

Abstract
Associative stimulation has been shown to enhance excitability in the human motor cortex (Stefan et al. 2000); however, little is known about the underlying mechanisms. An interventional paired associative stimulation (IPAS) was employed consisting of repetitive application of single afferent electric stimuli, delivered to the right median nerve, paired with single pulse transcranial magnetic stimulation (TMS) over the optimal site for activation of the abductor pollicis brevis muscle (APB) to generate approximately synchronous events in the primary motor cortex. Compared to baseline, motor evoked potentials (MEPs) induced by unconditioned, single TMS pulses increased after IPAS. By contrast, intracortical inhibition, assessed using (i) a suprathreshold test TMS pulse conditioned by a subthreshold TMS pulse delivered 3 ms before the test pulse, and (ii) a suprathreshold test TMS pulse conditioned by afferent median nerve stimulation delivered 25 ms before the TMS pulse, remained unchanged when assessed with appropriately matching test stimulus intensities. The increase of single-pulse TMS-evoked MEP amplitudes was blocked when IPAS was performed under the influence of dextromethorphan, an N-methyl-d-aspartate (NMDA) receptor antagonist known to block long-term potentiation (LTP). Further experiments employing the double-shock TMS protocol suggested that the afferent pulse, as one component of the IPAS protocol, induced disinhibition of the primary motor cortex at the time when the TMS pulse, as the other component of IPAS, was delivered. Together, these findings support the view that LTP-like mechanisms may underlie the cortical plasticity induced by IPAS.
Considerable evidence, including combined behavioral and physiological studies (Rioult-Pedotti et al. 2000), suggests that long-term potentiation (LTP) of synaptic efficacy and its counterpart long-term depression (LTD) are cellular mechanisms underlying learning and memory. LTP has been produced in vitro in neocortical slices derived from virtually any cortical region by different stimulation protocols. Between them, associative LTP may explain how inputs from local intracortical fibres, and cortico-cortical or thalamo-cortical afferents converging onto the same postsynaptic targets could interact to reshape local representational cortical patterns (e.g. Donoghue et al. 1996; Asanuma & Pavlides, 1997; Sanes & Donoghue, 2000). Associative LTP has been generated by pairing stimulation of cortical afferents with depolarization (Baranyi & Szente, 1987) or stimulation-induced firing (Baranyi & Feher, 1981) of the postsynaptic neuron, and by pairing stimulation of ‘vertical’ (thalamo-cortical as well as cortico-cortical fibres) with stimulation of ‘horizontal’ intracortical fibres in cortical layers II/III (Hess & Donoghue, 1994; Hess et al. 1996).
We have recently developed a protocol, shaped after models of associative LTP in experimental animals, to induce plasticity in the human motor cortex (Stefan et al. 2000). A rapidly evolving (< 30 min), long-lasting (duration > 60 min), reversible and topographically specific increase of corticomuscular excitability was induced when peripheral electric stimulation was paired with transcranial magnetic stimulation over the contralateral motor strip and timed to generate approximately synchronous events in the motor cortex. Experiments aimed at locating the level on the neuroaxis where this effect took place employed F-wave testing and electrical brainstem stimulation, which are sensitive to spinal excitability changes, but not to cortical excitability changes. These experiments showed that the excitability increase after interventional paired associative stimulation (IPAS) was generated at a supraspinal and therefore probably the cortical level (Stefan et al. 2000). Because, in addition, the increase of excitability was dependent on the synchronicity of activation of motor cortex output elements by each stimulation modality, we have conjectured that it might represent associative LTP or a closely related phenomenon in the human motor cortex (Stefan et al. 2000). This hypothesis leads to the following considerations which were experimentally tested in the present study: (1) The view that IPAS-induced plasticity is caused by increasing synaptic efficacy, would be supported if intracortical GABAA receptor-mediated inhibition, as one of the major alternative candidate mechanisms, were unchanged after IPAS. (2) Because LTP in the motor cortex depends upon activation of NMDA receptors (Aroniadou & Keller, 1995; Castro-Alamancos et al. 1995; Buonomano & Merzenich, 1998), a pharmacological blockade of these receptors should suppress the increase of IPAS-induced enhancement of motor cortical excitability. Some of the results have been published previously in abstract form (Stefan et al. 1999).
Methods
Subjects
The study was approved by the Ethics committee of the University of Rostock and all participants gave their written informed consent. Experiments were performed on 14 healthy volunteers (11 men, 3 women) aged 22–42 years (mean 27± 6 years). None had a history of physical or neurological illness. All volunteers were right handed, except two, who were left handed according to the Oldfield handedness inventory (Oldfield, 1971).
Stimulation
Focal transcranial magnetic stimulation (TMS) was performed using a flat figure-of-eight-shaped magnetic coil (outer diameter of each wing: 9.5 cm) connected with a Magstim 200 magnetic stimulator (Magstim, Whitland, Dyfed, UK). The coil was held tangentially to the skull with the handle pointing backward and laterally at a 45 deg angle to the sagittal plane. For the paired pulse experiments two Magstim 200 stimulators were connected to the same coil through a BISTIM module (Magstim). Electrical mixed nerve stimulation was performed with a Cantata electromyograph (Dantec Medical, Skovlunde, Denmark) using a standard stimulation block (cathode proximal) at a stimulation width of 200 μs.
Recording
Surface electromyographic activity (EMG) was recorded from the right abductor pollicis brevis muscle (APB) and using disposable silver-silver chloride surface electrodes (Dantec Medical, Skovlunde, Denmark) in a belly-tendon montage. Raw signals were amplified employing a Toennies amplifier (Toennies, Freiburg, Germany) and bandpass filtered between 20 and 2000 Hz. EMG signals were digitized at 5 kHz by an A/D converter (model 1401 plus, Cambridge Electronics Design, Cambridge, UK) and stored in a laboratory computer for display and later off-line analysis.
Experimental procedures
Subjects were seated comfortably in a reclining chair. At first the optimal position of the magnetic coil for eliciting motor evoked potentials (MEPs) in the resting right APB was assessed over the left motor cortex at a moderately suprathreshold stimulation intensity (usually around 60 % of the maximal stimulator output) and marked directly on the scalp with a soft-tip pen. At the optimal site, the resting motor threshold was determined as the minimum stimulator intensity needed to produce a response of at least 50 μV in the relaxed APB in at least 5 of 10 consecutive trials (Rossini et al. 1994). Thereafter, the stimulator intensity sufficient to evoke a peak-to-peak amplitude of 1 mV of the motor evoked potentials in the relaxed APB was determined (SI1mV).
In Expts 1 and 2 (see below) an interventional stimulation protocol was employed (Stefan et al. 2000). The intervention consisted of single electrical stimuli delivered to the right median nerve at the level of the wrist at 300 % of the perceptual threshold and followed by TMS at intensities sufficient to produce an unconditioned response amplitude of approximately 1 mV in the resting APB (SI1mV, as above). Ninety pairs were delivered at 0.05 Hz over 30 min at an interstimulus interval (ISI) of 25 ms. An ISI of 25 ms was used because this interval had been shown in previous experiments to be effective in inducing cortical plasticity in a high percentage of subjects (Stefan et al. 2000).
Complete muscle relaxation was continuously monitored by visual and auditory feedback. For amplitudes of motor evoked potentials of the resting target muscle (‘resting amplitudes’), 15–20 trials were collected both before, and immediately after intervention, using a stimulus intensity of SI1mV and a stimulation rate of about 0.1 Hz. Identical stimulus intensities were used before and after intervention.
Intracortical inhibition
Experiments 1a and b: paired-TMS-pulse inhibition (Kujirai et al. 1993)
A paired-pulse TMS technique was employed to probe intracortical inhibition. A subthreshold conditioning cortical magnetic stimulus attenuates the amplitude of the MEP following a test stimulus, delivered at 3 ms after the conditioning stimulus (Kujirai et al. 1993). Importantly, paired-pulse inhibition as produced by double-shock TMS has been shown to depend on the intensity of both the conditioning stimulus and the test stimulus (Kujirai et al. 1993). We used two complementary experimental approaches in order to be able to perform meaningful comparisons between paired-TMS-pulse inhibition following IPAS with that prior to IPAS.
Experiment 1a: paired-TMS-pulse inhibition following IPAS; influence of test stimulus efficacy
As a first step, the magnitude of paired-TMS-pulse inhibition at different test stimulus intensities was established in 12 subjects. Initially, the test stimulus intensities producing an unconditioned MEP response of approximately 1.0, 1.5 or 2.0 mV in the relaxed APB were determined (SI1mV-PRE, SI1.5mV-PRE, SI2mV-PRE). Preliminary experiments had shown that paired-TMS-pulse inhibition increased approximately linearly over this range of test stimulus intensities. Paired-TMS pulse inhibition was then assessed, for each of the three stimulus intensities separately, using two randomly intermixed blocks, consisting of 15 trials each. In one block, the test stimulus was applied alone and in the other block the test stimulus was preceded by a conditioning stimulus. For all test stimulus intensities, the intensity of the conditioning stimulus was set to 70 % of resting motor threshold, and, thus, at an intensity known to produce no changes of excitability at the level of the spinal cord (Kujirai et al. 1993; Di Lazzaro et al. 1998). At least 10 s elapsed between any two trials. The mean amplitude of the MEP responses obtained by a conditioning-test shock at an interstimulus interval of 3 ms was expressed as percentage of the mean amplitude of the unconditioned test responses. This value was termed CR3 (Werhahn et al. 1999). Paired-TMS pulse inhibition is then given by:
Paired-TMS-pulse inhibition = 100 % - CR3 (1)
(Werhahn et al. 1999). Paired-TMS pulse inhibition was assessed prior to, and following IPAS using a stimulus intensity of SI1mV-PRE.
Experiment 1b: paired-TMS pulse inhibition following IPAS; influence of conditioning stimulus intensity and test stimulus intensity
The magnitude of paired-TMS-pulse inhibition was established in six subjects prior to IPAS as a function of different conditioning stimulus intensities ranging from 50 to 90 % of resting motor threshold while keeping the test stimulus intensity constant at SI1mV-PRE. For each conditioning stimulus intensity, 20 paired TMS pulses were delivered. Additionally, 20 unconditioned TMS pulses were delivered to determine the magnitude of the unconditioned MEP response. After IPAS, the procedure was repeated and paired-TMS-pulse inhibition was redetermined using SI1mV-PRE at all five previously used conditioning stimulus intensities. Thereafter, the stimulus intensity was adjusted to yield a MEP amplitude of about 1 mV in the resting APB (SI1mV-POST), and paired-TMS-pulse inhibition was again determined, using SI1mV-POST at all previously used five conditioning stimulus intensities.
In four of the six subjects, resting motor thresholds were additionally reassessed following IPAS, using a magnetic coil connected to a single stimulator, to reconfirm previous findings that resting motor thresholds are unchanged following IPAS (Stefan et al. 2000).
Experiment 1c: intracortical inhibition of the primary motor cortex elicited by afferent input (Tokimura et al. 2000)
Because IPAS involves the pairing of median nerve stimulation with TMS at ISI = 25 ms, i.e. at an interval where short-latency inhibition by somatosensory input is assumed to represent an intracortical phenomenon (Tokimura et al. 2000), this type of intracortical inhibition can readily be assessed by comparing the size of the TMS-evoked MEP during IPAS with the unconditioned TMS responses recorded immediately before or after IPAS. Therefore, in addition to the testing of paired-TMS-pulse inhibition prior and following IPAS in Expt 1b, the first 20 and the last 20 MEPs evoked by the interventional stimulation (pairs consisting of median nerve stimulation followed by single-pulse TMS after 25 ms), were analysed. Additionally, 20 unconditioned MEPs and 20 MEPs conditioned by afferent median nerve stimulation were recorded after IPAS using SI1mV-POST.
Experiment 2: pharmacological intervention
We tested whether premedication with dextromethorphan modulated the IPAS-induced increase of resting amplitude. Six subjects participated in four sessions each using a double-blind protocol. In two of these four sessions, each subject received a single dose of 150 mg encapsulated dextromethorphan (Hustenstiller-Ratiopharm, Ratiopharm, Ulm, Germany), and in the other two sessions a placebo was given. Dextromethorphan at the dose used results in dextromethorphan brain concentrations in humans (Steinberg et al. 1996) similar to those that induce NMDA receptor block in vitro (Wong et al. 1988; Apland & Braitman, 1990). The order of the sessions was pseudorandom and counterbalanced across subjects. In order to avoid potential drug accumulation all sessions were separated by at least 2 days. Like the experiments, all analyses were done blind to the condition tested.
Dextromethorphan or placebo were administered 3 h before the beginning of an experimental session (Silvasti et al. 1987; Steinberg et al. 1996). Side effects consisted of mild drowsiness, occurred with equal frequency in the dextromethorphan and in the placebo sessions, and did not interfere with the subjects’ ability to complete the experiments.
For resting amplitudes, 20 trials were collected both before, and immediately after IPAS, using a stimulus intensity of SI1mV-PRE and a stimulation rate of 0.1 Hz.
Experiment 3: intracortical inhibition following afferent stimulation
Using the magnetic double-shock protocol, this experiment aimed at determining intracortical inhibition at 25 ms following the afferent median nerve stimulation, building on, and expanding experiments by Ridding & Rothwell (1999). The magnetic conditioning stimulus was delivered at 22 ms and the magnetic test stimulus was delivered at 25 ms following electric stimulation of the median nerve at 300 % of the perceptual threshold. As mentioned above, peripheral afferent stimulation at ISI = 25 ms may decrease the efficacy of a suprathreshold magnetic test pulse, due to cortical interactions (Tokimura et al. 2000). Because the magnitude of the paired-TMS-pulse inhibition may be influenced by the efficacy of the test pulse, we used several controls to perform meaningful comparisons between paired-TMS-pulse inhibition assessed in the presence or absence of an afferent cortical input.
First, CR3 was assessed at a test stimulus intensity of SI1mV and a conditioning stimulus intensity of 70 % resting motor threshold. Fifteen to twenty trials were collected.
In four experiments, the magnitude of the resting amplitudes was assessed when the magnetic test pulse was preceded by an afferent median nerve pulse at 25 ms. Thereafter, the intensity of the test stimulus was adjusted to produce a mean MEP amplitude of the APB of a similar magnitude as that obtained when a test stimulus of SI1mV was conditioned by median nerve stimulation. This intensity was termed SI0.6mV. CR3 was then reassessed using SI0.6mV.
In three experiments, the intensity of the test stimulus was adjusted to yield a MEP amplitude of ≈1 mV in the presence of an afferent stimulus (SI1mV-AFF). CR3 was reassessed using SI1mV-AFF.
Data analysis
Resting amplitudes were measured peak-to-peak in each individual trial. For each subject, resting amplitudes were averaged according to the different conditions described above, and entered into the final statistical analyses. If not stated otherwise, paired two-tailed t tests were employed for statistical analysis. Additionally, repeated measures analyses of variance (rmANOVA) and linear regression analysis were used in some experiments and are explained in the Results section. If not stated otherwise, all group data are given as means ± s.d. Effects were considered significant if P < 0.05.
Results
Intracortical inhibition
Experiment 1a
Mean test stimulus responses were 0.9 ± 0.2 mV at SI1mV-PRE, 1.4 ± 0.2 mV at SI1.5mV-PRE, and 1.8 ± 0.2 mV at SI2mV-PRE (Fig. 1). Within the range of test stimulus intensities applied, larger test stimulus amplitudes were associated with larger CR3 amplitudes (corresponding to a reduction of paired-TMS-pulse inhibition), in agreement with results by other investigators (Kujirai et al. 1993). The relation between test stimulus intensities and CR3 was best fitted by a linear regression (y = 23.104x + 13.37; regression analysis; Pearson's correlation coefficient R = 0.533; F = 13.53; P < 0.001; Fig. 1). In accordance with findings of a previous study (Stefan et al. 2000), resting amplitudes increased following IPAS (pre, 0.9 ± 0.2 mV; post, 1.4 ± 0.5 mV; mean increase, +52 %; P < 0.01), using SI1mV-PRE as probing stimulus intensity.
Figure 1. Paired-TMS pulse inhibition (Kujirai et al. 1993) following IPAS; influence of test stimulus efficacy.
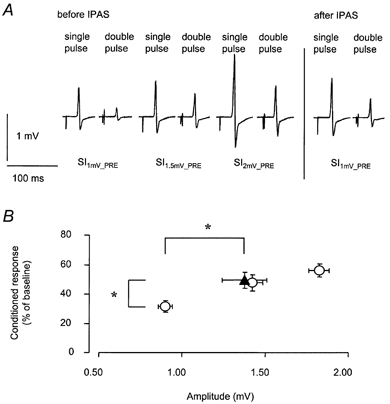
Before IPAS, test stimulus intensities were adjusted to produce an unconditioned MEP response of approximately 1.0, 1.5 mV or 2.0 mV in the relaxed APB (SI1mV-PRE, SI1.5mV-PRE, SI2mV-PRE). Paired-TMS pulse inhibition was assessed prior to, and following IPAS by conditioning the test pulse by a subthreshold stimulus at 3 ms. Larger test stimulus amplitudes were associated with larger CR3 amplitudes corresponding to a reduction of paired-TMS-pulse inhibition. A, data from a representative subject. Following IPAS, the relative size of the conditioned MEP elicited using a test stimulus intensity = SI1mV-PRE was larger (i.e. less paired-TMS-pulse inhibition) when compared with the conditioned MEP elicited using a test stimulus intensity = SI1mV-PRE before IPAS, and similar to the conditioned MEP elicited using a test stimulus intensity = SI1.5mV-PRE before IPAS. Each record shows the average of 15 trials. B, group data (means ± s.e.m.) from 12 subjects. CR3 (ordinate) is shown as a function of the magnitude of unconditioned test response (abscissa). ○, CR3 as assessed prior to IPAS at different test stimulus intensities producing an unconditioned MEP response of approximately 1.0, 1.5 or 2.0 mV. Following IPAS (▴), mean CR3 and resting amplitude, using SI1mV-PRE, increased when compared to CR3 or resting amplitude as assessed prior to IPAS. * Significant differences pre vs. post IPAS. For linear regression analysis see Results.
Following IPAS, CR3 increased from 31.7 ± 13.3 % to 49.6 ± 19.3 % (P < 0.01). This corresponds to a reduction of paired-TMS-pulse inhibition by 18 % points, according to eqn (1). However, the increase of CR3 did not exceed that expected by the increase in the unconditioned test amplitude (Fig. 1). For statistical evaluation we used the linear regression formula to calculate expected CR3 values, for all mean resting amplitudes obtained after IPAS. These values were compared to the actual CR3 values measured after IPAS using a paired t test (P = 0.472, not significant).
Experiment 1b
The results of Expt 1a suggest that the apparent decrease of paired-TMS-pulse inhibition following IPAS is due to the increased efficacy of the test stimulus producing a larger test response which is associated with less paired-TMS-pulse inhibition (Kujirai et al. 1993). This hypothesis predicts that paired-TMS-pulse inhibition could be restored to pre-interventional values by adjusting the intensity of the test stimulus so as to match the MEP size obtained before IPAS. Alternatively, the decrease in paired-TMS-pulse inhibition observed after IPAS could be due to a decreased efficacy of the conditioning stimulus, or to an interaction between changes in the efficacies of conditioning and test stimuli. These possibilities were explored in the following experiment.
The mean size of test stimulus responses was 1.0 ± 0.2 mV at SI1mV-PRE. In agreement with published reports, paired-TMS-pulse inhibition was maximal at a conditioning stimulus intensity of 70 - 80 % resting motor threshold (Fig. 2). Lower or higher conditioning stimulus intensities yielded a larger CR3 (corresponding to smaller paired-TMS-pulse inhibition) (Fig. 2). Following IPAS, resting motor threshold remained unaltered (pre, 31 ± 3 maximal stimulator output; post, 31 ± 3 % maximal stimulator output; n = 4 subjects), in agreement with our previous observations (Stefan et al. 2000). Resting amplitudes increased from 1.0 ± 0.2 mV to 1.5 ± 0.5 mV (mean increase, +40 %; P < 0.05; n = 6) when tested at SI1mV-PRE. With test stimulus intensity after IPAS reduced by, on average, 2 ± 1 % maximal stimulator output (= SI1mV-POST), mean resting amplitudes amounted to 0.9 ± 0.2 mV, and thus matched the size prior to IPAS.
Figure 2. Paired-TMS pulse inhibition (Kujirai et al. 1993) following IPAS; influence of conditioning stimulus intensity and adjustment of test stimulus intensity.
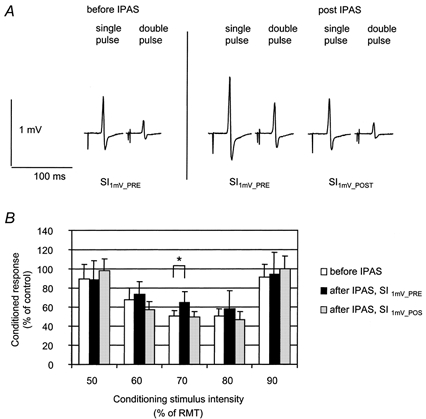
A, adjustment of test stimulus intensity. Data from a representative subject. Following IPAS, the relative size of the conditioned MEP elicited using a test stimulus intensity = SI1mV-PRE was larger (i.e. less paired-TMS-pulse inhibition) when compared with the conditioned MEP elicited using a test stimulus intensity = SI1mV-PRE before IPAS. When the test stimulus intensity was adjusted to produce a MEP amplitude of approximately 1 mV (SI1mV-POST), the conditioned MEP matched the size as before IPAS. Each record shows the average of 20 trials. B, influence of conditioning stimulus intensity and adjustment of test stimulus intensity. Data (means ± s.d.) from 6 subjects. CR3 (ordinate) is shown as a function of the intensity of the conditioning stimulus (abscissa). CR3 was assessed before (□) and after (▪ and ░) IPAS. Stimulus intensity was SI1mV-PRE (□ and ▪), or SI1mV-POST (░). Following IPAS, mean CR3 increased when using a conditioning stimulus intensity of 70 % of resting motor threshold and a test stimulus intensity of SI1mV-PRE. With test stimulus set to SI1mV-POST, CR3 matched that measured prior to IPAS. * Significant differences pre vs. post IPAS.
We performed a two-way rmANOVA (condition × CS-intensity, 3 × 5) with factors ‘condition’ (1, before IPAS; 2, after IPAS, SI1mV-PRE; 3, after IPAS, SI1mV-POST) and ‘CS-intensity’ (50, 60, 70, 80 and 90 % of resting motor threshold). We found a significant effect of CS-intensity (F = 18.63; P < 0.001) and a significant condition × CS-intensity interaction (F = 2.83; P < 0.02). Post hoc analysis of the latter interaction was done using paired t tests. CR3 increased from 50.0 ± 15.2 % to 65.0 ± 11.2 % (P < 0.05) when using SI1mV-PRE and a conditioning stimulus intensity of 70 % of the resting motor threshold. This corresponds to a reduction of paired-TMS-pulse inhibition by 15 % points, according to eqn (1), in good agreement with Expt 1a. With SI1mV-POST and the conditioning stimulus intensity set to 70 % of the resting motor threshold, CR3 amounted to 49.1 ± 15.8 %, similar to the mean CR3 as assessed prior to IPAS at SI1mV-PRE (50.0 ± 15.2 %).
Experiment 1c
The above experiments have shown that paired-TMS-pulse inhibition, when appropriately assessed, was unchanged following IPAS, while the response amplitude of MEPs evoked by single-pulse TMS was increased. The TMS-evoked MEP may be attenuated by stimulation of a contralateral afferent nerve (Tokimura et al. 2000). Within a small range of interstimulus intervals (19-25 ms), this attenuation has been shown to be mediated purely intracortically (Tokimura et al. 2000). Pharmacological evidence suggests that the interneuronal populations mediating the inhibition following somatosensory afferent stimulation are different from those mediating paired-TMS-pulse inhibition (Di Lazzaro et al. 2000b). Conceivably, the IPAS-induced increase of resting amplitudes could be related to a decrease of the intracortical inhibition that is evoked by somatosensory input. This question was addressed by analysing the ratio of the size of MEPs elicited during IPAS (i.e. by single-pulse TMS, conditioned by median nerve stimulation delivered 25 ms beforehand) and the size of their unconditioned controls. If afferent stimulation-induced intracortical inhibition were to be reduced by IPAS, this ratio should be increased after IPAS. IPAS did not induce an increase of the MEP amplitude ratio (Fig. 3). As with paired-TMS-pulse inhibition, it is possible that the inhibition induced by afferent input would be dependent on the magnitude of the test response. To account for this possibility, the test stimulus intensity was reduced to SI1mV-POST. The ratio of conditioned MEPs over unconditioned MEPs, elicited by SI1mV-POST, did not differ from that obtained with SI1mV-PRE. Statistical analysis of afferent input-induced inhibition using a one-way rmANOVA (condition 1, beginning of IPAS; condition 2, end of IPAS, SI1mV-PRE; condition 3, after IPAS, SI1mV-POST)) did not reveal differences between MEP ratios obtained for each of the three conditions (Fig. 3).
Figure 3. Short-latency inhibition by afferent input (Tokimura et al. 2000).
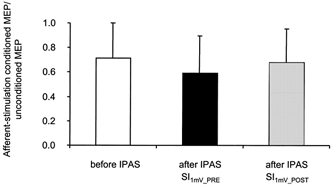
Short-latency inhibition before (□) and after (▪ and ░) IPAS. The size of MEPs conditioned by median nerve stimulation was normalized to the size of unconditioned MEPs elicited by magnetic pulses of the same intensity. MEPs were elicited with the test stimulus intensity set to SI1mV-PRE (□ and ▪), or SI1mV-POST (░). Data from 6 subjects. Data show means ± s.d.
Experiment 2: pharmacological intervention
The effects of IPAS on resting amplitude were tested in six subjects in four experimental sessions each, 2 for placebo, and 2 for dextromethorphan. Resting amplitudes were analyzed statistically using a three-way rmANOVA (block × drug × period, 2 × 2 × 2) with factors ‘block’ (1 and 2), ‘drug’ (placebo and dextromethorphan), and ‘period’ (pre and post). A significant effect was found for drug × period interaction (F = 50.24; P < 0.01), suggesting that the effect of period was dependent on the drug under which the experiment was performed. Both period (F = 9.00; P < 0.05) and drug (F = 19.20; P < 0.01) were found to be significant. Neither block (F = 0.30; P = 0.61), nor any of the other interaction terms was found to be significant. The results of blocks 1 and 2 were similar and were combined for further analysis. For all sessions, IPAS under the influence of placebo led to an increase in mean MEP amplitudes (pre, 1.1 ± 0.2 mV; post, 1.7 ± 0.4 mV; P < 0.001), on average, by 56 %. Under the influence of dextromethorphan, IPAS did not result in significant changes of resting amplitude; however, there was a trend for resting amplitudes to decrease (pre, 1.2 ± 0.2 mV; post, 1.0 ± 0.3 mV; P = 0.09; two-tailed paired t test; Fig. 4).
Figure 4. Effect of dextromethorphan on IPAS induced plasticity.
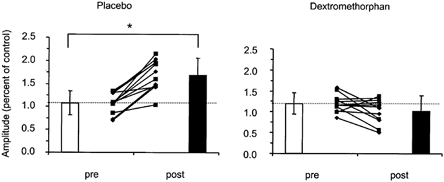
□, before IPAS; ▪, after IPAS. Data of two blocks of experiments, performed in 6 subjects each. Individual data from block 1 (▪) and block 2 (♦), are shown in the innermost part of each panel. Bars show means (± s.d.) of eight experimental sessions each. The ordinate of the right panel was scaled to match the control MEP size with that obtained in the placebo experiments, to facilitate comparison between the experiments. *Significant difference pre vs. post IPAS with placebo.
Experiment 3: intracortical inhibition during afferent stimulation
The question of which physiological effects were induced in the motor cortex by the afferent pulse, as one component of the IPAS protocol, was addressed in this experiment. Building on, and extending the experiments by Ridding & Rothwell (1999), paired-TMS-pulse inhibition was tested in the presence of an afferent stimulus while the magnitude of the test response was varied, by changing the intensity of the test stimulus. Results are summarized in Table 1.
Table 1.
Effect of afferent cortical input on paired-TMS-pulse inhibition

AFF: afferent median nerve stimulation at 300 % perceptual threshold, preceding the magnetic test pulse by 25 ms. CS+TS: conditioning shock at 70 % resting motor threshold followed after 3 ms by a suprathreshold magnetic test pulse at the intensity indicated in the column headings. RA: resting amplitude. Arrows indicate matching response sizes in the absence of a conditioning magnetic stimulus. For statistical evaluation, the entire set of CR3 values obtained in the absence of afferent stimulation (second row : CS+TS) were compared to the set of CR3 values obtained with afferent stimulation using test stimulus intensities producing test responses of the appropriate size (fourth row: AFF+CS+TS).
Using SI1mV, mean resting amplitudes were reduced to 35.2 ± 15.6 % of the control response. In the presence of an afferent stimulus, CR3 amounted to 105.4 ± 65.3 %, indicating that paired-TMS-pulse inhibition was completely abolished (n = 6 experiments). CR3 was additionally assessed at a test stimulus intensity producing a MEP response size of ≈0.6 mV, to match that obtained with afferent stimulation and SI1mV (n = 4 experiments). To achieve this, test stimulus intensity had to be lowered from SI1mV by 3 ± 1 % maximal stimulator output. At this test stimulus intensity, termed SI0.6mV, CR3 amounted to 42.9 ± 4.8 % of the control response, substantially less than 105.4 ± 65.3 %, obtained with conditioned SI1 mV in the presence of an afferent stimulus.
CR3 was assessed with the test stimulus intensity increased from SI1mV by 6 ± 1 % maximal stimulator output to produce a mean MEP response size of ≈1.0 mV in the presence of an afferent stimulus (SI1mV-AFF). With this test stimulus intensity and in the presence of an afferent stimulus, CR3 amounted to 65.1 ± 20.5 % of the control value.
For statistical evaluation, all CR3 values obtained in the presence of afferent stimulation were combined, and compared to the CR3 values obtained without afferent stimulation using test stimulus intensities producing test responses of the appropriate size (either 0.6 or 1 mV; t test, P < 0.02; Table 1).
Discussion
Excitability in the human motor system can be enhanced by a paired stimulation protocol (Stefan et al. 2000). Several lines of evidence suggest that IPAS-induced changes are generated at the level of the cortex (Stefan et al. 2000). Additionally, the increase of motor cortical excitability displays a number of remarkable properties: Rapid evolution, long duration, reversibility, topographical specificity, and timing dependency (requiring approximately synchronous activation of motor cortex output elements by each stimulation modality). Therefore, we have hypothesized that the increase of motor cortical excitability may represent associative LTP or a closely related phenomenon in the human motor cortex (Stefan et al. 2000). We will argue that the present findings provide strong support for this hypothesis.
Lack of evidence for lasting decrease in intracortical inhibition
Intracortical inhibition represents one of the principal candidate mechanisms underlying changes of cortical excitability (Jones, 1993; Donoghue et al. 1996). Following topical iontophoretic administration of the GABAA receptor antagonist bicuculline, receptive fields of neurons in the primary somatosensory cortex enlarge (Hicks & Dykes, 1983). In the rat primary motor cortex, movements that are normally exclusively represented in neighboring areas can be evoked by stimulation of adjacent areas after local inhibition has been blocked by bicuculline (Jacobs & Donoghue, 1991). These results suggest that pre-existing excitatory synapses onto local pyramidal neurons may become active when tonic inhibition impinging upon them is lifted. In principle, the increase in MEP amplitudes following IPAS could well be the consequence of a reduction in intracortical inhibition. Indeed, evidence for altered intracortical GABAergic inhibition underlying changes in cortical excitability has been found in other circumstances of human motor cortical plasticity (Chen et al. 1998; Liepert et al. 1998; Ziemann et al. 1998).
Intracortical inhibition has been probed in the present study by two paired stimulation protocols. The first one used a suprathreshold TMS pulse conditioned by a subthreshold conditioning pulse delivered 3 ms before (Kujirai et al. 1993). The second paired stimulation protocol used a suprathreshold TMS pulse conditioned by median nerve stimulation delivered 25 ms before (Tokimura et al. 2000).
There is evidence from multiple studies to suggest that paired-TMS-pulse inhibition (Kujirai et al. 1993) is dependent on the activity of inhibitory interneurons acting upon postsynaptic GABAA receptors (Ziemann et al. 1996; Hanajima et al. 1998; Werhahn et al. 1999; see, however, Boroojerdi et al. 2001). Pharmacological studies as well as a number of pathophysiological observations suggest that the inhibitory circuits that are accessible to neurophysiological testing by the paired-pulse TMS protocol are operational in modulating the excitability of the cortical output elements. Following IPAS we found paired-TMS-pulse inhibition to be decreased. At first sight this would support the concept of disinhibition as a mechanism underlying the IPAS-induced increase in cortical excitability. However, paired-TMS-pulse inhibition was not different from control values when the increased efficacy of the test stimulus after IPAS was taken into account. No difference in paired-TMS-pulse inhibition emerged when paired-TMS-pulse inhibition after IPAS was compared to a condition employing a similarly effective test stimulus intensity before IPAS (Expt 1a). In a complementary experiment, paired-TMS-pulse inhibition was unaltered when the amplitude of the test shock was adjusted to match the size of the unconditioned MEP amplitude following IPAS with that before IPAS (Expt 1b). Finally, there was no evidence for a change in the efficacy of the conditioning magnetic pulse (Expt 1b), and, therefore, no indication for a change in the recruitment of inhibitory interneuronal elements. Thus, the combined evidence from Expts 1a and b does not provide support for the concept that changes in the intracortical inhibition testable by paired TMS pulses underlie the excitability changes following IPAS. These findings are in excellent agreement with recent results obtained by Ridding & Taylor (2001). These authors repetitively paired a short train of afferent stimuli with TMS to induce lasting increases in excitability of the cortical representation of the first dorsal interosseus muscle similar to those obtained in the APB in the present study. Paired-TMS pulse inhibition was tested after intervention with the TMS pulse adjusted to evoke a MEP response of the same magnitude as before intervention. This condition, which used the same experimental design principle as that employed in Expt 1b of the present study, did not reveal any evidence for an alteration of paired-TMS-pulse inhibition by interventional stimulation. Our results extend the findings of Ridding and Taylor (2001) by showing that paired-TMS-pulse inhibition was unchanged at any relevant point on the function relating the efficacy of either the test or the conditioning stimulus to inhibition.
Intracortical inhibition may also be probed when a suprathreshold TMS pulse is conditioned by an afferent stimulation (Tokimura et al. 2000). The inhibitory interneurons subserving the inhibition that is produced by afferent input at short latencies, probably differ from those testable by double-shock stimulation, based on their different pharmacological (Di Lazzaro et al. 2000b) and physiological (Ridding & Rothwell, 1999; Tokimura et al. 2000) profile. The intracortical inhibition induced by somatosensory afferent input is far less well characterized than the paired-TMS-pulse inhibition. However, it is important to note that we found no indication for a decrease of this type of inhibition after IPAS (Experiment 1c).
Together, these findings provide evidence that IPAS-induced plasticity does not rely on GABAergic-dependent modulation of cortical excitability as a primary mechanism, and, therefore, probably relies on other cellular mechanisms.
Transient cortical disinhibition following afferent stimulation - a prerequisite for induction of associative plasticity?
LTP can be induced in cortical slices obtained from mammalian motor cortex when stimulation of ‘horizontal’ intracortical fibres is paired with stimulation of ‘vertical’, i.e. cortico-cortical or thalamo-cortical, afferents (Iriki et al. 1991; Hess & Donoghue, 1994; Hess et al. 1996). By contrast, stimulation of neither horizontal (Hess et al. 1996) nor vertical (Iriki et al. 1991; Hess et al. 1996) pathways alone is sufficient to induce LTP when applied at low frequencies. Importantly, the failure of exclusive horizontal pathway stimulation to induce LTP can be overcome when local intracortical inhibition is reduced pharmacologically, e.g. by iontophoretic application of the GABAA receptor antagonist bicuculline (Hess et al. 1996). Interestingly, intracortical inhibition is also reduced in cortical slices by stimulating afferent, i.e. thalamo-cortical and cortico-cortical pathways (Hess et al. 1996). Therefore, the evidence derived from cortical slice experiments suggests that stimulation of afferents enhances the excitability of the postsynaptic neuron, in addition to providing a synchronous signal to the postsynaptic target of horizontal pathways. We found paired-TMS-pulse inhibition to be reduced following an afferent pulse to the cortex. In the absence of single-neuron recordings we have no direct way of proving that this phenomenon reflects reduction of intracortical inhibition, rather than enhanced facilitation of excitatory input to pyramidal cells in the presence of an afferent stimulus. However, in view of the fact that single-pulse TMS evoked MEPs are attenuated by an afferent stimulus it seems unlikely that the same afferent stimulus would facilitate excitatory circuits contributing to the generation of an MEP exclusively in the presence of a conditioning TMS pulse. Therefore, we consider the most parsimonous explanation of our findings to be that cortical disinhibition is induced by stimulation of afferent nerve fibres. This conclusion is in agreement with previous findings both from this laboratory and from another group (Hess et al. 1999; Ridding & Rothwell, 1999) showing that electric stimulation of the median nerve is followed by a short-lived (duration of milliseconds) reduction of intracortical inhibition. Thus, the TMS pulse applied at 25 ms following the peripheral nerve stimulation operates on a motor cortex transiently disinhibited by the afferent volley as far as paired-TMS-pulse inhibition is concerned. Provided that paired-TMS-pulse inhibition is related to tonic inhibition under natural conditions, our observation bears on the interpretation of several studies which have shown that cortical plasticity may be blocked in the presence of substances enhancing postsynaptic GABA receptor activity. For example, training repetitive thumb movements in a stereotyped direction changes, temporarily, the direction of TMS-evoked thumb movements into the trained direction (Classen et al. 1998). This kind of cortical plasticity was prevented when training was performed under the influence of lorazepam, a drug that enhances GABAA receptor function (Bütefisch et al. 2000). The present findings raise the possibility that substances like benzodiazepines prevent cortical reorganization by impairing the ability of cortical afferents to induce a transient disinhibition. Strong local inhibition will, therefore, decrease the ability of local neurons to generate LTP and, thus, represents an important factor contributing to maintenance of cortical organization (homoiostasis). This supporting role of GABAA receptor mediated inhibition may be more important than a role for disinhibition as a carrier mechanism of cortical plasticity in humans. The above speculations also imply that the cortical interneurons tested with a paired-TMS pulse protocol (Kujirai et al. 1993) are much more involved in maintaining the general homoiostasis of cortical organization and modulation of synaptic strength in cortical connections targeting the pyramidal output cells than are the interneurons underlying the intracortical inhibition produced by an afferent input.
Role of NMDA receptors
Dextromethorphan blocked the effect of IPAS on MEP amplitudes. Because dextromethorphan blocks NMDA receptors non-competitively (Wong et al. 1988) this finding suggests that activation of NMDA receptors is a necessary step in IPAS-induced plasticity. Associative LTP in the motor cortex depends on the activation of NMDA receptors (Castro-Alamancos et al. 1995; Hess et al. 1996). Dextromethorphan has also been shown to block the induction of LTP in vitro (Krug et al. 1993), presumably as a consequence of its NMDA receptor blocking properties. Interestingly, IPAS in the presence of dextromethorphan produced a slight decrease of resting amplitudes, although the decrease did not quite reach significance in the two-tailed t test applied. This observation is reminiscent of the effects of 2-amino-5-phosphonovaleric acid (AP5), an NMDA receptor antagonist on the efficacy of protocols generating LTP in neocortical slices taken from experimental animals. AP5 reduces the probability of inducing LTP and, under some circumstances, can promote induction of LTD instead (Hirsch & Crepel, 1991).
Together with other properties of our protocol for inducing cortical plasticity, as described above, the results of the present paper further support the view that associative LTP-like mechanisms are involved in IPAS-induced plasticity in the human motor cortex.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft Cl 95/3-1 and by the FORUN-research program of the University of Rostock. L.G.C. was supported by a grant from the Humboldt-Foundation.
References
- Apland JP, Braitman DJ. Effects of non-opioid antitussives on epileptiform activity and NMDA responses in hippocampal and olfactory cortex slices. Brain Research. 1990;529:277–285. doi: 10.1016/0006-8993(90)90838-3. [DOI] [PubMed] [Google Scholar]
- Aroniadou VA, Keller A. Mechanisms of LTP induction in rat motor cortex in vitro. Cerebral Cortex. 1995;5:353–362. doi: 10.1093/cercor/5.4.353. [DOI] [PubMed] [Google Scholar]
- Asanuma H, Pavlides C. Neurobiological basis of motor learning in mammals. NeuroReport. 1997;8:I–VI. [PubMed] [Google Scholar]
- Baranyi A, Feher O. Synaptic facilitation requires paired activation of convergent pathways in the neocortex. Nature. 1981;290:413–415. doi: 10.1038/290413a0. [DOI] [PubMed] [Google Scholar]
- Baranyi A, Szente MB. Long-lasting potentiation of synaptic transmission requires postsynaptic modifications in the neocortex. Brain Research. 1987;423:378–384. doi: 10.1016/0006-8993(87)90867-5. [DOI] [PubMed] [Google Scholar]
- Boroojerdi B, Battaglia F, Muellbacher W, Cohen LG. Mechanisms influencing stimulus-response properties of the human corticospinal system. Clinical Neurophysiology. 2001;112:931–937. doi: 10.1016/s1388-2457(01)00523-5. [DOI] [PubMed] [Google Scholar]
- Buonomano DV, Merzenich MM. Cortical plasticity: from synapses to maps. Annual Review of Neuroscience. 1998;21:149–186. doi: 10.1146/annurev.neuro.21.1.149. [DOI] [PubMed] [Google Scholar]
- Bütefisch CM, Davis BC, Wise SP, Sawaki L, Kopylev L, Classen J, Cohen LG. Mechanisms of use-dependent plasticity in the human motor cortex. Proceedings of the National Academy of Sciences of the USA. 2000;97:3661–3665. doi: 10.1073/pnas.050350297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Donoghue JP, Connors BW. Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. Journal of Neuroscience. 1995;15:5324–5333. doi: 10.1523/JNEUROSCI.15-07-05324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Corwell B, Yaseen Z, Hallett M, Cohen LG. Mechanisms of cortical reorganization in lower-limb amputees. Journal of Neuroscience. 1998;18:3443–3450. doi: 10.1523/JNEUROSCI.18-09-03443.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen J, Liepert A, Wise SP, Hallett M, Cohen LG. Rapid plasticity of human cortical movement representation induced by practice. Journal of Neurophysiology. 1998;79:1117–1123. doi: 10.1152/jn.1998.79.2.1117. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero A, Meglio M, Cioni B, Toniali P, Rothwell JC. Direct demonstration of the effect of lorazepam on the excitability of the human motor cortex. Clinical Neurophysiology. 2000a;111:794–799. doi: 10.1016/s1388-2457(99)00314-4. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero A, Profice P, Pennisi MA, Di Giovanni S, Zito G, Tonali P, Rothwell JC. Muscarinic receptor blockade has differential effects on the excitability of intracortical circuits in the human motor cortex. Experimental Brain Research. 2000b;135:455–461. doi: 10.1007/s002210000543. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A, MaONEZZ P, Tonali P, Rothwell JC. Magnetic transcranial stimulation at intensities below active motor threshold activates intracortical inhibitory circuits. Experimental Brain Research. 1998;119:265–268. doi: 10.1007/s002210050341. [DOI] [PubMed] [Google Scholar]
- Donoghue JP, Hess G, Sanes JN. Substrates and mechanisms for learning in motor cortex. In: Bloedel J, Ebner T, Wise SP, editors. Acquisition of Motor Behavior in Vertebrates. Cambridge, MA, USA: MIT Press; 1996. pp. 363–386. [Google Scholar]
- Hanajima R, Ugawa Y, Terao Y, Sakai K, Furubayashi T, Machii K, Kanazawa I. Paired-pulse magnetic stimulation of the human motor cortex: differences among I waves. Journal of Physiology. 1998;509:607–618. doi: 10.1111/j.1469-7793.1998.607bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess A, Kunesch E, Classen J, Hoeppner J, Stefan K, Benecke R. Task-specific afferent modulation of intracortical inhibition. Experimental Brain Research. 1999;124:321–330. doi: 10.1007/s002210050629. [DOI] [PubMed] [Google Scholar]
- Hess G, Aizenman CD, Donoghue JP. Conditions for the induction of long-term potentiation in layer II/III horizontal connections of the rat motor cortex. Journal of Neurophysiology. 1996;75:1765–1778. doi: 10.1152/jn.1996.75.5.1765. [DOI] [PubMed] [Google Scholar]
- Hess G, Donoghue JP. Long-term potentiation of horizontal connections provides a mechanism to reorganize cortical motor maps. Journal of Neurophysiology. 1994;71:2543–2547. doi: 10.1152/jn.1994.71.6.2543. [DOI] [PubMed] [Google Scholar]
- Hicks TP, Dykes RW. Receptive field size for certain neurons in primary somatosensory cortex is determined by GABA-mediated intracortical inhibition. Brain Research. 1983;274:160–164. doi: 10.1016/0006-8993(83)90533-4. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Crepel F. Blockade of NMDA receptors unmasks a long-term depression in synaptic efficacy in rat prefrontal neurons in vitro. Experimental Brain Research. 1991;85:621–624. doi: 10.1007/BF00231747. [DOI] [PubMed] [Google Scholar]
- Iriki A, Pavlides C, Keller A, Asanuma H. Long-term potentiation of thalamic input to the motor cortex induced by coactivation of thalamocortical and corticocortical afferents. Journal of Neurophysiology. 1991;65:1435–1441. doi: 10.1152/jn.1991.65.6.1435. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Donoghue JP. Reshaping the cortical motor map by unmasking latent intracortical connections. Science. 1991;251:944–947. doi: 10.1126/science.2000496. [DOI] [PubMed] [Google Scholar]
- Jones EG. GABAergic neurons and their role in cortical plasticity in primates. Cerebral Cortex. 1993;3:361–372. doi: 10.1093/cercor/3.5.361-a. [DOI] [PubMed] [Google Scholar]
- Krug M, Matthies R, Wagner M, Brodemann R. Non-opioid antitussives and methadone differentially influence hippocampal long-term potentiation in freely moving rats. European Journal of Pharmacology. 1993;231:355–361. doi: 10.1016/0014-2999(93)90110-4. [DOI] [PubMed] [Google Scholar]
- Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, Wroe S, Asselman P, Marsden CD. Corticocortical inhibition in human motor cortex. Journal of Physiology. 1993;471:501–519. doi: 10.1113/jphysiol.1993.sp019912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepert J, Classen J, Cohen LG, Hallett M. Task-dependent changes of intracortical inhibition. Experimental Brain Research. 1998;118:421–426. doi: 10.1007/s002210050296. [DOI] [PubMed] [Google Scholar]
- Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia. 1971;9:97–113. doi: 10.1016/0028-3932(71)90067-4. [DOI] [PubMed] [Google Scholar]
- Ridding MC, Rothwell JC. Afferent input and cortical organisation: a study with magnetic stimulation. Experimental Brain Research. 1999;126:536–544. doi: 10.1007/s002210050762. [DOI] [PubMed] [Google Scholar]
- Ridding MC, Taylor JL. Mechanisms of motor-evoked potential facilitation following prolonged dual peripheral and central stimulation in humans. Journal of Physiology. 2001;537:623–631. doi: 10.1111/j.1469-7793.2001.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioult-Pedotti MS, Friedman D, Donoghue JP. Learning-induced LTP in neocortex. Science. 2000;290:533–536. doi: 10.1126/science.290.5491.533. [DOI] [PubMed] [Google Scholar]
- Rossini PM, Barker AT, Berardelli A, Caramia MD, Caruso G, Cracco RQ, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord and roots: basic principles and procedures for routine clinical application. Report of an IFCN committee. Electroencephalography and Clinical Neurophysiology. 1994;91:79–92. doi: 10.1016/0013-4694(94)90029-9. [DOI] [PubMed] [Google Scholar]
- Sanes JN, Donoghue JP. Plasticity and primary motor cortex. Annual Review of Neuroscience. 2000;23:393–415. doi: 10.1146/annurev.neuro.23.1.393. [DOI] [PubMed] [Google Scholar]
- Silvasti M, Karttunen P, Tukiainen H, Kokkonen P, Hanninen U, Nykanen S. Pharmacokinetics of dextromethorphan and dextrorphan: a single dose comparison of three preparations in human volunteers. International Journal of Clinical Pharmacology and Therpeutic Toxicology. 1987;25:493–497. [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Mechanisms of plasticity induced by paired associative stimulation in the human motor cortex. Society for Neuroscience Abstracts. 1999;25:787. [Google Scholar]
- Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123:572–584. doi: 10.1093/brain/123.3.572. [DOI] [PubMed] [Google Scholar]
- Steinberg G, Bell T, Yenari M. Dose escalation safety and tolerance study of the N-methyl-D aspartate antagonist dextromethorphan in neurosurgery patients. Journal of Neurosurgery. 1996;84:860–866. doi: 10.3171/jns.1996.84.5.0860. [DOI] [PubMed] [Google Scholar]
- Tokimura H, Di LaAROZZ V, Tokimura Y, Oliviero A, Profice P, Insola A, MaONEZZ P, Tonali P, RothwelL JC. Short latency inhibition of human hand motor cortex by somatosensory input from the hand. Journal of Physiology. 2000;523:503–513. doi: 10.1111/j.1469-7793.2000.t01-1-00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werhahn KJ, Kunesch E, Noachtar S, Benecke R, Classen J. Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. Journal of Physiology. 1999;517:591–597. doi: 10.1111/j.1469-7793.1999.0591t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BY, Coulter DA, Choi DW, Prince DA. Dextrorphan and dextromethorphan, common antitussives, are antiepileptic and antagonize N-methyl-D-aspartate in brain slices. Neuroscience Letters. 1988;85:261–266. doi: 10.1016/0304-3940(88)90362-x. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Hallett M, Cohen LG. Mechanisms of deafferentation-induced plasticity in human motor cortex. Journal of Neuroscience. 1998;18:7000–7007. doi: 10.1523/JNEUROSCI.18-17-07000.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann U, LÖnecker S, Steinhoff BJ, Paulus W. The effect of lorazepam on the motor cortical excitability in man. Experimental Brain Research. 1996;109:127–135. doi: 10.1007/BF00228633. [DOI] [PubMed] [Google Scholar]