

Abstract
This study tested the hypothesis that store-operated channels (SOCs) exist as a discrete population of Ca2+ channels activated by depletion of intracellular Ca2+ stores in cerebral arteriolar smooth muscle cells and explored their direct contractile function. Using the Ca2+ indicator fura-PE3 it was observed that depletion of sarcoplasmic reticulum (SR) Ca2+ by inhibition of SR Ca2+-ATPase (SERCA) led to sustained elevation of [Ca2+]i that depended on extracellular Ca2+ and slightly enhanced Mn2+ entry. Enhanced background Ca2+ influx did not explain the raised [Ca2+]i in response to SERCA inhibitors because it had marked gadolinium (Gd3+) sensitivity, which background pathways did not. Effects were not secondary to changes in membrane potential. Thus SR Ca2+ depletion activated SOCs. Strikingly, SOC-mediated Ca2+ influx did not evoke constriction of the arterioles, which were in a resting state. This was despite the fura-PE3-indicated [Ca2+]i rise being greater than that evoked by 20 mm [K+]o (which did cause constriction). Release of endothelial vasodilators did not explain the absence of SOC-mediated constriction, nor did a change in Ca2+ sensitivity of the contractile proteins. We suggest SOCs are a discrete subset of Ca2+ channels allowing Ca2+ influx into a ‘non-contractile’ compartment in cerebral arteriolar smooth muscle cells.
The L-type voltage-gated Ca2+ channel is a major pathway for Ca2+ entry in smooth muscle cells of most blood vessels, including arterioles in the cerebral circulation (Brandt et al. 1981; Rosenblum, 1984; Takayasu et al. 1988; Hill et al. 2001). Ca2+ entry through this channel couples closely with the contractile state of the smooth muscle cells and is linked with the regulation of gene expression and refilling of depleted SR (McCarron et al. 2000; Stevenson et al. 2001). Many blood vessels, including cerebral arterioles, also have contractile responses that are resistant to L-type Ca2+ channel blockers (Haws & Heistad, 1984; Uski et al. 1984; Edwards & Trizna, 1990; Pierre & Davenport, 1999) and there are several other less well-defined types of Ca2+ channel in vascular smooth muscle. Directly relevant to this study is the hypothesis that there is a specialised subset of Ca2+ channels that open in response to a signal from Ca2+-depleted SR. These are referred to as SOCs, or CCE channels (capacitative Ca2+ entry channels). 45Ca2+ flux experiments on rabbit ear artery first indicated the existence of such a pathway in response to SR depletion induced by noradrenaline (Casteels & Droogmans, 1981). The pathway was resistant to L-type Ca2+ channel antagonists such as methoxyverapamil (D600) but inhibited by manganese ions (Mn2+). More recent studies have employed SERCA inhibitors to deplete SR Ca2+, circumventing signalling mechanisms associated with membrane receptors and strengthening the case for a ‘receptor-independent’ link between SR Ca2+ content and SOCs (Fellner & Arendshorst, 1999; Loutzenhiser & Loutzenhiser, 2000; Trepakova et al. 2001). A complication of experiments involving SERCA inhibitors is that SERCA is a critical element of SR function and the specialised superficial buffer barrier of smooth muscle cells (van Breemen et al. 1985). Sustained [Ca2+]i elevation caused by SERCA inhibitors may not indicate SOC activation, but instead reduced buffering of background Ca2+ entry. Alternatively, it may result from enhanced background Ca2+ entry in response to store depletion, rather than activation of a discrete subset of specialised Ca2+ channels (i.e. SOCs).
Although in many experiments L-type Ca2+ channel antagonists inhibit contraction in cerebral arterioles there is evidence that smooth muscle cells in these vessels also have SOCs (Guibert & Beech, 1999). We recently showed that TRPC1 is a membrane protein in these cells and this protein is associated with SOC-like activity in some cell types (Li & Montell, 2000; Xu & Beech, 2001; Brough et al. 2001). From the involvement of TRP protein it is interesting to speculate that there is a specialised and discrete signalling complex linked with SOCs in vascular smooth muscle. This is indicated by the ‘signalplex’ of Drosophila TRP and by the co-immunoprecipitation of TRPC1 with caveolin and inositol 1,4,5-trisphosphate receptor (Liu et al. 2000; Lockwith et al. 2000; Rosado & Sage, 2001). If this is true in arterioles Ca2+ entry through SOCs may have a specialised function that is not directly linked to the contractile state of the cells or is simply involved in a separate cellular function. We first aimed to further explore the hypothesis that SOCs exist in native arteriolar smooth muscle cells as a discrete subset of Ca2+ channels linked to SR Ca2+ content. Having shown this, we explored the relationship between SOC-mediated Ca2+ entry and contraction. The data support the idea that specialised SOC proteins allow Ca2+ entry into a subcellular Ca2+ compartment in arteriolar smooth muscle cells.
Methods
Male Dutch dwarf rabbits (1-1.5 kg) were killed by an intravenous overdose of 70 mg kg−1 sodium pentobarbitone in accordance with the Code of Practice, UK Animals Scientific Procedures Act 1986. The brain was placed in ice-cold oxygenated Hanks’ solution and fragments of pial membrane dissected from across the cortical surface and incubated in Hanks’ solution containing 0.032 mg ml−1 protease (Sigma) and 0.2 mg ml−1 collagenase (type 1A, Sigma) for 10 min at 37 °C. The mixture was placed at 4 °C for 15 min and mechanically agitated to isolate fragments of arterioles. After centrifugation (1000 r.p.m.) for 5 min the supernatant was replaced with fresh Hanks’ solution. Arterioles were resuspended and dropped onto polylysine-coated coverslips and stored at 4 °C. Experiments were performed within 10 h. Arteriole fragments used in recordings had an external diameter of < 45 μm, and lacked visible adventitia or endothelial cells (Cheong et al. 2001).
For Ca2+ imaging experiments, isolated arterioles were pre-incubated with 1 μM fura-PE3 AM (acetoxymethyl ester form of fura-PE3; Vorndran et al. 1995) at 30–37 °C for 1 h in standard bath solution. This was followed by a 0.5-1 h wash period in standard bath solution at room temperature. The fura-PE3 AM incubation and wash periods, and all Ca2+ imaging experiments were in the presence of 10 μM methoxyverapamil (D600), unless [Ca2+]i elevation in response to raised [K+]o was studied. D600 (10 μM) completely blocks voltage-dependent Ca2+ entry in these arterioles (Guibert & Beech, 1999). In elevated [K+]o experiments, wortmannin (1 μM) was included in the fura-PE3 incubation solution to prevent contraction without effect on voltage-gated Ca2+ channels (Unno et al. 1998). Pre-incubation with 0.1 or 1 μM thapsigargin (TG) occurred for 0.5-1.5 h, and during the fura-PE3 loading and washing periods when applicable.
Fura-PE3 fluorescence was observed with an inverted epifluorescence microscope (Nikon TMD, Japan; or Zeiss Axiovert, Germany). A xenon arc lamp provided excitation light, the wavelength of which was selected by a monochromator (Till Photonics, Germany). Emission was collected via a 510 nm filter and sampled by a cooled CCD camera (Hamamatsu, Japan), producing images at 12-bit (Orca-ER camera) or 14-bit (H4880-82 camera) resolution. Every 10 or 20 s, images were sampled in pairs for the two excitation wavelengths (340 or 345, and 380 nm). With the Zeiss set-up we used 340 nm excitation, and with the Nikon set-up 345 nm because of different transmission and data capture efficiencies. Comparisons of absolute ratio changes were only made for the same recording set-up. Images were analysed offline using regions of interest (ROIs), which selected parts of the image frame corresponding to individual smooth muscle cells within arterioles. Three ROIs were also selected that were distant from the arteriole and used for background subtraction. [Ca2+]i is expressed as the ratio of the background-subtracted emission intensities for the two excitation wavelengths (340 or 345 nm)/(380 nm). When Mn2+ was quenching fura-PE3 fluorescence, excitation occurred at 360 nm. In some experiments (Fig. 1 and Fig. 2) spontaneous run-up/down of fura-PE3 ratio signals was subtracted according to a linear extrapolation. Imaging was controlled by Openlab 2 software (Image Processing & Vision Company Ltd, UK). Analysis and image presentation utilised Origin, Excel and Powerpoint software.
Figure 1. Responses of cerebral arteriolar smooth muscle cells to SR Ca2+ depletion.
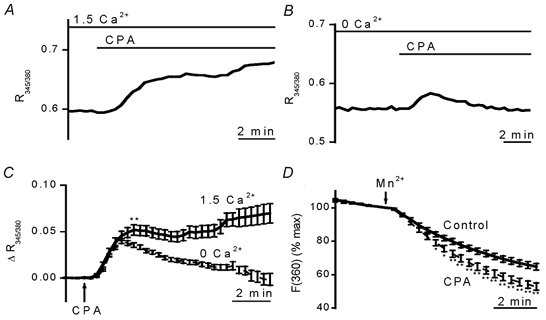
D600 (10 μM) was present in all experiments. A-C, SERCA inhibition with 10 μM CPA caused elevation of [Ca2+]i that depended on extracellular Ca2+. [Ca2+]i is given as the ratio of 345 and 380 nm fura-PE3 signals (R345/380). Example experiments in standard (1.5 mm Ca2+) bath solution (A) and Ca2+-free bath solution containing 0.4 mm EGTA (B). C, summary of data from experiments as shown in A and B. Data are mean ± s.e.m. changes in fura-PE3 ratio. For each data point, n ≥ 32 (total N = 14) and 41 (total N = 15) for 1.5 mm Ca2+ and Ca2+-free groups, respectively. P < 0.001 for the data point marked ** and all subsequent data points. D, SR Ca2+ depletion slightly enhanced Mn2+ influx. Mn2+ quenching of fura-PE3 (excited at 360 nm) was increased by 10 μM CPA. Mn2+ was applied at 0.5 mm in Ca2+-free solution without EGTA. Points are mean ± s.e.m. data normalised to the fluorescence intensity before addition of Mn2+. Control group, n/N = 65/13. CPA group, n/N = 45/9. * P < 0.05, ** P < 0.01.
Figure 2. SR Ca2+ depletion induces Gd3+ sensitivity.
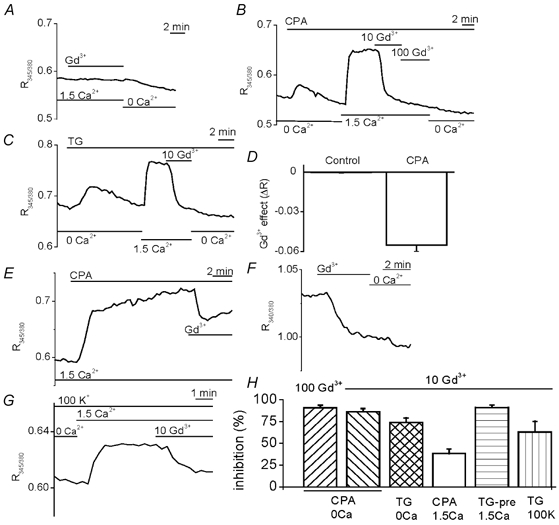
All experiments included 10 μM D600. A-F, the extracellular solution was either standard (1.5 mm Ca2+) or Ca2+-free (0.4 mm EGTA). A, no effect of 100 μM Gd3+ in control conditions. Ca2+-free solution reduced [Ca2+]i. B and C, effects of 10 and 100 μM Gd3+ after SR Ca2+ depletion with 10 μM CPA (B) or 0.1 μM TG (C) in Ca2+-free solution. D, mean ± s.e.m. 100 μM Gd3+-induced reduction of fura-PE3 ratio in the SR Ca2+-depleted group (as in B, n/N = 20/6) but not in the control (as in A, n/N = 43/10). E, 10 μM CPA-induced sensitivity to 10 μM Gd3+ without any exposure to Ca2+-free solution. F, 10 μM Gd3+ sensitivity, followed by Ca2+-free bath solution, after 1.5 h pre-treatment with 1 μM TG in standard bath solution. G, in a 0.1 μM TG-pre-treated arteriole, effects of 1.5 mm Ca2+ and 10 μM Gd3+ in the continuous presence of 100 mm K+ solution. H, mean ± s.e.m. effects of Gd3+ (10 or 100 μM) as a percentage relative to the fura-PE3 ratio before application of CPA (E) or Ca2+-free bath solution. CPA/0 Ca group: as shown in B for 10 Gd3+ (n/N = 10/3) and 100 Gd3+ (n/N = 20/6). TG/0 Ca group: as shown in C for 10 Gd3+ (n/N = 15/6). CPA/1.5 Ca group: as shown in E for 10 Gd3+ (n/N = 32/8). TG-pre/1.5 Ca group: as shown in F for 10 Gd3+ (n/N = 40/9). TG/100 K group: as shown in G for 10 Gd3+ (n/N = 13/3).
For diameter measurements arterioles were placed in a culture dish on the stage of an inverted trinocular microscope (Nikon TMS, Japan) with a CCD camera (Sony, Japan). External vessel diameter was measured using a video edge-detection system (Living Systems Instrumentation, Inc., Burlington, VT, USA) and signals were captured by an A/D converter (Picolog software, Pico Technology, Cambridge, UK) and stored on computer.
Membrane potential measurements were made using whole-cell patch recording applied to smooth muscle cells in arterioles (Cheong et al. 2001). The patch-clamp amplifier was an Axopatch 200A (Axon Instruments Inc., USA) and command- and data-sampling protocols were controlled by pCLAMP 6 (Axon Instruments). Patch pipettes were made from borosilicate glass tubing (Clark Electromedical Instruments, UK) and had resistances of 2–3 MΩ.
Agents were applied to arterioles using a continuous bath perfusion system with a flow rate of 4 ml min−1. All experiments were at room temperature except for the intact pial membrane experiments, which were at 37 °C (e.g. Fig. 3L).
Figure 3. Lack of a SOC-associated contractile response.
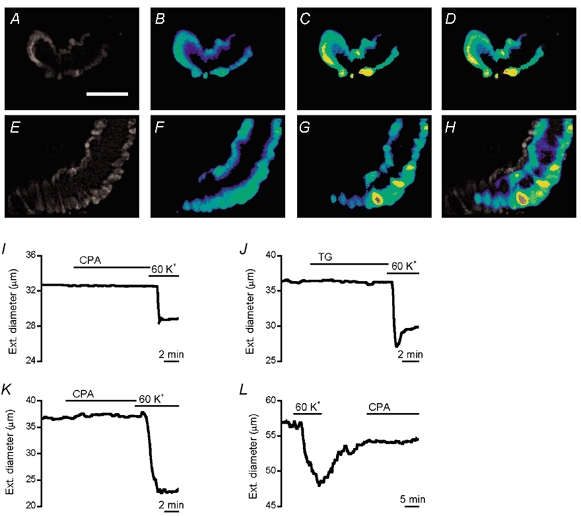
Images of fura-PE3 fluorescence from an isolated arteriole exposed to 10 μM CPA in the presence of 10 μM D600 (A-D) and an arteriole exposed to 60 mm K+ in the absence of D600 (E-H). Images A and E show fluorescence with excitation at 345 nm. The other images are ratios of the 345 nm/380 nm signals on a rainbow scale (blue, low [Ca2+]; red, high [Ca2+]). Images A and B were in the absence of extracellular Ca2+ and image C after the addition of 1.5 mm Ca2+. Images E and F were in standard bath solution and image G after addition of 60 mm K+. Image D is a merger of images A and C, and H a merger of E and G. Images A-D are for the experiment in Fig. 2B. The calibration bar in A is 40 μm and applies to all images. I-L, D600 was excluded. I-K, external diameters of isolated arterioles in standard bath solution at room temperature. In K, standard bath solution contained 0.3 mm l-NAME, 10 μM indomethacin, 100 nm apamin and 100 nm charbydotoxin. L, external diameter of an arteriole in intact pial membrane bathed in artificial CSF containing 0.3 mm l-NAME at 37 °C. There was bath application of 10 μM CPA, 1 μM TG, or 60 mm K+.
Hanks’ solution contained (mm): NaCl 137, NaH2PO4 0.34, KCl 5.4, K2HPO4 0.44, d-glucose 8, N-[2-hydroxyethyl]piperazine-N‘-2-ethanesulfonic acid (Hepes) 5, CaCl2 0.01. Standard bath solution contained (mm): NaCl 130, KCl 5, d-glucose 8, Hepes 10, MgCl2 1.2, CaCl2 1.5. Ca2+-free solution was standard bath solution in which 0.4 mm EGTA replaced the CaCl2. Artificial CSF contained (mm): NaCl 125, KCl 1.72, NaHCO3 24, MgSO4 1.74, KH2PO4 1.17, d-glucose 5.35, CaCl2 2.47, EDTA 0.023. When the extracellular K+ concentration was raised, the NaCl concentration in the bath solution was reduced by the equimolar amount. For membrane potential measurements, the patch pipette solution was (mm): KCl 130, NaCl 5, Hepes 10, EGTA 0.1, Na2ATP 3, MgCl2 2 (pH 7.4). All solutions except artificial CSF were titrated to pH 7.4 with the hydroxide of the dominant cation. Artificial CSF was bubbled with 5 % CO2 and 95 % O2.
In β-escin permeabilisation experiments, arterioles were washed in Ca2+-free solution (see above), incubated for 10 min in β-escin solution, 10 min in 10-EGTA solution without β-escin, and then in 10-EGTA solution containing increasing concentrations of added Ca2+ (5 min for each concentration). The β-escin and 10-EGTA solutions contained (mm): MgCl2 5, Na2ATP 5, NaGTP 0.1, creatine phosphate 5, Hepes 20 and leupeptin 0.001. The β-escin solution also contained (mm): β-escin 0.01, NaCl 25, KCl 95 and EGTA 2. The 10-EGTA solution also contained (mm): KCl 110, EGTA 10, FCCP 0.001 and ionomycin 0.0005. The concentrations of CaCl2 (mm) added to 10-EGTA solution (calculated free ionised Ca2+ concentrations are in parentheses, in μM units): 1.201 (0.03), 3.124 (0.1), 5.764 (0.3), 8.197 (1), 9.335 (3), 9.857 (10) and 10.132 (30). All solutions were titrated to a final pH of 7.1 with KOH. Ion concentrations in the multiple equilibria were calculated using EQCAL software (Biosoft, UK). CPA application began with the β-escin solution, and TG with 10-EGTA solution; both remained present throughout. Control and test (CPA or TG) experiments were carried out alternately. Data sets were paired if the control occurred immediately before or after the test experiment.
Data sets are expressed as mean ± s.e.m. In patch-clamp and vessel diameter experiments, n was the number of arterioles from which measurements were made. In imaging experiments, n was the number (3-5) of cells (ROIs) from which measurements were made. Separate cells/ROIs from one arteriole may have some interdependence because of gap junction connections and haze in the images and so we also give the number of arterioles (N). Statistical comparisons between two groups used n values and Student's unpaired t tests in which a significant difference was assumed if P < 0.05. Some data sets were fitted with the Hill equation (Origin software, USA).
General salts were from BDH or Sigma. Adenosine 5′-triphosphate (ATP), apamin, carbonyl cyanide 4-trifluoromethoxyphenylhydrazone (FCCP), charybdotoxin, collagenase, creatine phosphate, β-escin, guanosine 5′-triphosphate (GTP), guanosine 5′-[γ-thio]triphosphate (GTP-γ-S), indomethacin, ionomycin, leupeptin, methoxyverapamil (D600), Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME), protease and thapsigargin (TG) were from Sigma. Cyclopiazonic acid (CPA) was from Affiniti Research Products Limited (UK) or Tocris Cookson Ltd (UK). Wortmannin was from Tocris Cookson Ltd (UK). Fura-PE3 AM was from Calbiochem. Thapsigargin, CPA and fura-PE3 AM were made up as stock solutions in 100 % dimethyl sulfoxide (DMSO) and the final DMSO concentration was < 0.2 %. Other chemicals were prepared in water/salt solution.
Results
In the continuous presence of extracellular Ca2+, application of CPA, a selective SERCA inhibitor (Seidler et al. 1989; Luoet al. 2000), evoked a sustained rise in [Ca2+]i (Fig. 1A and C). In the absence of extracellular Ca2+, CPA evoked only a transient rise in [Ca2+]i (Fig. 1B and C). The transient rise in [Ca2+]i presumably reflects Ca2+ leakage into the cytoplasm from the SR.
Does the sustained rise in [Ca2+]i occur because there is reduced buffering of passive Ca2+ entry via background Ca2+ channels, or CPA-induced hyperpolarisation enhancing passive Ca2+ entry? The latter was ruled out by whole-cell recordings of membrane potential in standard bath solution containing D600 (as for Ca2+ imaging experiments). In these experiments the initial resting membrane potential was quite depolarised (-27.2 ± 2.5 mV, n = 8) (cf. Guibert & Beech, 1999) and we presume this was due to block of voltage-gated K+ channels by D600 (Terada et al. 1987; Cheong et al. 2001). Nevertheless, 10 min exposures to 10 μM CPA had no significant effect on the membrane potential (-2.9 ± 3.0 mV change, n = 8). Two types of experiments were used to distinguish SOC activation from reduced buffering of passive Ca2+ entry. First, if there is extra channel activity following store depletion CPA might stimulate Mn2+ entry, which will quench fura-PE3 fluorescence. Mn2+ was applied in Ca2+-free solution to avoid competition between the ions or a sustained elevation of [Ca2+]i in response to CPA. There was substantial background entry of Mn2+ (Fig. 1D), which also occurred when Mn2+ was applied in Ca2+-containing solution (data not shown). Mn2+ influx was significantly greater in CPA-treated arterioles, although the effect was small (Fig. 1D).
The above experiments suggest that extra ion flux occurred across the plasma membrane when stores were depleted. However, they do not prove that the extra flux occurred through discrete SOCs, channels that are closed under basal conditions and open specifically in response to store depletion. Evidence for discrete SOCs came from experiments with gadolinium (Gd3+). Without SERCA inhibition, [Ca2+]i was resistant to 100 μM Gd3+ (Fig. 2A and D). In some arterioles, Ca2+-free bath solution subsequently lowered [Ca2+]i (Fig. 2A). In contrast, after store depletion by treatment with CPA or TG in Ca2+-free bath solution only 10 μM Gd3+ was required to produce a striking reduction in [Ca2+]i (Fig. 2B–D and H). Gd3+ sensitivity did not arise because there was pre-exposure to Ca2+-free solution (Fig. 2E). The smaller effect of Gd3+ without exposure to Ca2+-free solution probably reflected a slower depletion of Ca2+ from the SR. Prolonged exposure to SERCA inhibitor in Ca2+-containing solution re-established the large Gd3+ effect (Fig. 2F and H). The Gd3+ effect did not result from a change in membrane potential. In TG pre-treated arterioles, 10 μM Gd3+ remained effective despite 100 mm K+ in the bath solution (Fig. 2G and H). Although 100 mm K+ depolarises the membrane potential to near the reversal potential of CPA-induced current (0 mV, data not shown) the driving force on Ca2+ entry remains large. Recordings of membrane potential using standard bath solution containing D600 and CPA revealed no significant effect of 10 μM Gd3+ (0.4 ± 1.8 mV change, n = 7).
In Ca2+-imaging experiments, CPA or TG increased [Ca2+]i but no contractile response was evident. An example is shown for a CPA experiment in which the [Ca2+]i rise was large and yet there was no change in the external diameter of the vessel (Fig. 3A-D). In contrast, 60 mm K+ increased [Ca2+]i and caused contraction (Fig. 3E-H). CPA (n = 7) and TG (n = 5) also did not evoke contraction in the absence of D600 when arteriolar diameter was measured using the video edge-detection system (Fig. 3I and J). CPA-induced release of endothelial vasodilators would seem unlikely to explain the lack of contraction because the arteriolar fragments were short and lacked visible endothelium as examined by phase-contrast or fura-PE3 fluorescence microscopy (e.g. Fig. 3A and E). However, to address the potential role of endothelial vasodilators more rigorously we repeated the CPA experiments in the presence of 0.3 mm l-NAME, 10 μM indomethacin, 100 nm charybdotoxin and 100 nm apamin. These agents prevent activation of BKCa channels and the production of nitric oxide, prostacyclin and EDHF, factors known to be critical for endothelium-dependent vasodilatation (Edwards & Weston, 2001). Again there was no constrictor effect of CPA (n = 5) (Fig. 3K). In all of these CPA and TG experiments arterioles subsequently contracted to 60 mm K+ (Fig. 3I-K) which depolarises arterioles to about −20 mV in the absence of D600 (Quinn et al. 2000). Arterioles also did not constrict following a 1 h pre-incubation in 1 μM TG: mean ± s.e.m. external diameters were 25.7 ± 1.3 μm (control, n = 16) and 33.8 ± 3.1 μm (TG, n = 6). CPA also did not evoke contraction in arterioles in intact pial membrane exposed to artificial CSF at 37 °C (Fig. 3L, n = 7).
Ca2+ entry via SOCs may not have caused contraction because the Ca2+ sensitivity of the contractile proteins was depressed by CPA or TG. Ca2+ sensitivity was determined by permeabilising arterioles with β-escin and successful permeabilisation confirmed by a contractile response to the non-hydrolysable GTP analogue GTP-γ-S (Fig. 4A). Arterioles remained Ca2+ responsive despite the presence of CPA (Fig. 4B and C) or TG. Neither SERCA inhibitor had a significant effect on the Ca2+ sensitivity of the contractile proteins (Fig. 4D and E).
Figure 4. SERCA inhibitors do not affect the Ca2+ sensitivity of contractile proteins.
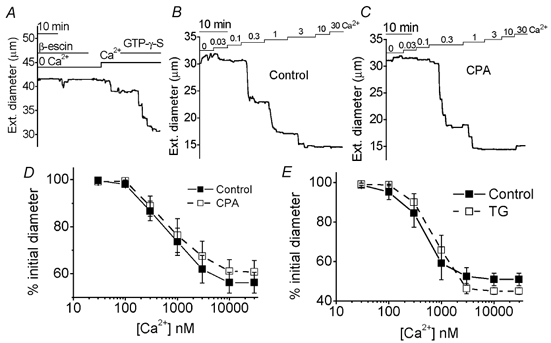
Measurements of the external diameter of β-escin-permeabilised arterioles. A and B, free ionised Ca2+ concentrations in the bath solution are given in μM. A, effect of GTP-γ-S (10 μM) in the presence of 0.1 μM Ca2+. B and C, for a paired experiment carried out on one day, the effects of increasing concentrations of Ca2+ on an arteriole in control conditions (B) and on another arteriole in the presence of 10 μM CPA (C). D and E, mean ± s.e.m. diameter of arterioles as a percentage of the initial diameter in the absence of Ca2+, shown for control and 10 μM CPA paired data (n = 11 for each), and control and 1 μM TG paired data (n = 6 for each).
Alternatively, SOC-associated Ca2+ entry may not have evoked contraction because the amplitude of the [Ca2+]i response failed to reach the activation threshold for the contractile proteins. This is unlikely because there is no discrete activation threshold, 100 nm Ca2+ causes contraction (Fig. 4A) and the average ‘pCa curve’ shows that any Ca2+ elevation above 100 nm will elicit additional contraction (Fig. 5A). The 100 nm Ca2+ value is significant because 100–150 nm is the resting free cytoplasmic Ca2+ concentration in vascular smooth muscle (Kuriyama et al. 1998). Nevertheless, we also compared the effects of CPA with those of elevated [K+]o, which increases [Ca2+]i and causes contraction by depolarising the membrane potential. There was a close correlation between -induced contraction and elevation of [Ca2+]i (Fig. 5B-D), in contrast to the observations with CPA and TG. Also, the sustained elevation of [Ca2+]i induced by CPA was greater than that evoked by 20 mm K+; the -dependent [Ca2+]i rise evoked by CPA was predicted to produce a contraction of the same amplitude as that produced by 34 mm K+ (Fig. 5D).
Figure 5. SOC-associated [Ca2+]i rise is not too small to cause contraction.
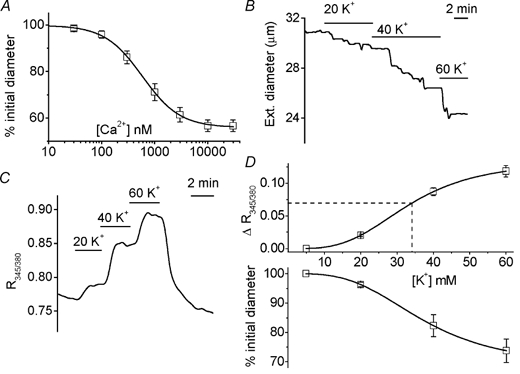
A, Ca2+ dependence of contraction in β-escin-permeabilised arterioles (n = 28). Mean ± s.e.m. data as described for control arterioles in Fig. 4D and E. The fitted curve is the Hill equation with a mid-point at 0.59 μM and a slope of 1.2. B-D, effects of elevating the K+ concentration (mm) of standard bath solution. D600 was excluded from all solutions. Wortmannin was included during [Ca2+]i measurements. B, external diameter measurement. C, [Ca2+]i indicated by fura-PE3. D, plotted against external K+ concentration, the mean ± s.e.m. external diameter as a percentage of that in standard bath solution (n = 7), and changes in fura-PE3 ratio (n/N = 29/8). The superimposed curves are fitted Hill equations with mid-points at 37.3 mm K+ (diameter) and 33.5 mm K+ (Ca2+). The dashed straight line is the mean change in fura-PE3 ratio (0.0697) in response to CPA in the presence of (from Fig. 1C), which is similar in amplitude to the effect of Gd3+ shown in Fig. 2D.
Discussion
The findings of this study support the hypothesis that SOCs exists as a discrete class of Ca2+ channel in cerebral arteriolar smooth muscle cells. Strikingly, Ca2+ entry associated with these SOCs was ‘non-contractile’, appearing to enter a subcellular Ca2+ compartment with restricted access to the contractile proteins.
A SOC-associated compartment is indicated because SERCA inhibitors cause a sustained SOC-dependent elevation of [Ca2+]i without causing any contraction and without preventing Ca2+ activation of the contractile proteins. Although the [Ca2+]i rise is of sufficient amplitude to activate the contractile proteins, it does not, suggesting it is compartmentalised. The effect contrasts with those of endothelin-1 and high [K+]o, which elevate [Ca2+]i by other mechanisms and cause contraction (Fig. 5) (Guibert & Beech, 1999). These other mechanisms may be specifically coupled or compartmentalised with the contractile proteins. If this is true, most SOCs must be elsewhere, in other ‘non-contractile’ compartments. The data in Fig. 1D, although only just statistically significant, seem to suggest that the total amount of Mn2+-quenchable fura-PE3 is greater after store depletion. This suggests that the SOC-associated compartment excluded background Mn2+ entry.
Rise of [Ca2+]i following SERCA inhibition is due both to passive Ca2+ release from SR and SOC activation. Neither of these effects causes contraction. To explain this we hypothesise that Ca2+ arising focally from either source enters the same subcellular compartment that is distant from the contractile proteins. The compartment may be sandwiched between adjacent plasma and SR membranes, with Ca2+ diffusing from the source being progressively extruded from the cell by Ca2+-ATPase and Na+-Ca2+ exchanger activities. If SERCA is not blocked, this Ca2+ will be taken directly into the SR. We suggest the compartment is cytoplasmic, firstly because it exists when SERCA is blocked and thus when Ca2+ cannot be taken into the SR. Secondly, fura-PE3 exhibits preferential localisation to the cytoplasm and is used as a cytoplasmic Ca2+ indicator in smooth muscle (Abe et al. 1995). Furthermore, we employed a relatively low concentration of fura-PE3 AM and a short loading period without pluronic acid, minimising the risk of fura-PE3 existing in intracellular organelles. The success of this strategy is indicated by the rise, rather than fall, in fura-PE3 ratio following depletion of SR (Fig. 1B) or mitochondria (C. Guibert & D. J. Beech, unpublished observations) and the monophasic Mn2+ quench response (Fig. 1D). Nevertheless, it cannot be excluded that the SOC-associated compartment involves mitochondria. There is electron microscopic evidence for mitochondria in a small superficial cytoplasmic space that is separated from contractile filaments in cerebral artery smooth muscle cells (Dahl et al. 1965). Electron microscopic studies of other types of smooth muscle cell have revealed cytoplasmic compartments 20–100 nm wide (van Breemen et al. 1995). These are beyond the resolution of the light microscope, which may explain why we have not resolved SERCA inhibitor-specific subcellular Ca2+ signals.
Non-contractile compartments in smooth muscle have been suggested previously based on disparities between tension and fura or aequorin signals (Abe et al. 1995; Tosun et al. 1998). Ca2+ sparks and their associated K+ or Cl− currents have been extensively studied and reflect [Ca2+]i transients arising from a point source in the SR (Bolton et al. 1999). Low frequencies of Ca2+ sparks are not contractile, but may evoke relaxation indirectly via K+ channel activation (Jaggar et al. 2000). SOC-mediated Ca2+ influx has a discrete effect on plasma membrane-localised adenylyl cyclase in C6-2B glioma cells (Fagan et al. 2000). Such localised signalling presumably serves to improve efficiency and speed, as suggested for the TRP ‘signalplex’ of Drosophila photoreceptors (Tsunoda & Zuker, 1999; Li & Montell, 2000). Intriguingly, the plasma membrane localisation of TRPC1 is clustered in cerebral arterioles (Xu & Beech, 2001), protein targeting that may be essential for the SOC-associated Ca2+ compartment.
Studies of several blood vessels show contractile effects of SERCA inhibitors (Gibson et al. 1998). We have observed contractile effects of CPA and TG on human left internal mammary and radial arteries studied by isometric tension recording, although these were modest in amplitude compared with contractions to U46619 or 60 mm K+ (R. Sadaba & D. J. Beech, unpublished observations). Previous studies of arterioles are few. Submucosal arterioles (external diameter 60–80 μm) did not constrict in response to CPA (Low et al. 1996), whereas pressurised skeletal muscle arterioles (internal diameter 60–100 μm) dilated and then constricted in response to TG (Potocnik & Hill, 2001). An important factor in these experiments is likely to have been the level of pre-existing ‘contractile’ Ca2+ influx induced by another stimulant, the contractile effect of which will be enhanced by SERCA inhibition as the superficial buffer barrier breaks down. In our experiments the arterioles were unstimulated by agonist or lumenal pressure, and in some experiments the L-type voltage-gated Ca2+ channels were blocked. Thus, the complicating pro-contractile effects of inhibiting the superficial buffer barrier were minimised. The hint of a contractile effect (< 0.5 μm) of CPA on arterioles in pial membrane (Fig. 3L) may have been because stretching of the membrane across the pin-hole of the recording chamber provided a stimulus for activation of L-type Ca2+ channels. Other complications can result from the presence of endothelium, the activation of SOCs in endothelial cells releasing relaxants such as nitric oxide. Again this complication was minimised by the use of short arteriolar fragments that lacked visible endothelium (Fig. 3A). Furthermore, CPA failed to induce significant contraction despite the presence of l-NAME, indomethacin, apamin and charybdotoxin (Fig. 3K).
The evidence for SOCs in pial arterioles is compelling. SERCA inhibition, resulting first in SR Ca2+ depletion, is followed by sustained elevation of [Ca2+]i that depends on extracellular Ca2+, increased Mn2+ entry and Gd3+ sensitivity of [Ca2+]i. The induction of Gd3+ sensitivity reveals a distinction from background Ca2+ entry. A TRPC1 blocking antibody inhibits Ca2+ entry in SR Ca2+-depleted but not control arterioles (Xu & Beech, 2001). Patch-clamp studies of aortic or pulmonary artery myocytes have revealed the activation of non-selective cation channels in response to SR Ca2+ depletion (McDaniel et al. 2001; Trepakova et al. 2001; Ng & Gurney, 2001). In preliminary experiments we have observed a similar current in rabbit pial arterioles in response to CPA (S. Z. Xu & D. J. Beech, unpublished observations). These non-selective channels appear similar to TRPC1 expressed in Sf9 cells (Sinkins et al. 1998). TRPC1 is also Gd3+ sensitive (Zitt et al. 1996). SOCs in choroidal arterioles and portal vein myocytes are more Ca2+ selective and thus appear to be a different type of SOC (Curtis & Scholfield, 2001; Albert & Large, 2002). A lack of cation selectivity will mean there will also be Na+ entry. Part of the [Ca2+]i elevation following SOC activation might, therefore, result from reduced Ca2+ extrusion via Na+-Ca2+ exchange (Arnon et al. 2000). Nevertheless, when the Na+ gradient was reduced (35 mm [Na+]o), Gd3+-sensitive Ca2+ entry was still observed (Fig. 2G).
Ionic currents through SOCs are small (Ng & Gurney, 2001; Trepakova et al. 2001; Albert & Large, 2002) and this may be a significant reason why CPA did not evoke depolarisation. The lack of a depolarising effect was shown not only by direct measurements of membrane potential but also by contraction studies in the absence of D600 in which depolarisation would elicit voltage-dependent Ca2+ entry and contraction. The SOC current may either be insufficient to affect the membrane potential because it is small relative to other ionic currents across the membrane, or it may be counter-balanced by a hyperpolarising Ca2+-activated current.
Because of the marked sensitivity of many cerebral arteriolar contractile responses to L-type Ca2+ channel antagonists it might be concluded that other types of Ca2+ channel are not expressed, or are expressed but have no function. This study supports the existence of SOCs as a discrete additional class of Ca2+ channel in smooth muscle cells of cerebral precapillary arterioles. Furthermore, we find that these SOCs are associated only with a subcellular Ca2+ compartment that is ‘non-contractile’. We speculate that such a compartment enables the efficient use of SOC-associated Ca2+ entry for the refilling of depleted SR, and perhaps also endoplasmic reticulum governing protein expression. Intriguingly, Gd3+-sensitive Ca2+ entry has recently been shown to be important for c-Jun N-terminal kinase activation (Kushida et al. 2001), perhaps indicating that SOCs are also coupled to transcription factors and gene expression. Subcellular Ca2+ compartmentalisation may be widespread in vascular smooth muscle and enable efficient use of Ca2+ signals as well as separation of Ca2+-dependent processes governing contractile and non-contractile functions.
Acknowledgments
We thank the Wellcome Trust and the British Heart Foundation for support.
REFERENCE
- Abe F, Mitsui M, Karaki H, Endoh M. Calcium compartments in vascular smooth muscle cells as detected by aequorin signal. British Journal of Pharmacology. 1995;116:3000–3004. doi: 10.1111/j.1476-5381.1995.tb15955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Large WA. A Ca2+-permeable non-selective cation channel activated by depletion of internal Ca2+ stores in single rabbit portal vein myocytes. Journal of Physiology. 2002;538:717–728. doi: 10.1113/jphysiol.2001.013101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon A, Hamlyn JM, Blaustein MP. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. American Journal of Physiology - Cell Physiology. 2000;278:C163–173. doi: 10.1152/ajpcell.2000.278.1.C163. [DOI] [PubMed] [Google Scholar]
- Bolton TB, Prestwich SA, Zholos AV, Gordienko DV. Excitation-contraction coupling in gastrointestinal and other smooth muscles. Annual Review of Physiology. 1999;61:85–115. doi: 10.1146/annurev.physiol.61.1.85. [DOI] [PubMed] [Google Scholar]
- Brandt L, Andersson KE, Edvinsson L, Ljunggren B. Effects of extracellular calcium and of calcium antagonists on the contractile responses of isolated human pial and mesenteric arteries. Journal of Cerebral Blood Flow Metabolism. 1981;1:339–347. doi: 10.1038/jcbfm.1981.37. [DOI] [PubMed] [Google Scholar]
- Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, Stevens T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB Journal. 2001;15:1727–1738. [PubMed] [Google Scholar]
- Casteels R, Droogmans G. Exchange characteristics of the noradrenaline-sensitive calcium store in vascular smooth muscle cells or rabbit ear artery. Journal of Physiology. 1981;317:263–279. doi: 10.1113/jphysiol.1981.sp013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong A, Dedman AM, Beech DJ. Expression and function of native potassium channel KVα1 subunits in terminal arterioles of rabbit. Journal of Physiology. 2001;534:691–700. doi: 10.1111/j.1469-7793.2001.00691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis TM, Scholfield CN. Nifedipine blocks Ca2+ store refilling through a pathway not involving L-type Ca2+ channels in rabbit arteriolar smooth muscle. Journal of Physiology. 2001;532:609–623. doi: 10.1111/j.1469-7793.2001.0609e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E, Flora G, Nelson E. Electron microscopic observations on normal human intracranial arteries. Neurology. 1965;15:120–140. doi: 10.1212/wnl.15.2.132. [DOI] [PubMed] [Google Scholar]
- Edwards G, Weston AH. EDHF - are there gaps in the pathway? Journal of Physiology. 2001;531:299. doi: 10.1111/j.1469-7793.2001.0299i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan KA, Smith KE, Cooper DM. Regulation of the Ca2+-inhibitable adenylyl cyclase type VI by capacitative Ca2+ entry requires localization in cholesterol-rich domains. Journal of Biological Chemistry. 2000;275:26530–26537. doi: 10.1074/jbc.M001369200. [DOI] [PubMed] [Google Scholar]
- Fellner SK, Arendshorst WJ. Capacitative calcium entry in smooth muscle cells from preglomerular vessels. American Journal of Physiology. 1999;277:F533–542. doi: 10.1152/ajprenal.1999.277.4.F533. [DOI] [PubMed] [Google Scholar]
- Gibson A, McFadzean I, Wallace P, Wayman CP. Capacitative Ca2+ entry and the regulation of smooth muscle tone. Trends in Pharmacogical Sciences. 1998;19:266–269. doi: 10.1016/s0165-6147(98)01222-x. [DOI] [PubMed] [Google Scholar]
- Guibert C, Beech DJ. Positive and negative coupling of the endothelin ETA receptor to Ca2+-permeable channels in rabbit cerebral cortex arterioles. Journal of Physiology. 1999;514:843–856. doi: 10.1111/j.1469-7793.1999.843ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haws CW, Heistad DD. Effects of nimodipine on cerebral vasoconstrictor responses. American Journal of Physiology. 1984;247:H170–176. doi: 10.1152/ajpheart.1984.247.2.H170. [DOI] [PubMed] [Google Scholar]
- Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. Journal of Applied Physiology. 2001;91:973–983. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. American Journal of Physiology - Cell Physiology. 2000;278:C235–256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- Kuriyama H, Kitamura K, Itoh T, Inoue R. Physiological features of visceral smooth muscle cells, with special reference to receptors and ion channels. Physiological Reviews. 1998;78:811–920. doi: 10.1152/physrev.1998.78.3.811. [DOI] [PubMed] [Google Scholar]
- Kushida N, Kabuyama Y, Yamaguchi O, Homma Y. Essential role for extracellular Ca2+ in JNK activation by mechanical stretch in bladder smooth muscle cells. American Journal of Physiology - Cell Physiology. 2001;281:C1165–1172. doi: 10.1152/ajpcell.2001.281.4.C1165. [DOI] [PubMed] [Google Scholar]
- Li HS, Montell C. TRP and the PDZ protein, INAD, form the core complex required for retention of the signalplex in Drosophila photoreceptor cells. Journal of Cell Biology. 2000;150:1411–1422. doi: 10.1083/jcb.150.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang W, Singh BB, Lockwich T, Jadlowiec J, O'Connell B, Wellner R, Zhu MX, Ambudkar IS. Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. Journal of Biological Chemistry. 2000;275:3403–3411. doi: 10.1074/jbc.275.5.3403. [DOI] [PubMed] [Google Scholar]
- Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. Journal of Biological Chemistry. 2000;275:11934–11942. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- Loutzenhiser K, Loutzenhiser R. Angiotensin II-induced Ca2+ influx in renal afferent and efferent arterioles: differing roles of voltage-gated and store-operated Ca2+ entry. Circulation Research. 2000;87:551–557. doi: 10.1161/01.res.87.7.551. [DOI] [PubMed] [Google Scholar]
- Low AM, Kotecha N, Neild TO, Kwan CY, Daniel EE. Relative contributions of extracellular Ca2+ and Ca2+ stores to smooth muscle contraction in arteries and arterioles of rat, guinea-pig, dog and rabbit. Clinical Experimental Pharmacology and Physiology. 1996;23:310–316. doi: 10.1111/j.1440-1681.1996.tb02829.x. [DOI] [PubMed] [Google Scholar]
- Luo D, Nakazawa M, Yoshida Y, Cai J, Imai S. Effects of three different Ca2+ pump ATPase inhibitors on evoked contractions in rabbit aorta and activities of Ca2+ pump ATPases in porcine aorta. General Pharmacology. 2000;34:211–220. doi: 10.1016/s0306-3623(00)00064-1. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Flynn ER, Bradley KN, Muir TC. Two Ca2+ entry pathways mediate InsP3-sensitive store refilling in guinea-pig colonic smooth muscle. Journal of Physiology. 2000;525:113–124. doi: 10.1111/j.1469-7793.2000.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JX. Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2001;280:L870–880. doi: 10.1152/ajplung.2001.280.5.L870. [DOI] [PubMed] [Google Scholar]
- Ng LC, Gurney AM. Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circulation Research. 2001;89:923–929. doi: 10.1161/hh2201.100315. [DOI] [PubMed] [Google Scholar]
- Pierre LN, Davenport AP. Blockade and reversal of endothelin-induced constriction in pial arteries from human brain. Stroke. 1999;30:638–643. doi: 10.1161/01.str.30.3.638. [DOI] [PubMed] [Google Scholar]
- Potocnik SJ, Hill MA. Pharmacological evidence for capacitative Ca2+ entry in cannulated and pressurized skeletal muscle arterioles. British Journal of Pharmacology. 2001;134:247–256. doi: 10.1038/sj.bjp.0704270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn K, Guibert C, Beech DJ. Sodium-potassium-ATPase electrogenicity in cerebral precapillary arterioles. American Journal of Physiology - Heart and Circulatory Physiology. 2000;279:H351–360. doi: 10.1152/ajpheart.2000.279.1.H351. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. Activation of store-mediated calcium entry by secretion-like coupling between the inositol 1,4,5-trisphosphate receptor type II and human transient receptor potential (hTrp1) channels in human platelets. Biochemical Journal. 2001;356:191–198. doi: 10.1042/0264-6021:3560191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum WI. Effects of calcium channel blockers on pial vascular responses to receptor mediated constrictors. Stroke. 1984;15:284–287. doi: 10.1161/01.str.15.2.284. [DOI] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. Journal of Biological Chemistry. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Sinkins WG, Estacion M, Schilling WP. Functional expression of TrpC1: a human homologue of the Drosophila Trp channel. Biochemical Journal. 1998;331:331–339. doi: 10.1042/bj3310331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson AS, Cartin L, Wellman TL, Dick MH, Nelson MT, Lounsbury KM. Membrane depolarization mediates phosphorylation and nuclear translocation of CREB in vascular smooth muscle cells. Experimental Cell Research. 2001;263:118–130. doi: 10.1006/excr.2000.5107. [DOI] [PubMed] [Google Scholar]
- Takayasu M, Bassett JE, Dacey RG., Jr Effects of calcium antagonists on intracerebral penetrating arterioles in rats. Journal of Neurosurgery. 1988;69:104–109. doi: 10.3171/jns.1988.69.1.0104. [DOI] [PubMed] [Google Scholar]
- Terada K, Kitamura K, Kuriyama H. Different inhibitions of the voltage-dependent K+ current by Ca2+ antagonists in the smooth muscle cell membrane of rabbit small intestine. Pflügers Archiv. 1987;408:558–564. doi: 10.1007/BF00581156. [DOI] [PubMed] [Google Scholar]
- Tosun M, Paul RJ, Rapoport RM. Coupling of store-operated Ca++ entry to contraction in rat aorta. Journal of Pharmacology and Experimental Therapeutics. 1998;285:759–766. [PubMed] [Google Scholar]
- Trepakova ES, Gericke M, Hirakawa Y, Weisbrod RM, Cohen RA, Bolotina VM. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. Journal of Biological Chemistry. 2001;276:7782–7790. doi: 10.1074/jbc.M010104200. [DOI] [PubMed] [Google Scholar]
- Tsunoda S, Zuker CS. The organization of INAD-signaling complexes by a multivalent PDZ domain protein in Drosophila photoreceptor cells ensures sensitivity and speed of signaling. Cell Calcium. 1999;26:165–171. doi: 10.1054/ceca.1999.0070. [DOI] [PubMed] [Google Scholar]
- Unno T, Beech DJ, Komori S, Ohashi H. Inhibitors of spasmogen-induced Ca2+ channel suppression in smooth muscle cells from small intestine. British Journal of Pharmacology. 1998;125:667–674. doi: 10.1038/sj.bjp.0702112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uski TK, Andersson KE, Brandt L, Ljunggren B. Characterization of the prostanoid receptors and of the contractile effects of prostaglandin F2 alpha in human pial arteries. Acta Physiologica Scandinavica. 1984;121:369–378. doi: 10.1111/j.1748-1716.1984.tb07468.x. [DOI] [PubMed] [Google Scholar]
- van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends in Pharmacological Sciences. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- Vorndran C, Minta A, Poenie M. New fluorescent calcium indicators designed for cytosolic retention or measuring calcium near membranes. Biophysical Journal. 1995;69:2112–2124. doi: 10.1016/S0006-3495(95)80082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circulation Research. 2001;88:84–87. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- Zitt C, Zobel A, Obukhov AG, Harteneck C, Kalkbrenner F, Luckhoff A, SchultZ G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron. 1996;16:1189–1196. doi: 10.1016/s0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]