 8.van Kranendonk J. Solid Hydrogen. New York: Plenum; 1983. [Google Scholar]
- 9.Driessen A, van der Poll E, Silvera I F. Phys Rev B. 1984;30:2517–2526. [Google Scholar]
- 10.Pedroni P, Meyer H, Weinhaus F, Haase D. Solid State Commun. 1974;14:279–281. [Google Scholar]
- 11.Jochemsen R. Ph.D. thesis. Univ. of Amsterdam; 1978. [Google Scholar]
- 12.Silvera I F, Berkhout P J, van Aernsbergen L M. J Low Temp Phys. 1979;35:611–625. [Google Scholar]
- 13.Ahlers G. J Chem Phys. 1964;40:3123–3124. [Google Scholar]
- 14.Hemley R J, Goncharov A F, Mao H K, Eggert J H, Karmon E. J Low Temp Phys. 1998;110:75–88. [Google Scholar]
- 15.Pravica M, Silvera I F. Phys Rev Lett. 1998;81:4180–4183. [Google Scholar]
- 16.Wijngaarden R J, Silvera I F. Phys Rev Lett. 1980;44:456–459. [Google Scholar]
- 17.Wijngaarden R J, Goldman V V, Silvera I F. Phys Rev B. 1993;27:5084–5087. [Google Scholar]
- 18.Wijngaarden R J. Ph.D. thesis. Univ. of Amsterdam; 1982. [Google Scholar]
- 19.Motizuki K, Nagamiya T. J Phys Soc Jpn. 1956;11:93–104. [Google Scholar]
- 20.Schmidt F. Ph.D. thesis. Free Univ. of Berlin; 1971. [Google Scholar]
- 21.Schmidt F. Phys Rev B. 1974;10:4480–4484. [Google Scholar]
- 22.Perrell L R, Haase D G. Am J Phys. 1984;52:831–833. [Google Scholar]
- 23.Driessen A. Ph.D. thesis. Univ. of Amsterdam; 1982. [Google Scholar]
- 24.Loubeyre P, LeToullec R, Häusermann D, Hanfland M, Hemley R J, Mao H K, Finger L W. Nature (London) 1996;383:702–704. [Google Scholar]
- 25.Berlinsky A J. Phys Rev B. 1975;12:1482–1486. [Google Scholar]
- 26.Lagendijk A, Silvera I F. Phys Lett. 1981;84A:28–31. [Google Scholar]