

Abstract
The current human genome was moulded and refined through generations of time. We propose that the basic framework for physiologic gene regulation was selected during an era of obligatory physical activity, as the survival of our Late Palaeolithic (50 000–10 000 BC) ancestors depended on hunting and gathering. A sedentary lifestyle in such an environment probably meant elimination of that individual organism. The phenotype of the present day Homo sapiens genome is much different from that of our ancient ancestors, primarily as a consequence of expressing evolutionarily programmed Late Palaeolithic genes in an environment that is predominantly sedentary. In this sense, our current genome is maladapted, resulting in abnormal gene expression, which in turn frequently manifests itself as clinically overt disease. We speculate that some of these genes still play a role in survival by causing premature death from chronic diseases produced by physical inactivity. We also contend that the current scientific evidence supports the notion that disruptions in cellular homeostasis are diminished in magnitude in physically active individuals compared with sedentary individuals due to the natural selection of gene expression that supports the physically active lifestyle displayed by our ancestors. We speculate that genes evolved with the expectation of requiring a certain threshold of physical activity for normal physiologic gene expression, and thus habitual exercise in sedentary cultures restores perturbed homeostatic mechanisms towards the normal physiological range of the Palaeolithic Homo sapiens. This hypothesis allows us to ask the question of whether normal physiological values change as a result of becoming sedentary. In summary, in sedentary cultures, daily physical activity normalizes gene expression towards patterns established to maintain the survival in the Late Palaeolithic era.
Introduction
The major goals of physiology as conceptualized by Guyton & Hall (1996) are to better explain the physical and chemical factors responsible for the origin, development and progression of life. This review will contrast exercise-deficient states (as seen in the present day sedentary individuals having little leisure-time or occupational physical activity) to that of the physically active lifestyle (as seen in the Late Palaeolithic and current hunter-gatherer societies) to deduce gene functions developed for exercise. (During the Late Palaeolithic period (50 000–10 000 BC) humans lived as hunter-gatherers, using rudimentary chipped stone tools, and are thus said to have lived in the ‘old stone age’; Eaton et al. 2002.) In a sense, physical inactivity is analogous to a loss of function resulting from a silencing of a gene, except that the missing element is not the gene but the environmental interaction of physical activity with the gene (Perusse & Bouchard, 1999; Booth & Vyas, 2001). However, it is possible to deduce function from a loss-of-function consideration. Therefore, this review will consider the viewpoints of Darwinian medicine and the ‘thrifty’ gene hypothesis better to elucidate functions for exercise-induced gene expressions.
Darwin's natural selection concept applied to physical activity: selection of our Late Palaeolithic ancestors’ genes determines levels of physical activity required for appropriate gene expression
The Darwinian concept of natural selection is that inheritable variations among the individuals of given types of organisms continually arise in nature and that some variations prove advantageous under prevailing conditions in that they enable the organism to leave relatively more surviving offspring (Cherow & Vallasi, 2002). The first chapter of Åstrand's Textbook of Work Physiology (1986) is devoted to the topic of evolution in which it stated that close to 100 % of the biologic existence of humans was adapted to an outdoor existence of hunting and foraging for foods. Later, Åstrand (1992) added that major adaptations for human survival ‘were consonant with habitual physical activity, including endurance and peak effort alternated with rest‘.
Trevathan et al. (1999) contend that 95 % of human biology, and presumably some of human behaviours, were naturally selected during the time period in which our ancestors lived as gatherers of wild food resources, but now these selections may be maladaptive. According to Gerber & Crews (1999), numerous alleles that evolved for function, selective advantage, and survival in the Late Palaeolithic era are now being exposed to sedentary lifestyles, fat-rich/fibre-poor diets, and an extended lifespan where they now put their carriers at a disadvantage with respect to chronic degenerative diseases and longevity with little impact on the genetic successes of their progeny. The assertion has been made that the portion of the human genome that determines basic anatomy and physiology has remained relatively unchanged over the past 10 000 years (Cavalli-Sforza et al. 1994; Cordain et al. 1998). Thus, most of the current human genome probably evolved in the physically active hunter-gatherer environment and remains unchanged to this day (Cordain et al. 1998). Consequently, this would lead to a dissonance between ‘Stone Age’ genes and ‘Space Age’ circumstances, with resulting disruption of ancient, complex homeostatic systems, as asserted by Eaton et al. (2002).
Estimates of physical activity in the Late Palaeolithic era and current physically active societies are much greater than in current sedentary lifestyles (Cordain et al. 1998). Cordain et al. (1998) published that daily Hominid energy expenditure declined from a value of 206 kJ kg−1 day−1 (49 kcal kg−1 day−1) that was present for much of the past 3.5 million years to 134 kJ kg−1 day−1 (32 kcal kg−1 day−1) for contemporary humans. This review will consider potential functions of exercise-deficiency-induced gene expression based upon the adaptive selection of human genes in an environment requiring physical activity for survival.
The concept of Darwinian (evolutionary) medicine applied to exercise deficiency
Nesse & Williams (1998) indicate that Darwinian medicine asks why the body is designed in a way that makes humans vulnerable to problems like atherosclerosis, a disease whose prevalence only increased in the last 100 years (Peery, 1975). Evolutionary medicine takes the view that many contemporary physical ills are related to incompatibility between lifestyles and environments in which humans currently live and the conditions under which human biology evolved (Trevathan et al. 1999). Observations made on populations in the 20th century support the contention made by Eaton et al. (1988) that there is now a mismatch between humans’ ancient, genetically controlled biology and certain important aspects of our lives such as exercise, nutrition, alcohol and tobacco. For example, Eaton et al. (2002) concluded that while current hunter-gatherer societies undergo similar (but slower) age-related losses in vision and hearing capacity than do sedentary societies, current hunter-gatherers rarely develop chronic degenerative disorders such as hypertension, obesity, sarcopenia, hypercholesterolaemia, non-occlusive atheromata and insulin resistance, as compared with their prevalence in similar-aged sedentary populations. Epidemiological reports indicate higher prevalences of breast cancer (22 % increase), mortality (41 % increase), coronary heart disease (43 % increase), gallstones (49 % increase), type 2 diabetes (85 % increase), colon cancer (85 % increase), diabetic coronary heart disease (92 % increase) and ischaemic stroke (117 % increase) in the Harvard Nurses Health participants who undertook less than 2.5 hours per week of moderate physical activity (e.g. brisk walking) as compared with cohorts who had more than 2.5 hours per week of physical activity (Hu et al. 1999, 2000, 2001; Leitzmann et al. 1999; Manson et al. 1999; Martinez et al. 1997; Rockhill et al. 1999, 2001). In the 1990s, Mexican Pima Indians expended 2100–2520 kJ day−1 more (500-600 more kcal day−1) in physical activity, had a diet lower in fat and higher in fibre content, weighed 26 kg less, and did not have the diabetes epidemic of their obese Arizonian Pima counterparts (Esparza et al. 2000). The changes in the prevalence of diabetes between two geographically separate but genetically similar Pima Indian tribes suggest the importance of environmental interactions with the genome (Pratley, 1998). For example, Gerber & Crews (1999) contend that reductions in physical activity in the past 100 years are being played out against a background of co-adapted gene complexes that show multiple epistatic, multifactorial and pleiotrophic relationships. This review will consider why exercise deficiency produces a dysfunction in gene expression (Fig. 1).
Figure 1. A simplified schematic illustration hypothesizing how environmental factors such as physical inactivity may influence gene expression and consequently genetic susceptibility.
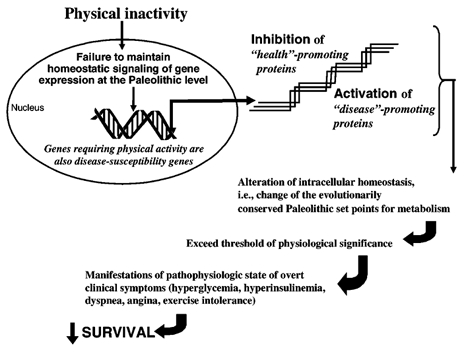
It is postulated that when a threshold of biological significance is exceeded, a cascade of potentially adverse consequences may result that engenders overt clinical symptoms for chronic health conditions, ultimately diminishing survival.
‘Thrifty’ gene concept applied to physical activity: our Late Palaeolithic ancestors’ genes determine our response to a sedentary lifestyle
Neel (1962) proposed that ‘thrifty’ genes were incorporated into the human genome because of their selective advantage over the less ‘thrifty’ ones during earlier phases of human evolution. According to Neel (1962), ‘thrifty’ genes would increase the efficiency of energy storage in periods of food availability as compared with those without these genes. Subsequently, during famines, individuals with ‘thrifty’ genes would have an advantage, using their larger previously stored energy to maintain homeostasis, while those without the ‘thrifty’ genes would be at a disadvantage and less likely to survive (Wendorf & Goldfine, 1991). Over 40 years later, Neel (1999) revised his original hypothesis with the statement: ‘It is now clear that the original “thrifty” genotype hypothesis, with its emphasis on feast or famine, presented an overly simplistic view of the physiological adjustments involved in the transition from the lifestyle of our ancestors to life in the high tech fast lane.’ Neel (1999) then wrote that the high conditioning of the muscle of our tribal, hunter-gatherer ancestors now needs to be considered as a part of his original thrifty gene hypothesis. Eaton et al. (2002) agree with Neel's contention that through nearly all of human evolution, physical exercise and food procurement were inextricably linked. The current review will also consider Neel's modified ‘thrifty’ gene hypothesis (1999) to explain function of exercise-induced adaptations in gene expression.
Exercise-induced changes in gene expression in specific organ systems
Skeletal muscle
Regulation of muscle size
Cordain et al. (1998) inferred that muscular loading must have been high for Homo erectus of 1 million years ago, as they did not use crafted tools of compound levers. Ruff (1993) reported that human bone robustness decreased from 50 000 years ago when human creativity and technological innovation increased dramatically and the efficiency of food gathering activities improved. Neel et al. (1998) wrote that although trained athletes retain the relative muscle mass of early humans (at least until competitions are over), modern humans are characterized by a ‘striking sarcopenia’. Thus, one can infer based on these statements that the relative size of human muscle mass in the Late Palaeolithic era was larger than today. Consequently what is called the mechanisms of skeletal muscle hypertrophy from sedentary levels today would probably be the maintenance of muscle mass in the Late Palaeolithic era. Given that the Late Palaeolithic culture of 10 000 BC demanded the lifting of heavy loads, larger masses of skeletal muscle may have favoured pre-reproductive survival. Perhaps, the remnants of such genomic programming is the observation of increased mortality rates seen in present day humans when skeletal muscle mass declines below a certain minimum secondary to physical frailty (Sherman, 2001), starvation (Kreiger, 1921; Winick, 1979) or AIDS (Kotler et al. 1989).
In order to better understand the molecular details of how increased muscle loading causes an increase in muscle mass, the skeletal α-actin gene is used as a model for examining such mechanisms. Perhaps the understanding of such molecular-genetic details may shed insight into how the environment may have influenced the expression of the skeletal α-actin gene, resulting in the observed differences in muscle mass in present-day sedentary individuals with those that are physically active, and in turn extrapolated to that of the Late Palaeolithic period.
Myofibrillar protein per whole muscle (Laurent et al. 1978), myofibrillar protein synthesis (Laurent et al. 1978) actin protein synthesis (Gregory et al. 1990), actin mRNA (Carson et al. 1995), and actin promoter activity (Carson et al. 1995) have all been shown to increase in an animal overload model, which leads to muscle hypertophy. One known hypertrophy regulatory site on the skeletal α-actin gene promoter is the serum response element 1 (SRE1; Carson et al. 1995). SREs are cis-acting DNA regulatory elements that respond to changes in the availability and activity of transcription factors that bind to the element (Lee et al. 1992). Deletion of the SRE1 element in the skeletal α-actin promoter eliminated the increased transcriptional activity of the α-actin promoter in overloaded skeletal muscles undergoing hypertrophy (Carson et al. 1995). Carson et al. (1995) concluded that SRE1 is a hypertrophy regulatory element that activates specific contractile protein genes to produce more mRNA in response to overload conditions. Serum response factor (SRF) is a transcription factor that homodimerizes to SRE1 on the skeletal α-actin gene (Lee et al. 1992). Increases in the quantity (Carson et al. 1995), and potentially the post-translational status (Fluck et al. 1999), of SRF were noted in hypertrophying muscles. An increase in SRF transcriptional activity should enhance transcription of skeletal α-actin mRNA. Therefore, it is likely that during the late phases of a hypertrophic stimulus, transcription of the α-actin gene is an important contributor to muscle hypertrophy.
However, the early adaptive changes in skeletal muscle during increased loading are probably also due to enhanced translation of existing mRNAs with such increases being significant enough to increase protein production after a few days of overloading. The increased translation is probably signalled, in part, through the enzyme p70S6K, which ultimately leads to enhanced protein synthesis (Baar & Esser, 1999). Intriguingly, the type of exercise stimuli governs the intricate balance of which signalling pathways are turned on or off, thereby providing for a regulation of phenotypic outcomes. For example, aerobic exercise involving predominantly endurance type of work does not phosphorylate p70S6K (Sherwood et al. 1999), whereas high-resistance loading type of exercise does (Bodine et al. 2001b). Further, Nadar & Esser (2001) also found that treadmill running did not have any effect on the phosphorylation status p70S6K. This suggests a differential regulation of signalling by the type of exercise and the possibility of different phenotypic outcomes.
As a further layer of complexity, the extrinsic microenvironment of the cell further governs the regulation of intracellular mediators such as AKT/PKB. For instance, insulin-like growth factor-I (IGF-I) is a well documented activator of AKT/PKB kinase activity, via the activation of phosphatidylinositol-3′-kinase (PI3′-K; for review see Butler et al. 1998). Therefore, altering the extracellular concentrations of IGF-I via autocrine/paracrine action ultimately results in a marked phenotypic change. Environmental cues such as increased loading of muscle by stretch have been shown to increase liver and muscle isoforms of IGF-I mRNA within muscle fibres (McKoy et al. 1999) and to increase IGF-I peptide in the whole muscle (Adams et al. 1999). Alternatively, IGF-I in muscle could also be artificially increased as achieved by adenovirally mediated gene transfer (Barton-Davis et al. 1998) or via direct application to the muscle by miniosmotic pumps (Chakravarthy et al. 2000), both of which resulted in a dramatic rescue of skeletal muscle from atrophy. In addition, it seems that activation of the AKT signalling pathway alone is sufficient to induce hypertrophy. For example, Bodine et al. (2001b) found that by using gene therapy methods to deliver a constitutively active form of AKT, it was possible to induce skeletal muscle hypertrophy, suggesting that this pathway plays a prominent role in muscle hypertrophy. Therefore, it is likely that the increased expression of IGF-I and activation of the AKT signalling pathway may significantly contribute to skeletal muscle hypertrophy. However, this is not to suggest that this is the only operative signalling pathway in muscle hypertrophy, since others have found that the activation of other pathways such as calcineurin (Dunn et al. 1999), and inhibition of certain ubiquitin ligases (Bodine et al. 2001a; Gomes et al. 2001), can enhance muscle mass.
Calcineurin is a Ca2+-calmodulin-dependent phosphatase that appears to be crucial in the signalling of functional overload-induced fibre hypertrophy. Dunn et al. (1999) have demonstrated the importance of calcineurin in muscle fibre hypertrophy with various pharmacological inhibitors of calcineurin. Calcineurin is probably activated in overloaded muscles via the chronic increases in intracellular calcium that occur under overloaded conditions as a result of a doubling of nerve-mediated muscle fibre activation and load-related increases in insulin-like growth factor (Dunn et al. 2000). Once activated, calcineurin may signal downstream genes involved in regulating muscle fibre size via dephosphorylation of its substrate transcription factors, nuclear factor of activated T cells (NFAT; Dunn et al. 2000). Various NFAT isoforms have been shown to be able to activate various genes, which have been implicated in the slow muscle fibre and muscle hypertrophy gene expression (Olson & Williams, 2000). Calcineurin has been shown to be required at only specific time points of muscle re-growth from a bout of muscle atrophy and these time points vary between fast and slow muscles (Mitchell et al. 2002).
In addition, a class of inhibitors also determines skeletal muscle size. Myostatin knockout mice had muscle hypertrophy (McPherron et al. 1997). Muscle unloading has been shown to increase myostatin protein, which was reversed when short periods of loading intervened during the unloading (Wehling et al. 2000). Sharma et al. (2001) concluded that there is strong circumstantial evidence for the role of myostatin regulating muscle growth after birth. More recently another negative regulator of skeletal muscle growth has been noted. Inhibition of glycogen synthase kinase-3β by a dominant negative mutant (Rommel et al. 2001) or by LiCl (Vyas et al. 2002) is associated with an enlargement of C2C12 myotubes in culture.
Capacity of skeletal muscle to oxidize fuels
A direct association between the duration of contractile activity and mitochondrial density of the contracting skeletal muscle has been long established (Holloszy & Booth, 1976). Cytochrome c protein is a marker for mitochondrial density and the capacity to oxidize fuels, which rise and fall during exercise and limb immobilization, respectively (Booth & Holloszy, 1977; Dudley et al. 1982). Increases and decreases in the cytochrome c mRNA concentration occur with exercise and physical inactivity, respectively (Morrison et al. 1987). Strikingly, identification of one potential signalling pathway for an exercise-induced mitochondrial biogenesis started with the 1996 report that AMP kinase (AMPK) activity was found to increase two- to three-fold in the deep red region of the quadriceps muscle within 5 min of the beginning of exercise and remained elevated for as long as the rat continued to run (Winder & Hardie, 1996). Evidence is accumulating for a role of AMPK in initiating the induction of some of the adaptations to endurance training, including the increase in muscle GLUT-4, hexokinase, uncoupling protein 3, and some of the mitochondrial oxidative enzymes (Winder & Hardie, 1996). The increase in AMPK activity during contraction increases NRF-1 (Bergeron et al. 2001; Murakami et al. 1998; Xia et al. 1997), a transcription factor, which, in turn, binds to the ALA synthase and mTFA gene promoters, resulting in increases in these proteins (Gordon et al. 2001) and consequently increases cytochrome c protein concentration and mitochondrial density (Hood, 2001). Increases in sarcoplasm Ca2+ concentrations are associated with increases in Ca2+-calmodulin-dependent protein kinase activity (Ojuka et al. 2002), which have been shown to increase PGC-1 and cytochrome c protein expression (Wu et al. 2002). Williams et al. (1986) observed increased concentrations of mRNAs from mitochondrial and nuclear genomes, as well as an increased mitochondrial DNA copy number, in skeletal muscles that underwent chronic electrical stimulation. Pilegaard et al. (2000) have demonstrated that recovery from exercise is associated with transient increases in the transcription of several metabolically related genes (uncoupling protein 3, pyruvate dehydrogenase kinase 4, haeme oxygenase-1, lipoprotein lipase, and carnitine pamitoyltransferase I) in human skeletal muscle. They have interpreted these findings to suggest that the transcriptional activation of target genes is a primary adaptive response to exercise within muscle cells and that the cumulative effects of transient increases in transcription during recovery from consecutive bouts of exercise probably underlie the kinetic basis for the cellular adaptations associated with exercise training.
A number of functions for exercise-induced increases in the concentrations of skeletal muscle mitochondria have been suggested. One function is that a smaller disruption in homeostasis occurs within contracting skeletal muscles with higher mitochondrial concentrations (Holloszy & Booth, 1976). For example, when the same human is tested at the same submaximal O2 consumption before and after endurance training, glycogen depletion and lactate concentrations in the quadriceps muscle are lower (Saltin & Karlsson, 1971). Trained muscles oxide more fatty acids (sparing the limited stores of glycogen) at the same absolute workload with a resultant protection against hypoglycaemia-induced fatigue and a longer exercise-time to exhaustion (Holloszy & Coyle 1984; Saltin & Åstrand, 1993). Endurance-trained skeletal muscles increase enzymes for β-oxidation and thus oxidize more fatty acids at a given absolute workload (Molé et al. 1971). Creatine phosphate concentrations are higher and inorganic phosphate ADP, AMP and lactate concentrations are lower in muscles of exercise-trained rats with higher mitochondrial concentrations as compared with untrained rats during the same contractile activity (Constable et al. 1987; Dudley et al. 1987). Constable et al. (1987) described this adaptation as ‘one aspect of the smaller disturbance in homeostasis that occurs in trained compared with untrained muscles performing the same activity.’ Thus, exercise-deficient skeletal muscles undergo a greater homeostatic disruption at the same absolute work intensity, which Eaton et al. (2002) attributed to discords ‘between „Stone Age” genes and „Space Age” circumstances, with resulting disruption of ancient, complex homeostatic systems‘.
Cardiovascular
Vascular biology
The vascular endothelium serves as an important modulator of vasomotor tone and function by synthesizing and releasing nitric oxide (NO) for flow-dependent dilatation of conduit arteries during periods of increased cardiac work (Kelm, 2002). However, endothelial function is dynamic and easily depressed by numerous factors. For example, postprandial lipaemia, hyperglycaemia, mental stress and/or physical inactivity can all lower NO expression levels in vessel walls (Abdu et al. 2001; Kelm, 2002). In addition, the coronary vascular response to acetylcholine depends on the integrity of the endothelium and the endothelial NO pathway (Kelm, 2002). Only if the endothelium is healthy can acetylcholine-mediated vasodilatation through NO occur. Patients with coronary endothelial dysfunction respond to acetylcholine by impaired production of endothelium-derived NO and a paradoxical vasoconstriction that is associated with diminished coronary blood flow (Hambrecht et al. 2000). Exercise training reversed the degree of endothelium-dependent vasoconstriction from acetylcholine in both epicardial coronary vessels and resistance vessels in patients with coronary artery disease (Hambrecht et al. 2000), suggesting a correlation between the health of coronary vessels and physical activity levels.
Interestingly, exercise of sedentary pigs enhances NO-mediated vasodilatation (Bowles et al. 2000). One of the mechanisms for this effect is via increased blood flow that occurs in the heart during exercise, which in turn produces shear stress in endothelial cells, ultimately resulting in enhanced NO levels and coronary endothelium-dependent relaxation (Muller et al. 1994). Exercise-mediated increases in NO levels are largely due to an up-regulation of endothelial nitric oxide synthase (ecNOS) mRNA (Sessa et al. 1994) and protein (Woodman et al. 1997) expression.
Many other mechanisms may underlie the maintenance of high NO levels during increased physical activity. For instance, extracellular membrane-bound superoxide dismutase (ecSOD) functions as a major cellular defence against oxygen free radicals (O2•−; Stroppolo et al. 2001). Extracellular SOD scavenges these free radicals and converts them into hydrogen peroxide, thereby preventing the formation of toxic metabolities such as peroxynitrite (Stroppolo et al. 2001). The prevention of these toxic metabolites is vital, since these metabolites can induce degradation of NO (Stroppolo et al. 2001). Exercise is associated with an increased level of ecSOD mRNAs in aortas of wild-type mice (Fukai et al. 2000). This adaptation was removed in transgenic mice lacking the eNOS gene (Fukai et al. 2000). The investigators interpreted their findings as suggesting that NO produced by endothelial cells stimulates increased ecSOD mRNA in adjacent smooth muscle cells, thus preventing O2•−-mediated degradation of NO as it traverses between the two cell types. Chronic aerobic exercise training selectively increases the levels of SOD-1 mRNA, protein and enzymatic activity in porcine coronary arterioles. This report (Rush et al. 2000) suggested that increased SOD-1 could contribute to the enhanced NO-dependent dilatation previously observed in coronary arterioles of exercised pigs by regulating the amount of superoxide in the vascular cell environment, thereby prolonging the biological half-life of NO.
Long-term exercise training increases the diameters of coronary blood vessels in exercise-trained monkeys (Kramsch et al. 1981). Kingwell et al. (2000) suggested that NO is one of the crucial signals for such adaptive changes in gene expression in the extracellular matrix that lead to the long-term increases in the vessel's structural diameter, resulting in enhanced coronary flow, and ultimately decreasing myocardial ischaemia. This adaptation would then lower shear stresses at a given blood flow and diminish a disruption in homeostasis.
Nitric oxide is also a potent anti-atherogenic agent, mediating its actions via vasodilatation, as well as inhibition of platelet aggregation, smooth muscle cell proliferation, and leucocyte adhesion to endothelial cells in the vessel wall (Wroblewski et al. 2000). A disturbance of endothelial function by the loss of NO and its consequent smooth muscle vasoconstriction is considered a key event in the development of atherosclerosis (Gielen et al. 2001).
Clinical and postmortem investigations of recent hunter-gatherer societies (artic Eskimos, Kenyan Kikuyu, Solomon Islanders, Navajo Indians, Masai pastoralists, Australian Aborigines, Kalahari San (Bushman), New Guinea highland natives and Congo Pygmies) reveal little or no heart disease (see Eaton et al. 1988 for references). Eaton et al. (1988) contend: ‘Like our Palaeolithic ancestors, they (recent hunter-gatherer societies) lacked tobacco, rarely had hypertension, and led lives characterized by considerable physical exercise. In addition, their serum cholesterol levels were low (Eaton et al. 1988). When individuals from hunter-gatherer societies became ‘westernized" (migrations of Japanese, Chinese and Samonans to the USA), their incidence of coronary heart disease rises (Eaton et al. 1988). Peery (1975) in his Ward Burdick Award address commented: ‘Ischaemic heart disease should be looked upon as new disease, largely due to greater consumption of meat and dairy products, and the more sedentary lifestyle that have been adopted in the United States as a result of our greater affluence.’ However, Cordain et al. (2002) found from field studies of thirteen 20th century hunter-gatherer societies that they consumed 65 % of their energy from animal food, yet were relatively free of signs and symptoms of cardiovascular disease. They suggest that qualitative differences in fat intake, high intakes of antioxidants, fibre, vitamins and phytochemicals, along with low salt intake may have operated synergistically with more exercise, less stress, and no smoking to prevent cardiovascular disease in the hunter-gatherers (Cordain et al. 2002).
Deficient physical activity in the current sedentary culture lowers NO production in endothelial cells of human coronary blood vessels producing vasoconstriction (Hambrecht et al. 2000). Hambrecht et al. 2000, concluded: ‘This finding provides a pathophysiologic framework for the elucidation of the positive effects of exercise on myocardial perfusion and emphasizes the therapeutic potential of endurance training for patients with stable coronary artery disease.’ The lack of exercise-induced blood flows producing nitric oxide could be a potential contributing factor to explain, in part, the Centers for Disease Prevention's finding that showed ‘no exercise’ accounted for 248 317 deaths from heart disease in the US in 1986 (34 % of total heart deaths) (Hahn et al. 1990).
Heart
There are two major categories of cardiac hypertrophy: one in which cardiac reserve (i.e. the maximum percentage that the cardiac output can increase above normal; Guyton & Hall, 1996) and contractility are enhanced (physiological hypertrophy associated with ‘athletes’); and the other in which contractility diminishes (pathological hypertrophy produced by pressure overload, such as hypertension, leading to congestive heart failure; Wikman-Coffelt et al. 1979). For example, according to Guyton & Hall (1996), cardiac reserve is 300–400 % in the healthy young adult, 500–600 % in the athletically trained person, and zero in heart failure. The significance of physiological cardiac hypertrophy is that it improves cardiac function by decreasing oxygen cost per unit of work, resting and submaximal heart rates, as well as increasing filling time, venous return and maximal cardiac output.
Does scientific evidence favour physiological cardiac hypertrophy or the sedentary healthy heart as the physiological norm in AD 2000? The current data seem to support the former possibility. On the basis of gross and microscopic examinations of the hearts of labourers, Linzbach (1947) coined the term ‘physiological left ventricular hypertrophy’. Physiological hypertrophy of cardiac myocytes cannot solely be explained by an inherited and fixed genome, but rather is attributable in part to the plastic nature of cardiac tissue, which in turn is influenced by a dynamic and changing microenvironment. For example, heart dimensions of sedentary young men rapidly increase with swim training and decrease with deconditioning (Ehsani et al. 1978). As Palaeolithic humans laboured for their survival, we speculate that they exhibited left ventricular hypertrophy and high cardiac reserves. Curiously, the term physiological cardiac hypertrophy is almost always now associated with ‘athletes’, rather than ‘labourers’. For example, the JAMA issue dedicated to the 1976 Olympic Games contained an article entitled ‘The Athletic Heart’ (Raskoff et al. 1976). In 2001, Iemitsu et al. wrote: ‘Chronic exercise training causes cardiac hypertrophy, which is defined as the athletic heart.’ This conversion of the population group with physiological cardiac hypertrophy from ‘labourers’ to ‘athletes’ illustrates a shift in the written physiologic norm, as well as a shift in the designation of the control group.
Another supporting set of data for the physiologic norm being physiological cardiac hypertrophy is the comparison between mice allowed to run on voluntary running wheels with their sedentary group housed without wheels. Mice housed with voluntary running wheels ran 3–5 km day−1 (Rothermel et al. 2001) or 7 km day−1 (Allen et al. 2001) with a resultant physiologic cardiac hypertrophy of 30 % and 18 %, respectively, as compared with the sedentary groups. We speculate that mice with running wheels more closely approximate the ‘wild’ or conditions under the environment that selected the genotype to survive. Based upon the phenotype determining survival in the Palaeolithic era, we believe that the physically active mice should be the control group. Our rationale is that mice in the ‘exercise’ group voluntarily ran, thereby suggesting that control must be considered as the voluntarily physically active group because the physiologic norm of their phenotype from their genotype is voluntarily running. Thus, we propose that the answer to the above question is that the scientific evidence favours physiological cardiac hypertrophy as the true norm for the genotype selected in an environment demanding physical activity for survival.
Dissection of the underlying mechanisms for this physiologic hypertrophy revealed a protein called calcineurin to have a likely role in the production of exercise-induced physiological hypertrophy in sedentary subjects. Rats who underwent voluntary running had a 250 % increase in myocardial calcineurin phosphatase activity (Eto et al. 2000). Another report found that MCIP1 overexpression in transgenic mice blocks calcineurin signalling and prevented about 50 % of the exercise-induced cardiac hypertrophy (Rothermel et al. 2001). Thus, the calcineurin-signalling pathway plays an important role in exercise-induced physiological hypertrophy. Potential roles for IGF-I and noradrenaline in physiological hypertrophy are inferred from the observations of a greater release of IGF-I and noradrenaline into the coronary venous blood of soccer players while at rest, as compared with sedentary controls (Neri Serneri et al. 2001). Approximately 50 % of isoprenaline-induced cardiac hypertrophy in mice was blocked by MCIP1 (Rothermel et al. 2001), which we interpret to mean that the aforementioned noradrenaline overspill reported in the heart of soccer players (Neri Serneri et al. 2001) could be signalling physiological hypertrophy.
A remarkably different gene expression pattern was noted between physiologic and pathologic cardiac hypertrophy. Rats permitted access to voluntary running wheels for 6 weeks had a 22 % increase in left ventricular weight to body weight ratio, and a subsequent 100 % selective increase in TGF-1 mRNA, with no associated changes in TGF-3, fibronectin, preprocollagen-1 or prepro-ANP (Calderone et al. 2001). Intriguingly, the latter four mRNAs were markedly upregulated in pathologic cardiac hypertrophy (Calderone et al. 2001). This selective expression implicates a potentially critical role for TGF-1 in myocardial remodelling, as suggested by Calderone et al. (2001). In addition, a greater accumulation of total collagen has been observed in hearts from pressure-overloaded rats than found in rats that ran on motor-driven treadmills (Burgess et al. 1996), thereby further corroborating the association of increased myocardial fibrosis with decreased compliance (Burlew & Weber, 2000).
In summary, these reports show that exercising produces a unique cardiac phenotype with superior physiological and clinical function. Nevertheless, the human heart of a sedentary subject is defined as ‘normal’ or ‘control’ according to current dogma, while physiological hypertrophy (i.e. an athlete's heart) is defined as an adaptation. We suggest that this designation may be incorrect based upon the fact that the human genotype selected in the Late Palaeolithic period probably favoured hearts with high physiologic capacity and high levels of physical activity, which were necessary for survival. We therefore suggest that the appropriate physiological control heart is from the physically active phenotype.
Ironically then, while the likely norm in 10 000 BC was physiological cardiac hypertrophy that facilitated survival, the prevalent form of cardiac enlargement in the present-day labour-free environment is pathological cardiac hypertrophy. Pathological cardiac hypertrophy reduces cardiac function with a progression to heart failure and shortened survival. This is yet another example whose conclusion is analogous to the thrifty gene hypothesis (Neel, 1999): a genotype that favoured survival in the physically active Palaeolithic era now fails to favour survival in a sedentary culture.
In sum, rather than considering cardiac hypertrophy as an adaptation to exercise, it may be more accurate to consider the notion that the true adaptation in AD 2000 may in fact be cardiac deconditioning due to a lack of exercise, i.e. a sedentary lifestyle. The physiological and clinical significance of this misnomer is that current research that is concerned with the genomic and proteomic adaptations of the compensated and failing heart to pressure overload makes comparisons with a sedentary ‘control’ group, when in fact the true ‘control’ group may be the physically active Late Palaeolithic heart. Thus, incorrect differentially expressed genes may be identified by a comparison of pathological hearts with sedentary hearts rather than the phenotype that determined the surviving genotype.
Endocrinology and metabolism
Insulin resistance
Contracting skeletal muscles increase their glucose uptake. Several studies have clearly demonstrated that proximal insulin-signalling steps are not components of the signalling mechanism by which exercise stimulates glucose uptake (Goodyear & Kahn, 1998). For example, contractile activity does not stimulate autophosphorylation of insulin receptors, IRS tyrosine phosphorylation, or PI3-kinase activity (Goodyear & Kahn, 1998). Furthermore, PI3-kinase inhibitors do not inhibit contraction-stimulated glucose transport in vitro. Goodyear & Kahn (1998) conclude that these signalling studies demonstrate that the underlying molecular mechanisms leading to the insulin- and exercise-induced stimulation of glucose uptake in skeletal muscle are distinct. AMP-activated protein kinase (AMPK) increases in fast red muscle at higher workloads in response to contraction (Winder, 2001). Downstream effects of AMPK activation probably include the stimulation of gene expression for glucose transporter 4 (GLUT4) and hexokinase (both for increased glucose uptake), and mitochondrial enzymes (for increased oxidative phosphorylation; Hardie & Hawley, 2001; Zheng et al. 2001). Intriguingly, activation of AMPK is not the only insulin-independent pathway by which exercise increases glucose uptake into skeletal muscle. Mu et al. (2001) demonstrated that the increase of glucose uptake into contracting skeletal muscle was attenuated by only ≈30 % in mice overexpressing a dominant negative form of AMPK. Increasing either AMPK activity or sarcoplasm Ca2+ concentrations in epitrochlearis muscles or L6 myocytes increased GLUT4 as well as MEF2A and MEF2D protein expression (Ojuka et al. 2002). MEF2A and MEF2D are transcription factors that activate the GLUT4 promoter (Mora & Pessin, 2000) and this same promoter has been shown to be activated by exercise (MacLean et al. 2002).
The failure to decrease muscle glycogen when skeletal muscles are inactive keeps AMPK activities low (Wojtaszewki et al. 2002). Furthermore, glucose transport from the extracellular space into the myocyte cytoplasm is mediated by GLUT4, which is the rate-limiting step for insulin- and exercise-stimulated glucose uptake in skeletal muscle (Thurmond & Pessin, 2001). Interestingly, GLUT4 transcription and mRNA and protein levels are decreased in inactive skeletal muscles (Vukovich et al. 1996).
Diabetes prevalence is 1.1 % in current hunter-gatherer, rudimentary horticultural, simple agricultural and pastoral societies (Eaton et al. 1988), while it is 6 % in developed countries (Black, 2002). Eaton et al. (2002) state that recent hunter-gatherers have largely been free of type 2 diabetes, implying that the underlying genetic factors probably had little adverse effect during the Late Palaeolithic era. The increased prevalence of type 2 diabetes upon ‘westernization’ also supports an environmental, rather than genetic, change for the type 2 diabetes epidemic. For example, the prevalence of type 2 diabetes is six times more for Arizona Pima Indians than for Mexican Pimas (Valencia et al. 1999). Arizona Pimas had five times less (< 5 h week−1) occupational physical activity than did Mexican Pimas (23 h week−1). Arizona Pimas consumed a typical US diet, while the diet of the Mexican Pimas was composed mainly of vegetable staples (Valencia et al. 1999). In the Pacific island of Naura, where diabetes was virtually unknown 50 years ago, it is now present in approximately 40 % of adults (Zimmet et al. 1990). Zimmet et al. concluded: ‘Apart from the heightened genetic susceptibility of certain ethic groups, environmental and behavioural factors such as sedentary lifestyle, nutrition and obesity are clearly important (for the epidemic of diabetes).’ Pre-diabetic conditions (decreased oral glucose tolerance with increased plasma glucose and insulin concentrations) occur within 5–10 days of reducing physical activity levels in healthy humans (Heath et al. 1983) and mice (Seider et al. 1982). Thus, reduced physical activity is associated with a rapid development of insulin resistance.
A potential evolutionary basis for promotion of type 2 diabetes by exercise deficiency was given by Wendorf & Goldfine (1991), who hypothesized that a: ‘selective insulin resistance in muscle would have the effect of blunting the hypoglycaemia that occurs during fasting but would allow energy storage in fat and liver during feeding. Both of these features could allow hunter-gatherers to have survival advantages during periods of food storage. However, in sedentary individuals allowed free access to food, this genotype would be disadvantageous; these individuals would become obese with concomitant secondary insulin resistance in fat and liver.’ Wendorf & Goldfine (1991) cite the Pima Indians, the spiny mouse and the Egyptian sand rat as examples to support their hypothesis as these groups do not have type 2 diabetes in their native state, but develop type 2 diabetes after residing in a sedentary environment with a constant food source. This review concurs with the Wendorf & Goldstein (1991) hypothesis. Ironically then, while thrifty genes probably have enhanced survival through reproduction during eras of famine and drought, these very same thrifty genes diminish survival in selected sedentary populations with continual access to food. For example, the lifespan of diabetics is shortened by an average of 12 years (American Diabetes Association, 1998).
Functioning of exercise-responsive genes in exercise deficiency
The current review has gone beyond a listing of changes in exercise-induced gene expression by contrasting phenotypes with high (hunter-gatherer societies) and low (i.e. cultures no longer requiring physical labour for food acquisition) physical activity levels. One stimulus for this comparison was the statement by Gerber & Crews (1999): ‘For those interested in the health and well-being of humankind, a basic understanding of evolutionary pressures that have shaped human physiological responses to the environment is a necessity‘.
Physical activity is one example of an environmental pressure that shaped the human genotype and phenotype. The differences in caloric expenditure are not trivial, and hence cannot be ignored. For example, Mexican Pima Indians have 21–31 kJ kg−1 day−1 more physical activity than do Arizonan Pima Indians, who are estimated to have geographically separated 700–1000 years ago (Esparza et al. 2000). Cordain et al. (1998) found that recently studied hunter-gatherers had 72 kJ kg−1 day−1 more physical activity than the typical US adult. An exercise deficiency of 72 kJ kg−1 day−1 is the work equivalent of a 70 kg human walking 19–33 more kilometers per day (12-21 more miles per day). Phenotypic changes associated with exercise deficiency are: decreased size and strength of skeletal muscle, lower capacity of skeletal muscle to oxidize carbohydrates and fats, higher insulin resistance, greater homeostatic disruption of cellular metabolism in skeletal muscle at a given absolute work load, lesser vasodilator capacity in perfusion vessels to the heart, smaller maximal cardiac outputs and stroke volumes, and sarcopenia (Holloszy & Booth, 1976; Heath et al. 1983; Åstrand & Rodahl, 1986; Åstrand, 1992; Kingwell, 2000; Tipton, 2001; McGuire et al. 2001). Examples of some of the changes and the mechanisms of such changes in gene expression that underlie the altered phenotypes are also delineated in this review. Trevathan et al. (1999) have asserted: ‘A better understanding of many modern health problems will emerge when we consider that most of human evolution took place when our ancestors were hunter-gatherers.’ Thus, the current review has employed an evolutionary approach to better understand the functions of genes in a high physical activity and exercise-deficient state.
The phenotype associated with exercise deficiency often shows that thresholds of biological significance have been surpassed by altered gene expression so that overt clinical conditions occur (Beaudet et al. 1995). For example, a deficiency in caloric expenditure of only 450 kJ day−1 (107 kcal day−1) from walking > 21 min day−1 to not walking at all is associated with increased prevalences of mortality and many chronic health conditions spanning from diabetes to cancer (Hu et al. 1999, 2000, 2001; Leitzmann et al. 1999; Manson et al. 1999; Martinez et al. 1997; Rockhill et al. 1999, 2001). Exercise deficiency also leads to an increased prevalence of obesity, hypertension, intermittent claudication, sarcopenia, osteoporosis and Alzeihmer's disease (Chakravarthy et al. 2002). Exercise deficiency contributed to 57 million US adults having a metabolic dysfunction (Syndrome X, the cluster of hypertension, atherosclerosis, truncal obesity and insulin resistance) in the US in 1990 (Ford et al. 2002). Conversely, a current dietary antioxidant deficiency, compared with hunter-gatherer diets, could also contribute to an increased prevalence of cardiovascular disease (Eaton & Konner, 1985; Cordain et al. 2002). Thus, although many other factors (for example, high fat/low fibre dietary habits, tobacco or free radicals) clearly contribute to the increased incidences of these disorders (Cordain et al. 2002; Eaton et al. 1988, 2002; Gerber & Crews, 1999; Trevathan et al. 1999), our experience has been that a lack of understanding by the general scientific, medical, judicial and legislative communities for the magnitude of the altered gene expression by exercise deficiency has led to their underestimation or non-consideration of the significance of the functions of exercise-induced gene expressions.
If the exercise-deficient phenotype did not contribute to overt clinical disorders, exercise-induced changes in gene expression would only be physiological phenomena (Booth et al. 2002). However, alterations in gene expression by exercise deficiency contribute to morbidity and mortality, which emphasizes the importance of using the evolutionary pressures that have shaped human physiological responses to define better the functions for exercise-induced changes in gene expression in both physiological and pathophysiological conditions.
Acknowledgments
This review was written with the support of NIH AR19393 (F. B.).
References
- Abru TA, Elhadd T, Pfeifer M, Clayton RN. Endothelial dysfunction in endocrine disease. Trends in Endocrinology and Metabolism. 2001;12:257–265. doi: 10.1016/s1043-2760(01)00425-8. [DOI] [PubMed] [Google Scholar]
- Adams GR, Haddad F, Baldwin KM. Time course of changes in markers of myogenesis in overloaded rat skeletal muscles. Journal of Applied Physiology. 1999;87:1705–1712. doi: 10.1152/jappl.1999.87.5.1705. [DOI] [PubMed] [Google Scholar]
- Allen DL, Harrison BC, Maass A, Bell ML, Byrnes WC, Leinwand LA. Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. Journal of Applied Physiology. 2001;90:1900–1908. doi: 10.1152/jappl.2001.90.5.1900. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. Economic consequences of diabetes mellitus in the US in (1997) Diabetes Care. 1998;21:296–309. doi: 10.2337/diacare.21.2.296. [DOI] [PubMed] [Google Scholar]
- Åstrand PO, Rodahl K. Textbook of Work Physiology. New York: McGraw Hill; 1986. pp. 1–11. [Google Scholar]
- Åstrand PO. J. B. Wolffe Memorial Lecture: ‘Why exercise?‘. Medical Science and Sports Exercise. 1992;24:153–162. [PubMed] [Google Scholar]
- Baar K, Esser K. Phosphorylation of p70(S6k). correlates with increased skeletal muscle mass following resistance exercise. American Journal of Physiology. Cell Physiology. 1999;276:C120–127. doi: 10.1152/ajpcell.1999.276.1.C120. [DOI] [PubMed] [Google Scholar]
- Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proceedings of the National Academy of Sciences of the USA. 1998;95:15603–15607. doi: 10.1073/pnas.95.26.15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudet AL, Scriver CR, Sly WS, Valle D. Genetics, biochemistry, and molecular basis of variant human phenotypes. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Stanbury JB, Wyngaarden JB, Fredrickson DS, editors. The Metabolic and Molecular Bases of Inherited Disease. 7. Vol. 1. New York: McGraw Hill; 1995. p. 79. [Google Scholar]
- Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. American Journal of Physiology. 2001;281:E1340–1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- Black SA. Diabetes, diversity, and disparity: what do we do with the evidence? American Journal of Public Health. 2002;92:543–548. doi: 10.2105/ajph.92.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, GonzaleZ M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nature Cell Biology. 2001b;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- Booth FW, Charavarthy MV, Gordon SE, Spangenburg EE. Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. Journal of Applied Physiology. 2002;93:3–30. doi: 10.1152/japplphysiol.00073.2002. [DOI] [PubMed] [Google Scholar]
- Booth FW, Holloszy JO. Cytochrome c turnover in rat skeletal muscles. Journal of Biological Chemistry. 1977;252:416–419. [PubMed] [Google Scholar]
- Booth FW, Vyas DR. Genes, environment, and exercise. Advances in Experimental Medicine and Biology. 2001;502:13–20. doi: 10.1007/978-1-4757-3401-0_3. [DOI] [PubMed] [Google Scholar]
- Bowles DK, Woodman CR, Laughlin MH. Coronary smooth muscle and endothelial adaptations to exercise training. Exercise Sport Science Review. 2000;28:57–62. [PubMed] [Google Scholar]
- Burgess ML, Buggy J, Price RL, Abel FL, Terracio L, Samarel AM, Borg TK. Exercise- and hypertension-induced collagen changes are related to left ventricular function in rat hearts. American Journal of Physiology. 1996;270:H151–159. doi: 10.1152/ajpheart.1996.270.1.H151. [DOI] [PubMed] [Google Scholar]
- Burlew BS, Weber KT. Connective tissue and the heart. Functional significance and regulatory mechanisms. Cardiology Clinics. 2000;18:435–442. doi: 10.1016/s0733-8651(05)70154-5. [DOI] [PubMed] [Google Scholar]
- Butler AA, Yakar S, Gewolb IH, Karas M, Okubo Y, Leroith D. Insulin-like growth factor-I receptor signal transduction: at the interface between physiology and cell biology. Comparative Biochemistry and Physiology. Part B, Biochemistry and Molecular Biology. 1998;121:19–26. doi: 10.1016/s0305-0491(98)10106-2. [DOI] [PubMed] [Google Scholar]
- Calderone A, Murphy RJ, Lavoie J, Colombo F, Beliveau L. TGF beta(1). and prepro-ANP mRNAs are differentially regulated in exercise-induced cardiac hypertrophy. Journal of Applied Physiology. 2001;91:771–776. doi: 10.1152/jappl.2001.91.2.771. [DOI] [PubMed] [Google Scholar]
- Carson JA, Yan Z, Booth FW, Coleman ME, SchwartZ RJ, Stump CS. Regulation of skeletal alpha-actin promoter in young chickens during hypertrophy caused by stretch overload. American Journal of Physiology. 1995;268:C918–924. doi: 10.1152/ajpcell.1995.268.4.C918. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza P. The History and Geography of Human Genes. Princeton: Princeton Publishers; 1994. [Google Scholar]
- Chakravarthy MV, Davis B, Booth FW. IGF-I restores satellite cell proliferative potential in immobilized old skeletal muscle. Journal of Applied Physiology. 2000;89:1365–1379. doi: 10.1152/jappl.2000.89.4.1365. [DOI] [PubMed] [Google Scholar]
- Chakravarthy MV, Joyner MJ, Booth FW. An obligation for primary care physicians to prescribe physical activity to sedentary patients to reduce the risk of chronic health conditions. Mayo Clinic Proceedings. 2002;77:165–173. doi: 10.4065/77.2.165. [DOI] [PubMed] [Google Scholar]
- Cherow BA, Vallasi GA. The Columbia Encyclopedia. 7. Boston: Columbia University Press; 2002. [Google Scholar]
- Constable SH, Favier RJ, McLane JA, Fell RD, Chen M, Holloszy JO. Energy metabolism in contracting rat skeletal muscle: adaptation to exercise training. American Journal of Physiology. 1987;253:C316–322. doi: 10.1152/ajpcell.1987.253.2.C316. [DOI] [PubMed] [Google Scholar]
- Cordain L, Eaton SB, Miller JB, Mann N, Hill K, Cordain L, Eaton SB, Miller JB, Mann N, Hill K. The paradoxical nature of hunter-gatherer diets: meat-based, yet non-atherogenic. European Journal of Clinical Nutrition. 2002;56(suppl. 1):S42–52. doi: 10.1038/sj.ejcn.1601353. [DOI] [PubMed] [Google Scholar]
- Cordain L, Gotshall RW, Eaton SB, Eaton SB., III Physical activity, energy expenditure and fitness: an evolutionary perspective. International Journal of Sports Medicine. 1998;19:328–335. doi: 10.1055/s-2007-971926. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Abraham WM, Terjung RL. Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. Journal of Applied Physiology: Respiratory, Environmental and Exercise Physiology. 1982;53:844–850. doi: 10.1152/jappl.1982.53.4.844. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Tullson PC, Terjung RL. Influence of mitochondrial content on the sensitivity of respiratory control. Journal of Biological Chemistry. 1987;262:9109–9114. [PubMed] [Google Scholar]
- Dunn SE, Chin ER, Michel RN. Matching of calcineurin activity to upstream effectors is critical for skeletal muscle growth. Journal of Cell Biology. 2000;151:663–672. doi: 10.1083/jcb.151.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. New England Journal of Medicine. 1985;312:283–289. doi: 10.1056/NEJM198501313120505. [DOI] [PubMed] [Google Scholar]
- Eaton SB, Konner M, Shostak M. Stoneagers in the fast lane: chronic degenerative diseases in evolutionary perspective. American Journal of Medicine. 1988;84:739–749. doi: 10.1016/0002-9343(88)90113-1. [DOI] [PubMed] [Google Scholar]
- Eaton SB, Strassman BI, Nesse RM, Neel JV, Ewald PW, Williams GC, Weder AB, Eaton SB, III, Lindeberg S, Konner MJ, Mysterud I, Cordain L. Evolutionary health promotion. Preventive Medicine. 2002;34:109–118. doi: 10.1006/pmed.2001.0876. [DOI] [PubMed] [Google Scholar]
- Ehsani AA, Hagberg JM, Hickson RC. Rapid changes in left ventricular dimensions and mass in response to physical conditioning and deconditioning. American Journal of Cardiology. 1978;42:52–56. doi: 10.1016/0002-9149(78)90984-0. [DOI] [PubMed] [Google Scholar]
- Esparza J, Fox C, Harper IT, Bennett PH, SchulZ LO, Valencia ME, Ravussin E. Daily energy expenditure in Mexican and USA Pima Indians: low physical activity as a possible cause of obesity. International Journal of Obesity and Related Metabolic Disorders. 2000;24:55–59. doi: 10.1038/sj.ijo.0801085. [DOI] [PubMed] [Google Scholar]
- Eto Y, Yonekura K, Sonoda M, Arai N, Sata M, Sugiura S, Takenaka K, Gualberto A, Hixon ML, Wagner MW, Aoyagi T. Calcineurin is activated in rat hearts with physiological left ventricular hypertrophy induced by voluntary exercise training. Circulation. 2000;101:2134–2137. doi: 10.1161/01.cir.101.18.2134. [DOI] [PubMed] [Google Scholar]
- Fluck M, Carson JA, SchwartZ RJ, Booth FW. SRF protein is upregulated during stretch-induced hypertrophy of rooster ALD muscle. Journal of Applied Physiology. 1999;86:1793–1799. doi: 10.1152/jappl.1999.86.6.1793. [DOI] [PubMed] [Google Scholar]
- Ford ES, Giles WH, DietZ WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. Journal of Clinical Investigation. 2000;105:1631–1639. doi: 10.1172/JCI9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber LM, Crews DE. Evolutionary perspectives on chronic diseases. In: Trevathan WR, Smith EO, McKenna JJ, editors. Evolutionary Medicine. New York: Oxford University Press; 1999. pp. 443–469. [Google Scholar]
- Gielen S, Schuler G, Hambrecht R. Exercise training in coronary artery disease and coronary vasomotion. Circulation. 2001;103:E1–6. doi: 10.1161/01.cir.103.1.e1. [DOI] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proceedings of the National Academy of Sciences of the USA. 2001;98(14):440–14 445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear LJ, Kahn BB. Exercise, glucose transport, and insulin sensitivity. Annual Review of Medicine. 1998;49:235–261. doi: 10.1146/annurev.med.49.1.235. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Rungi AA, Inagaki H, Hood DA. Effects of contractile activity on mitochondrial transcription factor A expression in skeletal muscle. Journal of Applied Physiology. 2001;90:389–396. doi: 10.1152/jappl.2001.90.1.389. [DOI] [PubMed] [Google Scholar]
- Gregory P, Gagnon J, Essig DA, Reid SK, Prior G, Zak R. Differential regulation of actin and myosin isoenzyme synthesis in functionally overloaded skeletal muscle. Biochemical Journal. 1990;265:525–532. doi: 10.1042/bj2650525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton AC, Hall JE. Textbook of Physiology. 9. Philadelphia: W. B. Saunders; 1996. [Google Scholar]
- Hahn RA, Teutsch SM, Rothenberg RB, Marks JS. Excess deaths from nine chronic diseases in the United States, 1986. JAMA. 1990;264:2654–2659. [PubMed] [Google Scholar]
- Hambrecht R, Wolf A, Gielen S, Linke A, Hofer J, Erbs S, Schoene N, Schuler G. Effect of exercise on coronary endothelial function in patients with coronary artery disease. New England Journal of Medicine. 2000;342:454–460. doi: 10.1056/NEJM200002173420702. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- Heath GW, Gavin JR, III, Hinderliter JM, Hagberg JM, Bloomfield SA, Holloszy JO. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. Journal of Applied Physiology: Respiratory, Environmental and Exercise Physiology. 1983;55:512–517. doi: 10.1152/jappl.1983.55.2.512. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Annual Review of Physiology. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. Journal of Applied Physiology: Respiratory, Environmental and Exercise Physiology. 1984;56:831–838. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- Hood DA. Invited review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. Journal of Applied Physiology. 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- Hu FB, Manson JE, Stampfer MJ, ColditZ G, Liu S, Solomon CG, Willett WC. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. New England Journal of Medicine. 2001;345:790–797. doi: 10.1056/NEJMoa010492. [DOI] [PubMed] [Google Scholar]
- Hu FB, Sigal RJ, Rich-Edwards JW, ColditZ GA, Solomon CG, Willett WC, Speizer FE, Manson JE. Walking compared with vigorous physical activity and risk of type 2 diabetes in women: a prospective study. JAMA. 1999;282:1433–1439. doi: 10.1001/jama.282.15.1433. [DOI] [PubMed] [Google Scholar]
- Hu FB, Stampfer MJ, ColditZ GA, Ascherio A, Rexrode KM, Willett WC, Manson JE. Physical activity and risk of stroke in women. JAMA. 2000;283:2961–2967. doi: 10.1001/jama.283.22.2961. [DOI] [PubMed] [Google Scholar]
- Iemitsu M, Miyauchi T, Maeda S, Sakai S, Kobayashi T, Fujii N, Miyazaki H, Matsuda M, Yamaguchi I. Physiological and pathological cardiac hypertrophy induce different molecular phenotypes in the rat. American Journal of Physiology. 2001;281:R2029–2036. doi: 10.1152/ajpregu.2001.281.6.R2029. [DOI] [PubMed] [Google Scholar]
- Kelm M. Flow-mediated dilatation in human circulation: diagnostic and therapeutic aspects. American Journal of Physiology. 2002;282:H1–5. doi: 10.1152/ajpheart.2002.282.1.H1. [DOI] [PubMed] [Google Scholar]
- Kingwell BA. Nitric oxide-mediated metabolic regulation during exercise: effects of training in health and cardiovascular disease. FASEB Journal. 2000;14:1685–1696. doi: 10.1096/fj.99-0896rev. [DOI] [PubMed] [Google Scholar]
- Kotler DP, Tierney AR, Wang J, Pierson RN., Jr Magnitude of body-cell-mass depletion and the timing of death from wasting in AIDS. American Journal of Clinical Nutrition. 1989;50:444–447. doi: 10.1093/ajcn/50.3.444. [DOI] [PubMed] [Google Scholar]
- Kramsch DM, Aspen AJ, AbramowitZ BM, Kreimendahl T, Hood WB. Reduction of coronary atherosclerosis by moderate conditioning exercise in monkeys on an atherogenic diet. New England Journal of Medicine. 1981;305:1483–1489. doi: 10.1056/NEJM198112173052501. [DOI] [PubMed] [Google Scholar]
- Kreiger M. Ueber die atrophie der menschlichen organe bei inanition. Zeitschrift fur angewandte Anatomie und Konstitutionslehre. 1921;7:87–134. [Google Scholar]
- Laurent GJ, Sparrow MP, Millward DJ. Turnover of muscle protein in the fowl. Changes in rates of protein synthesis and breakdown during hypertrophy of the anterior and posterior latissimus dorsi muscles. Biochemical Journal. 1978;176:407–417. doi: 10.1042/bj1760407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TC, Shi Y, SchwartZ RJ. Displacement of BrdUrd-induced YY1 by serum response factor activates skeletal alpha-actin transcription in embryonic myoblasts. Proceedings of the National Academy of Sciences of the USA. 1992;89:9814–9818. doi: 10.1073/pnas.89.20.9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitzmann MF, Rimm EB, Willett WC, Spiegelman D, Grodstein F, Stampfer MJ, ColditZ GA, Giovannucci E. Recreational physical activity and the risk of cholecystectomy in women. New England Journal of Medicine. 1999;341:777–784. doi: 10.1056/NEJM199909093411101. [DOI] [PubMed] [Google Scholar]
- Linzbach AJ. Mikrometrische und histologische analyse hypertropher menschlicher herzen. Virchows Archiv fur Pathologische Anatomie und Physiologie und fur Klinische Medizin. 1947;314:534–594. [PubMed] [Google Scholar]
- McGuire DK, Levine BD, Williamson JW, Snell PG, Blomqvist CG, Saltin B, Mitchell JH. A 30-year follow-up of the Dallas Bedrest and Training Study: II. Effect of age on cardiovascular adaptation to exercise training. Circulation. 2001;104:1358–1366. [PubMed] [Google Scholar]
- McKoy G, Ashley W, Mander J, Yang SY, Williams N, Russell B, Goldspink G. Expression of insulin growth factor-1 splice variants and structural genes in rabbit skeletal muscle induced by stretch and stimulation. Journal of Physiology. 1999;516:583–592. doi: 10.1111/j.1469-7793.1999.0583v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean PS, Zheng D, Jones JP, Olson AL, Dohm GL. Exercise induced transcription of the muscle glucose transporter (GLUT 4). gene. Biochemical Biophysical Research Communications. 2002;292:409–414. doi: 10.1006/bbrc.2002.6654. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Manson JE, Hu FB, Rich-Edwards JW, ColditZ GA, Stampfer MJ, Willett WC, Speizer FE, Hennekens CH. A prospective study of walking as compared with vigorous exercise in the prevention of coronary heart disease in women. New England Journal of Medicine. 1999;341:650–658. doi: 10.1056/NEJM199908263410904. [DOI] [PubMed] [Google Scholar]
- Martine ME, Giovannucci E, Spiegelman D, Hunter DJ, Willett WC, ColditZ GA. Leisure-time physical activity, body size, and colon cancer in women. Nurses'. Health Study Research Group Journal of the National Cancer Institute. 1997;89:948–955. doi: 10.1093/jnci/89.13.948. [DOI] [PubMed] [Google Scholar]
- Mitchell PO, Mills ST, Pavlath GK. Calcineurin differentially regulates maintenance and growth of phenotypically distinct muscles. American Journal of Physiology. 2002;282:C984–992. doi: 10.1152/ajpcell.00483.2001. [DOI] [PubMed] [Google Scholar]
- Molé PA, Oscai LB, Holloszy JO. Adaptations of muscle to exercise. Journal of Clinical Investigation. 1971;50:2323–2330. doi: 10.1172/JCI106730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora S, Pessin JE. The MEF2A isoform is required for striated muscle specific expression of the insulin-responsive GLUT4 glucose transporter. Journal of Biological Chemistry. 2000;275(16):323–16 328. doi: 10.1074/jbc.M910259199. [DOI] [PubMed] [Google Scholar]
- Morrison PR, Montgomery JA, Wong TS, Booth FW. Cytochrome c protein-synthesis rates and mRNA contents during atrophy and recovery in skeletal muscle. Biochemical Journal. 1987;241:257–263. doi: 10.1042/bj2410257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Molecular Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Muller JM, Myers PR, Laughlin MH. Vasodilator responses of coronary resistance arteries of exercise-trained pigs. Circulation. 1994;89:2308–2314. doi: 10.1161/01.cir.89.5.2308. [DOI] [PubMed] [Google Scholar]
- Murakami T, Shimomura Y, Yoshimura A, Sokabe M, Fujitsuka N. Induction of nuclear respiratory factor-1 expression by an acute bout of exercise in rat muscle. Biochimica et Biophysica Acta. 1998;1381:113–122. doi: 10.1016/s0304-4165(98)00018-x. [DOI] [PubMed] [Google Scholar]
- Nader GA, Esser KA. Intracellular signaling specificity in skeletal muscle in response to different modes of exercise. Journal of Applied Physiology. 2001;90:1936–1942. doi: 10.1152/jappl.2001.90.5.1936. [DOI] [PubMed] [Google Scholar]
- Neel JV. Diabetes mellitus: a ‘thrifty’ genotype rendered detrimental by ‘progress’? American Journal of Human Genetics. 1962;14:352–353. [PMC free article] [PubMed] [Google Scholar]
- Neel JV, Weder AB, Julius S. Type II diabetes, essential hypertension, and obesity as ‘syndromes of impaired genetic homeostasis’: the ‘thrifty genotype’ hypothesis enters the 21st century. Perspectives in Biology and Medicine. 1998;42:44–74. doi: 10.1353/pbm.1998.0060. [DOI] [PubMed] [Google Scholar]
- Neri Serneri GG, Boddi M, Modesti PA, Cecioni I, Coppo M, Padeletti L, Michelucci A, Colella A, Galanti G. Increased cardiac sympathetic activity and insulin-like growth factor-I formation are associated with physiological hypertrophy in athletes. Circulation Research. 2001;89:977–982. doi: 10.1161/hh2301.100982. [DOI] [PubMed] [Google Scholar]
- Nesse RM, Williams GC. Evolution and the origins of disease. Scientific American. 1998;279:86–93. doi: 10.1038/scientificamerican1198-86. [DOI] [PubMed] [Google Scholar]
- Olson EN, Williams RS. Remodeling muscles with calcineurin. Bioessays. 2000;22:510–519. doi: 10.1002/1521-1878(200011)22:11<1049::AID-BIES14>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Peery TM. The new and old diseases: A study of mortality trends in the United States, 1900–1969. Ward Burdick Award address. American Journal Clinical Pathology. 1975;63:453–474. doi: 10.1093/ajcp/63.4.453. [DOI] [PubMed] [Google Scholar]
- Perusse L, Bouchard C. Genotype-environment interaction in human obesity. Nutrition Reviews. 1999;57:S31–37. doi: 10.1111/j.1753-4887.1999.tb01785.x. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Ordway GA, Saltin B, Neufer PD. Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. American Journal of Physiology. 2000;279:E806–814. doi: 10.1152/ajpendo.2000.279.4.E806. [DOI] [PubMed] [Google Scholar]
- Pratley RE. Gene-environment interactions in the pathogenesis of type 2 diabetes mellitus: lessons learned from the Pima Indians. Proceedings of the Nutrition Society. 1998;57:175–181. doi: 10.1079/pns19980029. [DOI] [PubMed] [Google Scholar]
- Raskoff WJ, Goldman S, Cohn K. The ‘athletic heart’. Prevalence and physiological significance of left ventricular enlargement in distance runners. JAMA. 1976;236:158–162. doi: 10.1001/jama.236.2.158. [DOI] [PubMed] [Google Scholar]
- Robin ED. Claude Bernard and the Internal Environment: A Memorial Symposium. New York: Dekker; 1979. [Google Scholar]
- Rockhill B, Willett WC, Hunter DJ, Manson JE, Hankinson SE, ColditZ GA. A prospective study of recreational physical activity and breast cancer risk. Archives of Internal Medicine. 1999;159:2290–2296. doi: 10.1001/archinte.159.19.2290. [DOI] [PubMed] [Google Scholar]
- Rockhill B, Willett WC, Manson JE, Leitzmann MF, Stampfer MJ, Hunter DJ, ColditZ GA. Physical activity and mortality: a prospective study among women. Amercian Journal of Public Health. 2001;91:578–583. doi: 10.2105/ajph.91.4.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel C, Bodine SC, Clarke BA, Rossman R, NuneZ L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nature Cell Biology. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- Rothermel BA, McKinsey TA, Vega RB, Nicol RL, Mammen P, Yang J, Antos CL, Shelton JM, Bassel-Duby R, Olson EN, Williams RS. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proceedings of the National Academy of Sciences of the USA. 2001;98:3328–3333. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff CB, Trinkaus E, Walker A, Larsen CS. Postcranial robusticity in Homo. I: Temporal trends and mechanical interpretation. Amercian Journal of Physical Anthropology. 1993;91:21–53. doi: 10.1002/ajpa.1330910103. [DOI] [PubMed] [Google Scholar]
- Rush JW, Laughlin MH, Woodman CR, Price EM. SOD-1 expression in pig coronary arterioles is increased by exercise training. American Journal of Physiology. 2000;279:H2068–2076. doi: 10.1152/ajpheart.2000.279.5.H2068. [DOI] [PubMed] [Google Scholar]
- Saltin B, Åstrand PO. Free fatty acids and exercise. American Journal of Clinical Nutriton. 1993;57(suppl. 5):752–757S. doi: 10.1093/ajcn/57.5.752S. [DOI] [PubMed] [Google Scholar]
- Saltin B, Karlsson J. Muscle ATP, CP, and lactate during exercise after physical conditioning. In: Pernow B, Saltin B, editors. Muscle Metabolism During Exercise. New York: Plenum Press; 1971. pp. 395–399. [Google Scholar]
- Seider MJ, Nicholson WF, Booth FW. Insulin resistance for glucose metabolism in disused soleus muscle of mice. American Journal of Physiology. 1982;242:E12–18. doi: 10.1152/ajpendo.1982.242.1.E12. [DOI] [PubMed] [Google Scholar]
- Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circulation Research. 1994;74:349–353. doi: 10.1161/01.res.74.2.349. [DOI] [PubMed] [Google Scholar]
- Sharma M, Langley B, Bass J, Kambadur R. Myostatin in muscle growth and repair. Exercise Sport Science Reviews. 2001;29:155–158. doi: 10.1097/00003677-200110000-00004. [DOI] [PubMed] [Google Scholar]
- Sherman S. Preventing and treating osteoporosis: strategies at the millennium. Annals of the New York Academy of Sciences. 2001;949:188–197. [PubMed] [Google Scholar]
- Sherwood DJ, Dufresne SD, Markuns JF, Cheatham B, Moller DE, Aronson D, Goodyear LJ. Differential regulation of MAP kinase, p70(S6K), and Akt by contraction and insulin in rat skeletal muscle. American Journal of Physiology. 1999;276:E870–878. doi: 10.1152/ajpendo.1999.276.5.E870. [DOI] [PubMed] [Google Scholar]
- Stroppolo ME, Falconi M, Caccuri AM, Desideri A. Superefficient enzymes. Cellular and Molecular Life Sciences. 2001;58:1451–1460. doi: 10.1007/PL00000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond DC, Pessin JE. Molecular machinery involved in the insulin regulated fusion of GLUT4-containing vesicles with plasma membrane. Molecular Membrane Biology. 2001;18:237–245. doi: 10.1080/09687680110082400. [DOI] [PubMed] [Google Scholar]
- Tipton KD. Muscle protein metabolism in the elderly: influence of exercise and nutrition. Canadian Journal of Applied Physiology. 2001;26:588–606. doi: 10.1139/h01-033. [DOI] [PubMed] [Google Scholar]
- Trevathan WR, Smith EO, McKenna JJ. Introduction. In: Trevathan WR, Smith EO, McKenna JJ, editors. Evolutionary Medicine. New York: Oxford University Press; 1999. pp. 3–6. [Google Scholar]
- Valencia ME, Bennett PH, Ravussin E, Esparza J, Fox C, SchulZ LO. The Pima Indians in Sonora, Mexico. Nutrition Reviews. 1999;57:S55–57. doi: 10.1111/j.1753-4887.1999.tb01789.x. [DOI] [PubMed] [Google Scholar]
- Vukovich MD, Arciero PJ, Kohrt WM, Racette SB, Hansen PA, Holloszy JO. Changes in insulin action and GLUT-4 with 6 days of inactivity in endurance runners. Journal of Applied Physiology. 1996;80:240–244. doi: 10.1152/jappl.1996.80.1.240. [DOI] [PubMed] [Google Scholar]
- Vyas D, Spangenburg EE, Abraha TW, Childs TE, Booth FW. GSK3B negatively regulates skeletal myotube hypertrophy. American Journal of Physiology - Cell Physiology. 2002;283:C545–551. doi: 10.1152/ajpcell.00049.2002. [DOI] [PubMed] [Google Scholar]
- Wehling M, Cai B, Tidball JG. Modulation of myostatin expression during modified muscle use. FASEB Journal. 2000;14:103–110. doi: 10.1096/fasebj.14.1.103. [DOI] [PubMed] [Google Scholar]
- Wendorf M, Goldfine ID. Archaeology of NIDDM. Excavation of the ‘thrifty’ genotype. Diabetes. 1991;40:161–165. doi: 10.2337/diab.40.2.161. [DOI] [PubMed] [Google Scholar]
- Wikman-Coffelt J, Parmley WW, Mason DT. The cardiac hypertrophy process. Analyses of factors determining pathological vs. physiological development. Circulation Research. 1979;45:697–707. doi: 10.1161/01.res.45.6.697. [DOI] [PubMed] [Google Scholar]
- Williams RS, Salmons S, Newsholme EA, Kaufman RE, Mellor J. Regulation of nuclear and mitochondrial gene expression by contractile activity in skeletal muscle. Journal of Biological Chemistry. 1986;261:376–380. [PubMed] [Google Scholar]
- Winder WW. Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. Journal of Applied Physiology. 2001;91:1017–1028. doi: 10.1152/jappl.2001.91.3.1017. [DOI] [PubMed] [Google Scholar]
- Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. American Journal of Physiology. 1996;270:E299–304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- Winick M. Hunger Disease - Studies by Jewish Physicians in the Warsaw Ghetto. New York: John Wiley & Sons; 1979. [Google Scholar]
- Wojtaszewki JF, Jorgensen SB, Hellsten Y, Hardie DG, Richter EA. Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA). riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes. 2002;51:284–292. doi: 10.2337/diabetes.51.2.284. [DOI] [PubMed] [Google Scholar]
- Woodman CR, Muller JM, Laughlin MH, Price EM. Induction of nitric oxide synthase mRNA in coronary resistance arteries isolated from exercise-trained pigs. American Journal of Physiology. 1997;273:H2575–2579. doi: 10.1152/ajpheart.1997.273.6.H2575. [DOI] [PubMed] [Google Scholar]
- Wroblewski Lissin L, Cooke JP. Maintaining the endothelium: preventive strategies for vessel integrity. Preventive Cardiology. 2000;3:172–177. doi: 10.1111/j.1520-037x.2000.803811.x. [DOI] [PubMed] [Google Scholar]
- Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- Xia Y, Buja LM, Scarpulla RC, McMillin JB. Electrical stimulation of neonatal cardiomyocytes results in the sequential activation of nuclear genes governing mitochondrial proliferation and differentiation. Proceedings of the National Academy of Sciences of the USA. 1997;94(11):399–11 404. doi: 10.1073/pnas.94.21.11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Maclean PS, Pohnert SC, Knight JB, Olson AL, Winder WW, Dohm GL. Regulation of muscle GLUT-4 transcription by AMP-activated protein kinase. Journal of Applied Physiology. 2001;91:1073–1083. doi: 10.1152/jappl.2001.91.3.1073. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Dowse G, Finch C, Serjeantson S, King H. The epidemiology and natural history of NIDDM - lessons from the South Pacific. Diabetes/Metabolism Reviews. 1990;6:91–124. doi: 10.1002/dmr.5610060203. [DOI] [PubMed] [Google Scholar]