

Abstract
Experiments were performed to determine whether capacitative Ca2+ entry (CCE) can be activated in canine pulmonary and renal arterial smooth muscle cells (ASMCs) and whether activation of CCE parallels the different functional structure of the sarcoplasmic reticulum (SR) in these two cell types. The cytosolic [Ca2+] was measured by imaging fura-2-loaded individual cells. Increases in the cytosolic [Ca2+] due to store depletion in pulmonary ASMCs required simultaneous depletion of both the inositol 1,4,5-trisphosphate (InsP3)- and ryanodine (RY)-sensitive SR Ca2+ stores. In contrast, the cytosolic [Ca2+] rises in renal ASMCs occurred when the SR stores were depleted through either the InsP3 or RY pathways. The increase in the cytosolic [Ca2+] due to store depletion in both pulmonary and renal ASMCs was present in cells that were voltage clamped and was abolished when cells were perfused with a Ca2+-free bathing solution. Rapid quenching of the fura-2 signal by 100 μM Mn2+ following SR store depletion indicated that extracellular Ca2+ entry increased in both cell types and also verified that activation of CCE in pulmonary ASMCs required the simultaneous depletion of the InsP3- and RY-sensitive SR Ca2+ stores, while CCE could be activated in renal ASMCs by the depletion of either of the InsP3- or RY-sensitive SR stores. Store depletion Ca2+ entry in both pulmonary and renal ASMCs was strongly inhibited by Ni2+ (0.1–10 mM), slightly inhibited by Cd2+ (200–500 μM), but was not significantly affected by the voltage-gated Ca2+ channel (VGCC) blocker nisoldipine (10 μM). The non-selective cation channel blocker Gd3+ (100 μM) inhibited a portion of the Ca2+ entry in 6 of 18 renal but not pulmonary ASMCs. These results provide evidence that SR Ca2+ store depletion activates CCE in parallel with the organization of intracellular Ca2+ stores in canine pulmonary and renal ASMCs.
The sarcoplasmic reticulum (SR) of smooth muscle cells possesses two types of Ca2+ release channels: ryanodine (RY)-sensitive channels that are activated by rises in the [Ca2+], and inositol 1,4,5-trisphosphate (InsP3)-sensitive Ca2+ channels that are activated by InsP3, which is produced downstream of neuro-humoral stimulation of G-protein-coupled or tyrosine-coupled membrane bound receptors (Bootman & Berridge, 1995). Recent contractile and Ca2+ imaging studies from our laboratory demonstrated that structural differences exist in the SR Ca2+ stores of pulmonary and renal arterial smooth muscle cells (ASMCs) (Jabr et al. 1997; Janiak et al. 2001). The contractile data demonstrated that in pulmonary arterial rings phenylephrine (PE) caused contraction through release from InsP3-sensitive stores (Somlyo & Somlyo, 1994). This initial contraction could be inhibited without affecting subsequent contraction due to release from RY-sensitive stores with caffeine (CAF) (Jabr et al. 1997) when the InsP3 stores were depleted in the presence of the sarcoplasmic-endoplasmic reticulum Ca2+ ATPase (SERCA) blocker cyclopiazonic acid (CPA) (Goeger et al. 1988). Similarly, depletion of RY-sensitive stores by exposing cells to CAF in the presence of RY did not affect subsequent contraction due to release from the InsP3-sensitive stores. These experiments along with recent Ca2+ imaging experiments on individual smooth muscle cells dispersed from pulmonary and renal arteries (Janiak et al. 2001) provided good evidence that, in canine pulmonary ASMCs, the InsP3- and RY-sensitive Ca2+ stores are independent, whereas in canine renal ASMCs the InsP3 and RY receptors lie on an overlapping SR Ca2+ store.
Depletion of SR Ca2+ stores in many cell types activates Ca2+ permeable store-operated channels (SOCs) on the plasma membrane, which replete the empty stores through a process known as capacitative Ca2+ entry (CCE) (Putney, 1986). The mechanisms linking store depletion to CCE activation are diverse. CCE can be activated through stimulation of SR-bound InsP3 receptors that couple to SOCs (Ma et al. 2000). Decreases in the endoplasmic reticulum (ER) luminal Ca2+ content can also activate CCE independent of direct InsP3 receptor stimulation (Hofer et al. 1998). A capacitative influx factor (CIF) can also be released, presumably from the ER, due to ER [Ca2+] decreases, which then activates SOCs on the plasma membrane (Putney & Bird, 1993; Randriamampita & Tsien, 1993).
CCE is common to many cell types including vascular smooth muscle cells from high resistance arteries; depletion of the intracellular Ca2+ stores activates a CCE pathway in smooth muscle cells from preglomerular arteriolar vessels (Fellner & Arendshorst, 1999), rat pulmonary arteries (Robertson et al. 2000; Ng & Gurney, 2001), human pulmonary arteries (Golovina, 1999) and swine renal arteries (Utz et al. 1999). Although a recent report demonstrated that exposing vascular smooth muscle cells to CIF activates CCE (Trepakova et al. 2000), the mechanism of CCE activation and the underlying conductances are not well understood (Gibson et al. 1998). A better understanding of the functional organization of SR Ca2+ stores and CCE pathways in pulmonary ASMCs is particularly important since release of Ca2+ from SR stores and activation of CCE pathways have been implicated in the unique constrictor response of pulmonary arteries to hypoxia (Jabr et al. 1997; Robertson et al. 2000; Dipp & Evans, 2001).
Because SR stores are coupled to capacitative entry and the organization of the SR stores of pulmonary and renal ASMCs are different, the present study was designed to test the following hypotheses: (1) that SR Ca2+ store depletion activates CCE in both pulmonary and renal ASMCs and (2) that activation of this pathway parallels the differential organization of the RY and InsP3 intracellular Ca2+ stores in these ASMCs (Janiak et al. 2001). Some of this work has appeared in abstract form (Wilson et al. 2001).
METHODS
Cell isolation
Smooth muscle cells were isolated from high resistance canine pulmonary and renal arteries as previously described (Janiak et al. 2001). Mongrel dogs of either sex were killed with pentobarbital sodium (45 mg kg−1i.v.) and ketamine (15 mg kg−1i.v.), as approved by the University of Nevada at Reno Institutional Animal Care and Use Committee. The heart and lungs were excised en bloc, while the kidneys were excised separately. The third and fourth branches of pulmonary and renal arteries were dissected at 5 °C to decrease cellular metabolic activity. The main pulmonary and renal arteries were flushed with a low-Ca2+ physiological saline solution (PSS) containing (mm): 125 NaCl; 5.36 KCl; 0.336 Na2HPO4; 0.44 K2HPO4; 11 Hepes; 1.2 MgCl2; 0.05 CaCl2; 10 glucose; 2.9 sucrose; pH 7.4 (adjusted with Tris); osmolarity, 300 mosmol l−1 (adjusted with sucrose). The PSS solution was continuously bubbled with 100 % O2 during dissections. Arteries were cleaned of connective tissue, cut into small pieces and placed in a tube containing fresh PSS. Tissue was immediately digested or cold stored in the refrigerator (5 °C) for up to 24 h. To disperse cells, tissue was placed in low-Ca2+ PSS containing enzymes. Pulmonary and renal tissues were digested differently. Pulmonary arterial tissue was incubated with a solution containing (in mg ml−1): 0.5 collagenase type XI; 0.03 elastase type IV and 0.5 bovine serum albumin (fat-free) for 14–16 h at 5 °C. The tissue was then washed several times with 5 °C low-Ca2+ PSS solution and triturated with a fire-polished Pasteur pipette. Renal arterial tissue was incubated with low-Ca2+ PSS solution containing (in mg ml−1): 1.67 collagenase type XI; 0.13 elastase type IV and 0.67 bovine serum albumin (fat free) for 18–23 min at 34 °C. The tissue was then washed several times in warm (34 °C) low-Ca2+ PSS and subsequently triturated with a fire-polished Pasteur pipette. The resulting dispersed pulmonary or renal ASMCs were cold stored at 5 °C for up to 8 h until experiments were performed.
Fluorescence imaging
Global [Ca2+] measurements
The cytosolic [Ca2+] was measured in pulmonary and renal ASMCs loaded with the ratiometric Ca2+-sensitive dye fura-2 AM (Molecular Probes, Eugene, OR, USA) using a dual excitation digital Ca2+ imaging system (IonOptix Inc., Milton, MA, USA) equipped with an intensified CCD camera. The imaging system was mounted on an inverted microscope (Nikon) outfitted with a × 40 (NA 1.3, Nikon Inc., Melville, NY, USA) oil-immersion objective. Fura-2 AM was dissolved in DMSO and added from a 1 mm stock to the cell suspension at a final concentration of 10 μM. Cells were loaded with fura-2 AM for 15 min at 34 °C and an additional 20 min in a perfusion chamber (Warner Instruments, Hamden, CT, USA or Medical Systems Corp., Greenvale, NY, USA) at room temperature in the dark. In some experiments, cells were loaded with fura-2 AM for 20–30 min in the dark at room temperature. Cells were then washed for 30 min to allow for dye esterification at 2 ml min−1 with a balanced salt solution of the following composition (mm): 126 NaCl; 5 KCl; 0.3 NaH2PO4; 10 Hepes; 1 MgCl2; 2 CaCl2; 10 glucose; pH 7.4 (adjusted with NaOH), 285–305 mosmol l−1. This range of osmolarities is under 10 %, and is far less than the 20 % decrease in osmolarity that has been shown to affect Ca2+ influx (Welsh et al. 2000). Cells were illuminated with a xenon arc lamp at 340 ± 15 and 380 ± 12 nm (Omega Optical, Brattleboro, VT, USA) and emitted light was collected from regions that encompassed single cells with a CCD camera at 510 nm (Nikon Inc.). If cells contracted, the experiment was paused and the regions of interest resized. In most experiments, images were acquired at 1 Hz and stored on either compact disk or magnetic media for later analysis. Although it is difficult to accurately measure intracellular calcium ([Ca2+]i) (Baylor & Hollingworth, 2000), estimates were made from the relation:
The values for the denominator maximum (Sf2), denominator minimum (Sb2), minimum ratio (Rmin) and maximum ratio (Rmax) were determined from in situ calibrations of fura-2 for each cell. The Kd for fura-2 was assumed to be 224 nm (Grynkiewicz et al. 1985). Specifically, at the end of each experiment, cells were dialysed with 1 μM ionomycin. To determine Rmax, cells were perfused with a balanced salt solution that contained 10 mm Ca2+ while the Rmin balanced salt solution did not have any added Ca2+ and contained 10 mm EGTA. During the Ca2+ calibration, 5 mm 2,3-butanedione monoxime was added to the bathing solution to inhibit smooth muscle contraction (Waurick et al. 1999).
The pharmacology of the extracellular Ca2+ entry pathway was studied in cells that had their intracellular Ca2+ stores maximally depleted by exposure to a cocktail including 10 μM CPA and 10 μM RY followed by brief exposure(s) to 1 μM angiotensin II (ANG II) (pulmonary), 1–10 μM PE (renal), and/or 10 mm CAF, while being perfused with a Ca2+-free bathing solution. Once the intracellular Ca2+ stores were depleted, cells were then re-exposed to 2 mm extracellular Ca2+ and changes in the cytosolic [Ca2+] were monitored in the absence or presence of pharmacological inhibitors of Ca2+ entry pathways. An elevation in cytosolic Ca2+ levels above basal values during Ca2+ re-addition was used as a marker of store depletion-induced extracellular Ca2+ entry.
Measurements of the cytosolic [Ca2+] before and during CCE and pharmacological inhibition were made once the fura-2 fluorescence ratio had stabilized. The amplitudes of the increase in cytosolic [Ca2+] due to SR Ca2+ store depletion are expressed relative to baseline values. The Ca2+-free balanced salt solution was prepared by substituting MgCl2 for CaCl2 and adding 1 mm EGTA. Experimental temperature was maintained at 29–32 °C with a dual automatic temperature controller (Warner Instruments or Medical Systems Corp.). Ni2+ was used as a non-selective inhibitor of extracellular Ca2+ entry via SOCs (Lewis, 1999), voltage-gated Ca2+ channels (VGCCs) (McDonald et al. 1994), non-selective cation channels (NSCCs) (Inoue, 1991) and Na+-Ca2+ exchange (Hobai et al. 1997). The lanthanide (Inoue, 1991) Gd3+ was used because it too inhibits NSCCs (Hescheler & Schultz, 1993) and SOCs (Utz et al. 1999). Nisoldipine was used to inhibit VGCCs (McDonald et al. 1994) while Cd2+ was used to inhibit VGCCs, Na+-Ca2+ exchange (Kimura et al. 1987; Hobai et al. 1997; Blaustein & Lederer, 1999) and NSCCs (Inoue et al. 2001). In experiments in which cells were exposed to Cd2+, measurements of the cytosolic [Ca2+] were made within ≈2 min because Cd2+ inhibits Na+-Ca2+ exchange within several seconds (Hobai et al. 1997), but enters cells more slowly binding fura-2 and thus increasing the ratio (Hinkle et al. 1992).
Mn2+ quench
Mn2+ is a commonly used probe for studying Ca2+ influx changes because many Ca2+ selective channels are permeable to Mn2+ and because it quenches the fura-2 fluorescence emission (Missiaen et al. 1990). Also, Mn2+ cannot be transported out of the cytosol into intracellular compartments or extruded from the cell (Gomes & Madeira, 1986). The rate of Mn2+ quench of fura-2 was determined by regression analysis of fluorescence intensity over time for cells excited at 357 ± 10 nm. Cells were analysed only if the rate of fura-2 quench by Mn2+ in the presence of ionomycin was at least 4-fold greater than the basal rate. Background fluorescence was collected automatically and subtracted from the acquired fluorescence video images during each experiment. The Ca2+-free balanced salt solution used in the Mn2+ quench experiments did not have any added Ca2+ or EGTA. Experimental temperature was 22–25 °C.
Electrophysiology
Voltage clamp currents were measured using the amphotericin B perforated-patch clamp technique (Rae et al. 1991; Ahn & Hume, 1997). Amphotericin B was mixed as a stock solution at 6 mg (100 μl)−1 in DMSO and used at a final concentration of 0.3 mg ml−1 of pipette solution. Thin-walled borosilicate capillaries (Sutter Instruments Inc., USA) were used to manufacture pipettes with resistances of 3–6 MΩ when filled with a pipette solution containing (mm): 90 potassium aspartate; 50 KCl; 1 MgCl2; 0.2 NaGTP; 2 MgATP; and 10 Hepes; pH 7.2 (adjusted with KOH); 285–305 mosmol l−1 (adjusted with mannitol). Cells were continuously bathed with a balanced salt solution (as described above). The voltage offset between the patch pipette and the bath solution was nulled immediately before patch formation. Cells were voltage clamped at -40 mV, near to the resting membrane potential for arterial smooth muscle cells (Kuriyama et al. 1998). In separate experiments the resting membrane potential was measured to be -21 ± 2 mV in pulmonary (n = 8) and -25 ± 4 mV (n = 6) in renal ASMCs that were perforated-patch voltage clamped. Membrane currents were recorded, filtered through a four-pole Bessel filter at 5 kHz, and membrane capacity currents nulled with the patch clamp amplifier (Axopatch 1B; Axon Instruments, Union City, CA, USA) connected to a computer via an analog-to-digital converter (Ionoptix, Milton, MA, USA). Experimental temperature was 22–25 °C.
Chemicals and drugs
Ionomycin free acid was purchased from Calbiochem (San Diego, CA, USA) and all other chemicals were purchased from Sigma (St Louis, MO, USA).
Statistical analysis
All data are presented as means ± s.e.m. Statistical difference within groups was determined with Student's two-tailed paired t test and between groups with a one-way analysis of variance with a Student-Newman-Keuls (SNK) multiple comparison procedure or two-tailed unpaired Student's t test. In some instances a repeated measures analysis of variance was used with Dunnett's multiple comparison procedure. In cases where the data were not normally distributed, a Wilcoxon signed rank sum test was used to test for differences within groups and a Kruskal-Wallis one-way ANOVA on ranks with Dunn's multiple comparison procedure between groups or a Friedman repeated measures ANOVA on ranks with Dunnett's multiple comparison procedure. The specific test used for each data set is noted in the legend to each figure. A P value of < 0.05 was accepted as statistically significant. The n values reported reflect the total number of cells tested. Multiple trials were performed and cells isolated from multiple dogs for each experimental paradigm. For the pulmonary experiments shown in Fig. 3, 36 trials were performed on cells isolated from 17 dogs while 25 trials were performed on renal cells isolated from 17 dogs. For the experiments shown in Fig. 4B, nine trials were performed on cells isolated from seven dogs while six trials were performed on renal ASMCs isolated from five dogs. For the experiments shown in Fig. 5D, 19 trials were performed on cells isolated from eight dogs. For the experiments shown in Fig. 6D, 19 trials were performed on cells isolated from nine dogs. For the experiments shown in Fig. 8, 25 trials were performed on cells isolated from 10 dogs. For the experiments shown in Fig. 10, 19 trials were performed on cells isolated from nine dogs.
Figure 3. Effect of release or depletion of sarcoplasmic reticulum Ca2+ stores on the basal cytosolic [Ca2+] in pulmonary and renal ASMCs.

▪, pulmonary ASMCs; □, renal ASMCs. Bars indicate the change in the cytosolic [Ca2+] from resting levels. Error bars represent ± s.e.m. Numbers of independent experiments are shown in parentheses. Thaps, thapsigargin. * Significantly different from 10 mm CAF and 1 μM ANG II pulmonary ASMC groups (P < 0.05); † significantly different from 10 μM PE and 10 mm CAF renal ASMC groups (P < 0.05); Kruskal-Wallis one-way ANOVA on ranks and Dunn's multiple comparison procedure.
Figure 4. Effect of sarcoplasmic reticulum Ca2+ store depletion on the cytosolic [Ca2+] in pulmonary and renal ASMCs voltage clamped at -40 mV.
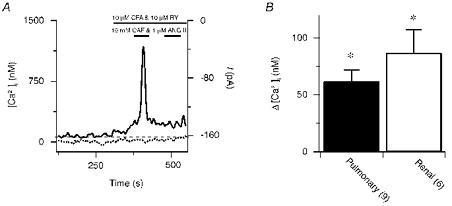
A, 10 mm CAF and 1 μM ANG II-induced [Ca2+] (continuous line) and net membrane current (dotted line) responses in the presence of 10 μM RY and 10 μM CPA in a single pulmonary ASMC. The dashed line shows the resting cytosolic [Ca2+]. B, change in the cytosolic [Ca2+] from resting levels in pulmonary (▪) and renal (□) ASMCs. Error bars represent ± s.e.m. Numbers of independent experiments are shown in parentheses. * Mean value significantly different from control; paired two-tailed t test.
Figure 5. Effect of sarcoplasmic reticulum Ca2+ store depletion on the rate of Mn2+ entry in pulmonary ASMCs.
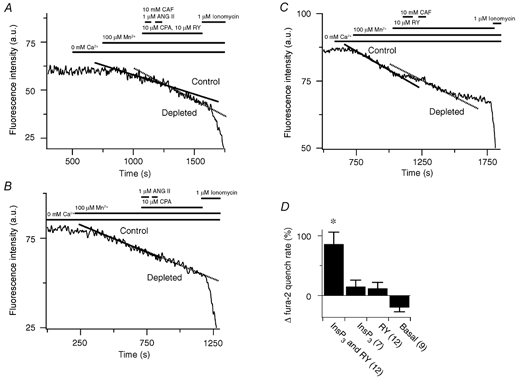
A, 10 mm CAF and 1 μM ANG II effect on fura-2 quench in the presence of 10 μM RY and 10 μM CPA. a.u., arbitrary units. B, 1 μM ANG II effect on fura-2 quench in the presence of 10 μM CPA. C, 10 mm CAF effect on fura-2 quench in the presence of 10 μM RY. D, percentage change in fura-2 quench compared to control. Error bars represent ± s.e.m. Numbers of independent experiments are shown in parentheses. * Significantly different from the change in basal, InsP3 store-depleted and RY store-depleted quench rates (P < 0.05); one-way ANOVA with a SNK multiple comparison procedure.
Figure 6. Effect of sarcoplasmic reticulum Ca2+ store depletion on the rate of Mn2+ entry in renal ASMCs.
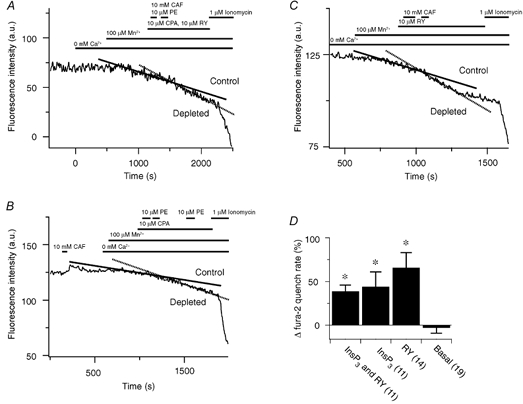
A, 10 mm CAF and 10 μM PE effect on fura-2 quench in the presence of 10 μM RY and 10 μM CPA. B, 10 μM PE effect on fura-2 quench in the presence of 10 μM CPA. C, 10 mm CAF effect on fura-2 quench in the presence of 10 μM RY. D, percentage change in fura-2 quench compared to control. Error bars represent ± s.e.m. Numbers of independent experiments are shown in parentheses. * Significantly different from the change in the basal quench rate (P < 0.05); one-way ANOVA with a SNK multiple comparison procedure.
Figure 8. Pharmacological inhibition of store depletion-induced Ca2+ entry in pulmonary ASMCs.
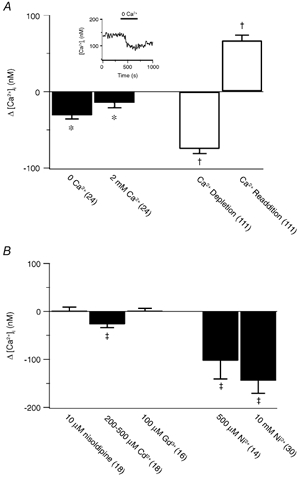
A, change in the cytosolic [Ca2+] compared to the resting cytosolic [Ca2+] for control (▪) and store-depleted (□) experiments. Inset, effect of Ca2+ removal on the resting cytosolic [Ca2+] in an unstimulated cell. B, change in the cytosolic [Ca2+] from that measured during CCE. Error bars represent ± s.e.m. Numbers of independent experiments are shown in parentheses. * Mean significantly different from control (P < 0.05); repeated measures ANOVA with Dunnett's multiple comparison procedure. † Mean significantly different from control (P < 0.05); Friedman repeated measures ANOVA on ranks with Dunnett's multiple comparison procedure. ‡ Mean significantly different from control (P < 0.05); Wilcoxon signed rank test.
Figure 10. Pharmacological inhibition of store depletion-induced Ca2+ entry in renal ASMCs.
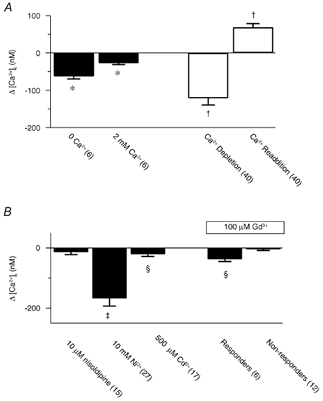
A, change in the cytosolic [Ca2+] compared to the resting cytosolic [Ca2+] for control (▪) and store-depleted (□) experiments. B, change in the cytosolic [Ca2+] from that measured during CCE. Error bars represent ± s.e.m. Numbers of independent experiments are shown in parentheses. * Mean significantly different from control (P < 0.05); repeated measures ANOVA with Dunnett's multiple comparison procedure. † Mean significantly different from control (P < 0.05); Friedman repeated measures ANOVA on ranks with Dunnett's multiple comparison procedure. ‡ and § Mean significantly different from control (P < 0.05) by Wilcoxon signed rank test or paired t test, respectively.
RESULTS
Depletion of intracellular Ca2+ stores activates extracellular Ca2+ entry in canine pulmonary and renal ASMCs
Figure 1A shows the estimated cytosolic [Ca2+] over time for a single pulmonary ASMC measured with the Ca2+-sensitive dye fura-2. Exposure of the cell to 10 mm CAF caused a rapid, transient rise in the cytosolic [Ca2+] that decayed back to baseline following agonist washout. Figure 1B shows that continuous exposure to 10 μM RY and three separate exposures to 10 mm CAF for 30 s depleted RY-sensitive Ca2+ stores in this cell as evidenced by the degradation and abolition of CAF-induced Ca2+ release. The basal cytosolic [Ca2+] did not change with RY-sensitive Ca2+ store depletion. Figure 1C shows that 1 μM ANG II induced a rapid, transient rise in the cytosolic [Ca2+] that fell slowly back to baseline. Figure 1D shows that SERCA inhibition with 10 μM CPA and brief exposures to 1 μM ANG II depleted InsP3-sensitive Ca2+ stores (Janiak et al. 2001). The initial ANG II exposure caused a transient rise in the cytosolic [Ca2+], while the second exposure did not, indicating that the InsP3 Ca2+ stores were depleted. This depletion protocol did not appreciably increase the basal [Ca2+]. However, Fig. 1E shows that simultaneous depletion of RY- and InsP3-sensitive Ca2+ stores caused the cytosolic [Ca2+] to remain elevated. During continuous 10 μM CPA and 10 μM RY administration, a brief 10 mm CAF exposure elicited a rapid, transient rise in the intracellular [Ca2+], much like that induced by 10 mm CAF alone (Fig. 1A). However, the cytosolic [Ca2+] did not return to baseline after agonist washout, but stabilized at 60 nm above the basal [Ca2+] (arrow). Figure 1F shows that when extracellular Ca2+ was removed and Ca2+ uptake inhibited, 10 mm CAF caused a rapid transient rise in the cytosolic [Ca2+] that fell slowly back to baseline.
Figure 1. Effect of sarcoplasmic reticulum Ca2+ store release or depletion on the cytosolic [Ca2+] in pulmonary ASMCs.
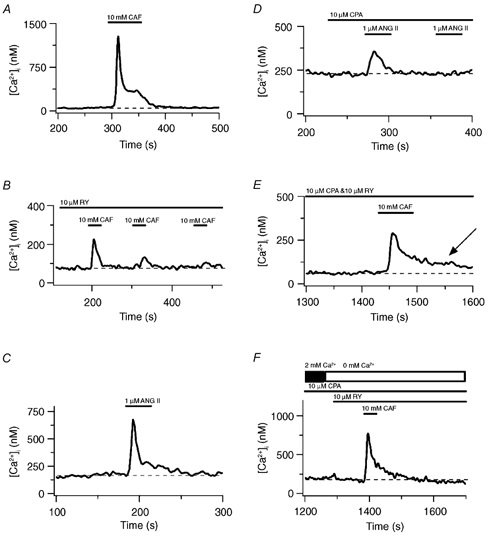
A, 10 mm CAF-induced Ca2+ transient. B, 10 mm CAF-induced Ca2+ transients in the presence of 10 μM RY. C, 1 μM ANG II-induced Ca2+ transient. D, 1 μM ANG II-induced Ca2+ transients in the presence of 10 μM CPA. E, 10 mm CAF-induced Ca2+ transient and the basal cytosolic [Ca2+] increase in the presence of 10 μM RY and 10 μM CPA. F, 10 mm CAF-induced Ca2+ transient during perfusion with Ca2+-free bathing solution in the presence of 10 μM RY and 10 μM CPA. Dashed lines show the resting cytosolic [Ca2+]. The arrow indicates an elevated cytosolic [Ca2+].
Extracellular Ca2+ entry pathways activated by store depletion were then investigated in renal ASMCs. Figure 2A shows that 10 mm CAF caused a rapid, transient increase in the cytosolic [Ca2+] that fell back to baseline following agonist washout in a single renal ASMC. Figure 2B shows that three 30 s 10 mm CAF exposures in the continuous presence of 10 μM RY depleted the Ca2+ stores as evidenced by the degradation of CAF-induced Ca2+ release. However, the cytosolic [Ca2+] remained elevated at 100 nm above baseline (arrow). Figure 2C shows that release of InsP3 stores did not cause any sustained increase in the cytosolic [Ca2+]. Exposure to 10 μM PE caused a rapid, transient rise in the cytosolic [Ca2+] that fell slowly back to basal following agonist washout. Figure 2D shows that 1 μM thapsigargin induced a rise in the cytosolic [Ca2+] indicating that block of Ca2+ sequestration unmasks a tonic Ca2+ release pathway (e.g. a passive leak). Moreover, two 30 s 10 μM PE exposures caused intracellular Ca2+ store depletion via InsP3-sensitive pathways as demonstrated by the abolition of Ca2+ release by the second 10 μM PE exposure. Depletion of InsP3-sensitive intracellular Ca2+ stores caused a 55 nm elevation in the basal cytosolic [Ca2+] (arrow).
Figure 2. Effect of sarcoplasmic reticulum Ca2+ store release or depletion on the cytosolic [Ca2+] in renal ASMCs.
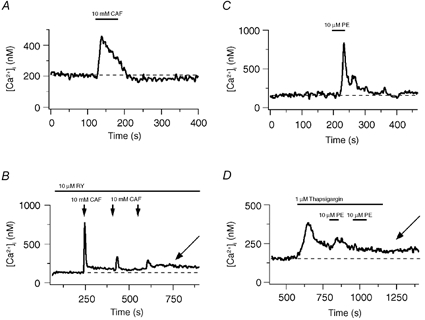
A, 10 mm CAF-induced Ca2+ transient. B, 10 mm CAF-induced Ca2+ transients in the presence of 10 μM RY. C, 10 μM PE-induced Ca2+ transient. D, 10 μM PE-induced Ca2+ transients and the basal cytosolic [Ca2+] increase in the presence of 1 μM thapsigargin. Dashed lines show the resting cytosolic [Ca2+]. Arrows indicate elevated cytosolic Ca2+.
The summarized data in Fig. 3 show that in pulmonary ASMCs the basal cytosolic [Ca2+] increases depend on how the intracellular Ca2+ stores are released or depleted. For the cells analysed in Fig. 3, the basal [Ca2+] of pulmonary ASMCs was 112 ± 13 nm while in renal ASMCs it was 187 ± 23 nm. Although the basal [Ca2+] was significantly greater in renal ASMCs (P < 0.05, two-tailed unpaired t test) these values are close to and in the normal range of 100–150 nm for the basal [Ca2+] in arterial SMCs (Kuriyama et al. 1998). Simultaneous depletion of InsP3- and RY-sensitive Ca2+ stores by continuous exposure to 10 μM CPA and 10 μM RY and one to three 30 s 10 mm CAF exposures caused the basal [Ca2+] to rise by an average of 98 ± 21 nm. Depletion of the intracellular Ca2+ stores of cells perfused with a Ca2+-free extracellular bathing solution did not cause any significant change in the cytosolic [Ca2+]. Depletion of the RY-sensitive Ca2+ stores alone by continuously exposing cells to 10 μM RY and exposing cells 1–3 times to 10 mm CAF for 30 s did not alter the cytosolic [Ca2+]. Likewise, depletion of InsP3-sensitive Ca2+ stores alone with 10 μM CPA and exposing cells 1–4 times to 1 μM ANG II for 30 s did not cause any change in the cytosolic [Ca2+]. Moreover, neither CAF (10 mm for 30 s) nor ANG II (1 μM for 30 s) increased the basal cytosolic [Ca2+] significantly following agonist washout.
Individual Ca2+ store depletion elevates the basal cytosolic [Ca2+] in renal ASMCs. As summarized in Fig. 3, depletion of InsP3-sensitive Ca2+ stores with continuous 1 μM thapsigargin and one to three 30 s 1–10 μM PE exposures increased the cytosolic [Ca2+] by 40 ± 8 nm. Similarly, RY-sensitive Ca2+ store depletion with continuous 10 μM RY and 30 s 10 mm CAF exposures increased the basal cytosolic [Ca2+] by 48 ± 7 nm. Simultaneous depletion of intracellular Ca2+ stores through InsP3- and RY-sensitive pathways with continuous 10 μM CPA, 10 μM RY and 30 s 1–10 μM PE and 10 mm CAF exposures increased the cytosolic [Ca2+] by 24 ± 3 nm. In experiments in which intracellular stores were depleted, removal of extracellular Ca2+ caused the cytosolic [Ca2+] to fall by 86 ± 9 nm (n = 17, P < 0.05) compared to that measured during store depletion-induced extracellular Ca2+ entry. Furthermore, 10 mm CAF or 1–10 μM PE for 30 s caused transient increases in the cytosolic [Ca2+] but did not significantly alter the basal cytosolic [Ca2+] following agonist washout.
Cells were voltage clamped while their intracellular [Ca2+] was measured to determine whether the rises in the cytosolic [Ca2+] were due to changes in membrane permeability or changes in driving force due to alterations in the membrane potential. Figure 4A illustrates that Ca2+ store depletion caused the cytosolic [Ca2+] to increase in an individual pulmonary ASMC when the membrane potential was held constant at -40 mV. The Ca2+ stores were depleted by exposing the cell twice to 10 mm CAF and 1 μM ANG II in the continuous presence of 10 μM RY and 10 μM CPA. Depletion of the Ca2+ stores was confirmed by the lack of agonist-induced Ca2+ release during the second exposure. The initial agonist exposure caused a transient [Ca2+] increase that remained 94 nm above baseline. In these experiments no attempts were made to isolate store depletion-induced Ca2+ currents from the total membrane current. Figure 4B shows summary data illustrating that in pulmonary ASMCs the cytosolic [Ca2+] increased significantly by 62 ± 10 nm when the driving force for Ca2+ was fixed and the intracellular Ca2+ stores were depleted. Depletion of the intracellular Ca2+ stores of renal ASMCs that were voltage clamped caused the cytosolic [Ca2+] to also increase significantly by 87 ± 20 nm.
Changes in membrane Ca2+ permeability were assessed by measuring the rate of Mn2+ quench of fura-2. Figure 5A shows the fluorescence intensity over time measured at 510 nm at an excitation wavelength of 357 nm in a single pulmonary ASMC. Removal of extracellular Ca2+ did not cause any decline in the fluorescence intensity. However, 100 μM Mn2+ caused the fluorescence intensity to decrease at a rate of 0.026 a.u. s−1. Following simultaneous depletion of both the InsP3- and RY-sensitive SR Ca2+ stores the fura-2 quench rate by Mn2+ increased by 49 % to 0.038 a.u. s−1. Figure 5B shows that selective depletion of the InsP3-sensitive SR Ca2+ store did not increase the Mn2+ flux. Prior to InsP3 store depletion the rate of quench was 0.031 a.u. s−1 and following depletion the rate increased by 8 % to 0.033 a.u. s−1. Similarly, selective depletion of RY-sensitive Ca2+ stores also did not increase the Mn2+ quench rate. Figure 5C shows that prior to store depletion the rate of quench was 0.029 a.u. s−1 and following depletion decreased by 28 % to 0.021 a.u. s−1. Figure 5D summarizes these results showing that depletion of both stores caused an 86 ± 20 % increase in the Mn2+ quench of fura-2 compared to the rate prior to store depletion, which was significantly greater than the quench rate following the selective depletion of RY or InsP3 stores or compared to control experiments. The control experiments in comparison, which were performed over the same time period as the store depletion experiments, had a decline in the basal Mn2+ quench rate of 28 ± 7 %. Additionally, selective depletion of either InsP3- or RY-sensitive Ca2+ stores did not significantly increase the fura-2 quench rate over that observed in control experiments.
Figure 6 shows that in renal ASMCs depletion of the common SR Ca2+ store increases the Mn2+ quench rate of fura-2. Figure 6A illustrates that depletion of the SR Ca2+ store through activation of both the InsP3 and RY pathways caused the Mn2+ quench rate to increase by 32 % from 0.0356 to 0.047 a.u. s−1. Figure 6B shows that selective depletion of the stores through InsP3-sensitive pathways increased the Mn2+ quench rate by 170 % from 0.0091 to 0.0246 a.u. s−1. Figure 6C demonstrates that Ca2+ store depletion through RY-sensitive pathways doubled the Mn2+ quench rate, which increased from 0.02 to 0.041 a.u. s−1. Figure 6D summarizes these results illustrating that, in renal ASMCs, Ca2+ store depletion through InsP3- and RY-sensitive pathways caused a 39 ± 7 % increase in fura-2 quench rate, while depletion through InsP3-sensitive pathways alone caused a 44 ± 17 % increase and depletion via RY-sensitive pathways alone caused a 67 ± 17 % increase. All three groups had significantly greater increases in the fura-2 quench rate than that in control experiments where the basal Mn2+ quench rate declined by 3 ± 6 % over equivalent time periods.
Pharmacological properties of Ca2+ store depletion-induced extracellular Ca2+ entry in pulmonary and renal ASMCs
Figure 7A shows that in a single pulmonary ASMC 10 mm CAF caused a rapid, transient rise in the cytosolic [Ca2+] that fell back to basal values. The SR Ca2+ stores were then fully depleted by perfusing with a Ca2+-free bathing solution in the continuous presence of 10 μM CPA, 10 μM RY and 100 μM CAF and exposing the cell twice for 30 s to 10 mm CAF and 1 μM ANG II. Subsequent addition of extracellular Ca2+ while in the continued presence of 10 μM CPA, 10 μM RY and 100 μM CAF caused the cytosolic [Ca2+] to rise 138 nm above basal values. Removal of extracellular Ca2+ at the end of the experiment caused the cytosolic [Ca2+] to fall 174 nm, 36 nm below baseline.
Figure 7. Store-operated channels mediate extracellular Ca2+ entry in pulmonary ASMCs when sarcoplasmic reticulum Ca2+ stores are depleted.
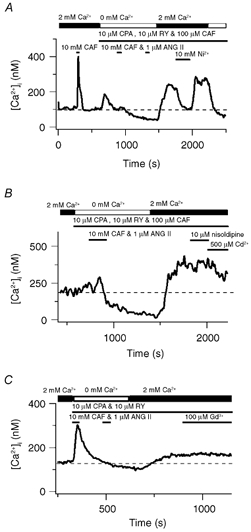
A, effect of 10 mm Ni2+ and extracellular Ca2+ removal on the cytosolic [Ca2+] during CCE. B, effect of 10 μM nisoldipine and 500 μM Cd2+ on the cytosolic [Ca2+] during CCE. C, effect of 100 μM Gd3+ during CCE. Dashed lines show the resting cytosolic [Ca2+].
The pharmacological properties of Ca2+ store depletion-induced Ca2+ entry were examined. Ni2+ reduced the elevated cytosolic [Ca2+] to the resting cytosolic [Ca2+] (Fig. 7A). Figure 7B shows that following SR Ca2+ store depletion extracellular Ca2+ re-addition raised the cytosolic [Ca2+] 166 nm above basal values. This cytosolic [Ca2+] increase was not decreased by 10 μM nisoldipine while 500 μM Cd2+ (Inoue, 1991) produced a small (90 nm) reduction in the cytosolic [Ca2+]. Figure 7C shows a representative experiment illustrating that depletion of intracellular Ca2+ stores and subsequent Ca2+ reintroduction caused the cytosolic [Ca2+] to increase 36 nm above the resting concentration. The depletion-induced cytosolic [Ca2+] increase was unaffected by 100 μM Gd3+.
The summarized data in Fig. 8A demonstrate that in pulmonary ASMCs depletion of intracellular Ca2+ stores activates extracellular Ca2+ entry. Bathing cells in a Ca2+-free solution that contained CPA, RY, CAF and/or ANG II caused a 75 ± 6 nm decrease in the cytosolic [Ca2+] compared to basal values. Subsequent addition of 2 mm Ca2+ to the bathing solution elicited a significant, 67 ± 7 nm rise in the cytosolic [Ca2+] above basal values. Removal of extracellular Ca2+ in control experiments (Fig. 8, inset) caused the cytosolic [Ca2+] to decrease by 31 ± 5 nm over the same time period. With subsequent addition of 2 mm extracellular Ca2+ the cytosolic [Ca2+] was 15 ± 6 nm below basal values.
Figure 8B summarizes data showing that Ni2+ can inhibit CCE in pulmonary ASMCs. The cytosolic [Ca2+] was decreased by 145 ± 26 nm by 10 mm Ni2+ during CCE, which is 70 ± 14 nm below basal [Ca2+] values. This 70 nm decrease below basal levels was accounted for in control experiments where 10 mm Ni2+ caused a 79 ± 16 nm (n = 13) decrease in the cytosolic [Ca2+]. Comparable decreases in the cytosolic [Ca2+] during CCE were induced by 500 μM Ni2+ (103 ± 38 nm), indicating that the Ni2+-induced decrease in the cytosolic [Ca2+] is not due to a non-specific inhibition of all Ca2+ conducting pathways by a super maximal Ni2+ exposure. During CCE the cytosolic [Ca2+] was significantly reduced by 200–500 μM Cd2+ (27 ± 7 nm), a reduction notably smaller than that caused by Ni2+. In control experiments 500 μM Cd2+ did not change the cytosolic [Ca2+] (3 ± 5 nm, n = 3). Furthermore, during CCE the cytosolic [Ca2+] was unaltered by 10 μM nisoldipine or 100 μM Gd3+. In control experiments 10 μM nisoldipine caused a small but not significant increase in the cytosolic [Ca2+] of 14 ± 6 nm (n = 3) while Gd3+ caused a slight, but not significant, decrease in the [Ca2+] of 19 ± 17 nm (n = 7).
Parallel experiments were performed in renal ASMCs. Depletion-induced Ca2+ entry was activated in renal ASMCs by exposing cells to a cocktail including 10 μM CPA and 10 μM RY and briefly exposing cells to 10 mm CAF and/or 1–10 μM PE and/or, in some cases, 1 μM ANG II while perfusing the cells with a Ca2+-free bathing solution. Figure 9A shows that depletion of intracellular Ca2+ stores activated extracellular Ca2+ influx, which was reversibly inhibited by 10 mm Ni2+. The cytosolic [Ca2+] rose above basal values by 43 nm with the addition of 2 mm extracellular Ca2+ following store depletion, while Ni2+ caused the [Ca2+] to decrease below basal values by 29 nm. Figure 9B illustrates that 100 μM Gd3+ caused the cytosolic [Ca2+] to decrease by 29 nm during CCE. Figure 9C shows that 10 μM nisoldipine did not inhibit the sustained Ca2+ plateau in renal ASMCs, but did cause a transient [Ca2+] decrease in the cell shown in this panel and overall in a total of 4 of 15 cells. This transient decrease is unlike the dihydropyridine-induced sustained inhibition of CCE in skeletal muscle (Hopf et al. 1996) and that of non-selective cation channels involved in store refilling in vascular smooth muscle (Curtis & Scholfield, 2001). Figure 9D shows that during CCE 500 μM Cd2+ reduced the cytosolic [Ca2+] by 48 nm, back to baseline.
Figure 9. Store-operated channels mediate extracellular Ca2+ entry in renal ASMCs when sarcoplasmic reticulum Ca2+ stores are depleted.
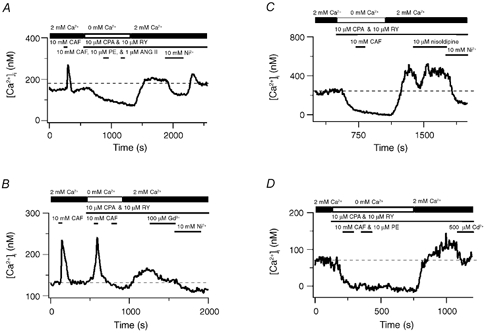
A, effect of 10 mm Ni2+ on the cytosolic [Ca2+] during CCE. B, effect of 100 μM Gd3+ on the cytosolic [Ca2+] during CCE. C, effect of 10 μM nisoldipine on the cytosolic [Ca2+] during CCE. D, effect of 500 μM Cd2+ on the cytosolic [Ca2+] during CCE. Dashed lines show the resting cytosolic [Ca2+].
Figure 10 summarizes CCE and pharmacological data in renal ASMCs. Figure 10A shows that extracellular Ca2+ removal caused a 63 ± 7 nm decrease in the cytosolic [Ca2+] while after Ca2+ re-addition the cytosolic [Ca2+] was 28 ± 4 nm below basal values. Depletion of the intracellular Ca2+ stores by placing cells in a Ca2+-free bathing solution and exposing them to 10 μM CPA and 10 μM RY followed by brief exposure(s) to 1–10 μM PE and/or 10 mm CAF caused the basal cytosolic [Ca2+] to decrease significantly, by 122 ± 18 nm, while subsequent Ca2+ addition caused the cytosolic [Ca2+] to rise significantly above basal values by 69 ± 10 nm.
CCE in renal ASMCs, like pulmonary ASMCs, was due to activation of SOCs. Figure 10B summarizes data showing that depletion-induced extracellular Ca2+ entry was strongly inhibited by Ni2+ and only slightly inhibited by Cd2+ or Gd3+. The cytosolic [Ca2+] was decreased by 168 ± 25 nm by 10 mm Ni2+ compared to that measured during CCE, which was 98 ± 17 nm below basal levels. This 98 nm decrease in the cytosolic [Ca2+] below basal levels was largely accounted for in control experiments where 10 mm Ni2+ caused a 76 ± 12 nm (n = 14) decrease in the basal cytosolic [Ca2+]. Furthermore, during CCE, 500 μM Cd2+ caused a 21 ± 7 nm [Ca2+] decrease and 100 μM Gd3+ caused a 38 ± 7 nm [Ca2+] decrease in 6 of the 18 cells tested. In control experiments neither 500 μM Cd2+ (1 ± 3 nm, n = 6) nor 100 μM Gd3+ (11 ± 6 nm, n = 8) reduced the basal cytosolic [Ca2+]. Additionally, 10 μM nisoldipine failed to affect the CCE-induced cytosolic [Ca2+] increase (14 ± 8 nm decrease) or basal cytosolic [Ca2+] (10 ± 7 nm decrease, n = 6).
Discussion
The current study provides evidence that depletion of SR Ca2+ stores activates CCE in both canine pulmonary and renal ASMCs. The finding that there is CCE in vascular smooth muscle cells is not surprising, and complements work by other researchers (Robertson et al. 1995; Golovina, 1999; Utz et al. 1999; Trepakova et al. 2000; Ng & Gurney, 2001). However, the finding that activation of store depletion-induced extracellular Ca2+ entry in canine pulmonary and renal ASMCs parallels the organization of the SR stores in these cell types is novel (Jabr et al. 1997; Janiak et al. 2001). In pulmonary ASMCs, where the RY- and InsP3-sensitive Ca2+ stores can be depleted separately, only simultaneous depletion of the two stores caused an elevation in the resting cytosolic [Ca2+], which was dependent on the presence of extracellular Ca2+. However, in renal ASMCs, where the RY and InsP3 stores exhibit significant overlap, depleting intracellular Ca2+ stores through either pathway alone or through both pathways simultaneously caused an elevation in the cytosolic [Ca2+]. These increases in the cytosolic [Ca2+] following depletion of the SR Ca2+ stores still occurred in cells that were voltage clamped. This indicates that the increases in the cytosolic [Ca2+] with store depletion were due to changes in the membrane permeability to Ca2+ and were not artifacts due to changes in the Ca2+ driving force (Stojilkovic et al. 1992).
The equilibrium relationship between the basal [Ca2+] and plasma membrane Ca2+ fluxes reflects the balance of influx and efflux across the cell plasma membrane (Keizer et al. 1995). Therefore increases in the basal [Ca2+] are attributable to enhanced extracellular Ca2+ influx (through VGCCs and SOCs) or decreased Ca2+ efflux (through re-uptake of cytosolic Ca2+ by SR and plasma membrane Ca2+ pumps). The superficial buffer barrier uses SR proximal to the plasma membrane for vectoral transport of Ca2+ out of the cell. Short-circuiting the SR, either by opening Ca2+ release channels or inhibiting Ca2+ uptake, blocks this vectoral transport of Ca2+. The cytosolic [Ca2+] then rises because Ca2+ extrusion is slowed while influx remains constant. Given that Mn2+ is not transported through Ca2+ pumps, the rate of Mn2+ quench of the fura-2 signal can be used to test for the presence of a superficial buffer barrier. In cells that have a superficial buffer barrier, depletion of intracellular stores induces rises in the cytosolic [Ca2+] that may not be coincident with increases in Mn2+ quench (Chen et al. 1992; Chen & van Breemen, 1993). In contrast, depletion of the SR stores in both pulmonary and renal ASMCs caused an increase in Mn2+ quench, which is common in cells where store depletion activates CCE (Hopf et al. 1996; Bennett et al. 1998), indicating that store depletion enhances Ca2+ influx. Thus, the store depletion-induced rise in the basal [Ca2+] in both pulmonary and renal ASMCs is due to increases in influx and not decreases in efflux due to the operation of a superficial buffer barrier (Chen & van Breemen, 1993).
The mechanism of activation for CCE is complex in both canine pulmonary and renal arterial smooth muscle cells. The luminal Ca2+ content may regulate CCE activation in either canine pulmonary or renal ASMCs by inducing a conformational change in InsP3 receptors (Ma et al. 2000) or through activation of RY receptors (Bennett et al. 1998) leading to opening of SOCs. Alternatively, the decreased luminal [Ca2+] may cause the release of a CIF factor that activates SOCs and CCE (Trepakova et al. 2000). In canine pulmonary ASMCs, simultaneous InsP3 and RY store depletion activates CCE. In contrast, depletion of the Ca2+ stores with CPA alone is sufficient to activate CCE in rat pulmonary ASMCs (Ng & Gurney, 2001). The canine renal ASMCs are similar to rat pulmonary ASMCs in that depletion of the common Ca2+ pool through either RY or InsP3 pathways activates CCE. Interestingly, one might hypothesize that in canine pulmonary ASMCs depletion of one of the two independent stores would still allow for Ca2+ to be sequestered into a functional Ca2+ store. For example, depletion of the RY store could potentially activate a CCE pathway that would be masked through Ca2+ sequestration by the functional InsP3 store. However, equilibrium analysis of compartmental models of Ca2+ responses suggests that Ca2+ sequestration by the SR cannot influence the basal cytosolic [Ca2+]. Although SR Ca2+ release and reuptake can affect the time required to reach equilibrium, the basal [Ca2+] is ultimately determined by the balance of influx and efflux of Ca2+ across the plasma membrane (Keizer & De Young, 1993; Keizer et al. 1995; Smith et al. 1996). The buffering capacity of the functional SR would therefore not affect the final cytosolic [Ca2+] when one of the two independent SR stores in the pulmonary ASMCs was depleted. Moreover, in experiments in which CPA was applied, all SERCA-mediated Ca2+ sequestration was probably inhibited (Jabr et al. 1997; Janiak et al. 2001). For that reason Ca2+ released by the InsP3 store could not be sequestered by the RY store. Our observation that simultaneous InsP3 and RY depletion procedures were necessary to activate CCE in canine pulmonary ASMCs suggests a convergence and integration of the signal(s) that ‘report’ InsP3 and RY store depletion to plasma membrane Ca2+ influx pathways. In comparison, the CCE activation process in rat pulmonary ASMCs (Ng & Gurney, 2001) and canine renal ASMCs does not require this integrated response.
The pharmacology of CCE inhibition leads to the conclusion that Ni2+- and Cd2+-sensitive pathways mediate Ca2+ entry in pulmonary ASMCs. Additionally, Ni2+-, Cd2+- and Gd3+-sensitive pathways are activated in renal ASMCs. Based on the pharmacological criteria alone the identity of the channel underlying CCE in canine pulmonary and renal ASMCs cannot be identified with absolute certainty since gadolinium, nickel and cadmium inhibit many different Ca2+ permeable channels (Lewis, 1999). However, VGCCs do not contribute to Ca2+ entry as nisoldipine had no effect on Ca2+ entry either in unstimulated cells or following store depletion. It is possible that the reduction in the cytosolic [Ca2+] in the presence of Ni2+, Cd2+ and Gd3+ was due to an indirect effect of these ions on the membrane potential, thus reducing the driving force for Ca2+ entry. However, our observations argue against this. When the cells were voltage clamped and the driving force was held constant we still observed CCE following store depletion in both pulmonary and renal ASMCs. Further, the Ni2+-induced decrease in the cytosolic [Ca2+] below basal values in store-depleted pulmonary and renal ASMCs was accounted for by the Ni2+-induced decrease in the cytosolic [Ca2+] in unstimulated cells. The cytosolic [Ca2+] was also unaffected by Cd2+ and Gd3+ in unstimulated pulmonary or renal ASMCs.
We have recently reported that members of the short family of transient receptor potential (TRPC) channels, a class of non-selective cation channels that may constitute CCE, are expressed in canine pulmonary and renal ASMCs (Walker et al. 2001). TRPC4, TRPC6 and TRPC7 are expressed in both canine pulmonary and renal ASMCs while TRPC3 is expressed only in renal ASMCs; TRPC1, TRPC2 and TRPC5 are not expressed in either cell type. These TRPC channel isoforms are inhibited by various divalent transition metals, including Cd2+ and lanthanides, such as Gd3+ (Okada et al. 1999; Inoue et al. 2001; Minke & Cook, 2002). TRPC4 may underlie CCE in both pulmonary and renal ASMCs because depletion of intracellular Ca2+ stores activates TRPC4 (McKay et al. 2000), whereas stimulation of G-protein-coupled receptors and diacylglycerols activates TRPC3, TRPC6 and TRPC7 independent of Ca2+ store depletion (Hofmann et al. 1999; Okada et al. 1999; McKay et al. 2000).
There are several potential physiological functions for CCE in pulmonary and renal ASMCs. Typically, CCE acts to replenish empty intracellular Ca2+ stores (Putney & McKay, 1999), and our studies suggest that this process occurs in pulmonary and renal ASMCs. CCE was recently shown to promote agonist-induced contraction of rat pulmonary artery rings (McDaniel et al. 2001). Moreover, there is evidence that CCE is involved in hypoxic-induced vasoconstriction of high-resistance pulmonary arteries (Jabr et al. 1997; Robertson et al. 2000). The 50–100 nm CCE-mediated increases in the cytosolic [Ca2+] that were observed are unlikely to induce, but could facilitate, smooth muscle cell contraction or could indirectly alter sarcolemmal conductances (Kuriyama et al. 1998). CCE is also associated with cell proliferation as well as excitation-contraction coupling and is greater in proliferating human pulmonary arterial myocyte cultures compared to those that are growth arrested (Golovina, 1999).
In conclusion, CCE is common to pulmonary and renal ASMCs and the mechanism of activation parallels the differential organization of the intracellular Ca2+ stores. Potentially, one or more of the different TRPC isoforms may encode for the channel responsible for CCE in these two smooth muscle cell types. Future experiments are required to elucidate the activation mechanism(s), molecular candidates for, and the physiological role(s) of store depletion Ca2+ entry in these smooth muscle cells.
Acknowledgments
We would like to thank Dr Linda Ye, Phillip Keller and Shen Xiao-Ming for technical assistance and Eric V. Leaver for insightful discussions. This work was supported by National Heart Lung and Blood Institute grants HL-48254 (J.R.H.), HL-10476 (S.M.W.) and ASU CLAS Faculty grant-in-aid (G.D.S.).
References
- Ahn DS, Hume JR. pH regulation of voltage-dependent K+ channels in canine pulmonary arterial smooth muscle cells. Pflügers Archiv. 1997;433:758–765. doi: 10.1007/s004240050342. [DOI] [PubMed] [Google Scholar]
- Baylor SM, Hollingworth S. Measurement and interpretation of cytoplasmic [Ca2+] signals from calcium-indicator dyes. News in Physiological Sciences. 2000;15:19–26. [PubMed] [Google Scholar]
- Bennett DL, Bootman MD, Berridge MJ, Cheek TR. Ca2+ entry into PC12 cells initiated by ryanodine receptors or inositol 1,4,5-trisphosphate receptors. Biochemical Journal. 1998;329:349–357. doi: 10.1042/bj3290349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiological Reviews. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Chen Q, Cannell M, van Breemen C. The superficial buffer barrier in vascular smooth muscle. Canadian Journal of Physiology and Pharmacology. 1992;70:509–514. doi: 10.1139/y92-066. [DOI] [PubMed] [Google Scholar]
- Chen Q, van Breemen C. The superficial buffer barrier in venous smooth muscle: sarcoplasmic reticulum refilling and unloading. British Journal of Pharmacology. 1993;109:336–343. doi: 10.1111/j.1476-5381.1993.tb13575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis TM, Scholfield CN. Nifedipine blocks Ca2+ store refilling through a pathway not involving L-type Ca2+ channels in rabbit arteriolar smooth muscle. Journal of Physiology. 2001;532:609–623. doi: 10.1111/j.1469-7793.2001.0609e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipp M, Evans AM. Cyclic ADP-ribose is the primary trigger for hypoxic pulmonary vasoconstriction in the rat lung in situ. Circulation Research. 2001;89:77–83. doi: 10.1161/hh1301.093616. [DOI] [PubMed] [Google Scholar]
- Fellner SK, Arendshorst WJ. Capacitative calcium entry in smooth muscle cells from preglomerular vessels. American Journal of Physiology. 1999;277:F533–542. doi: 10.1152/ajprenal.1999.277.4.F533. [DOI] [PubMed] [Google Scholar]
- Gibson A, McFadzean I, Wallace P, Wayman CP. Capacitative Ca2+ entry and the regulation of smooth muscle tone. Trends in Pharmacological Sciences. 1998;19:266–269. doi: 10.1016/s0165-6147(98)01222-x. [DOI] [PubMed] [Google Scholar]
- Goeger DE, Riley RT, Dorner JW, Cole RJ. Cyclopiazonic acid inhibition of the Ca2+-transport ATPase in rat skeletal muscle sarcoplasmic reticulum vesicles. Biochemical Pharmacology. 1988;37:978–981. doi: 10.1016/0006-2952(88)90195-5. [DOI] [PubMed] [Google Scholar]
- Golovina VA. Cell proliferation is associated with enhanced capacitative Ca2+ entry in human arterial myocytes. American Journal of Physiology. 1999;277:C343–349. doi: 10.1152/ajpcell.1999.277.2.C343. [DOI] [PubMed] [Google Scholar]
- Gomes DC, Madeira VM. Magnesium and manganese ions modulate Ca2+ uptake and its energetic coupling in sarcoplasmic reticulum. Archives of Biochemistry and Biophysics. 1986;249:199–206. doi: 10.1016/0003-9861(86)90575-8. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hescheler J, Schultz G. Nonselective cation channels: physiological and pharmacological modulations of channel activity. EXS. 1993;66:27–43. doi: 10.1007/978-3-0348-7327-7_2. [DOI] [PubMed] [Google Scholar]
- Hinkle PM, Shanshala ED, Nelson EJ. Measurement of intracellular cadmium with fluorescent dyes. Further evidence for the role of calcium channels in cadmium uptake. Journal of Biological Chemistry. 1992;267:25553–25559. [PubMed] [Google Scholar]
- Hobai IA, Bates JA, Howarth FC, Levi AJ. Inhibition by external Cd2+ of Na/Ca exchange and L-type Ca channel in rabbit ventricular myocytes. American Journal of Physiology. 1997;272:H2164–2172. doi: 10.1152/ajpheart.1997.272.5.H2164. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Fasolato C, PoANZZ T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+] Journal of Cell Biology. 1998;140:325–334. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hopf FW, Reddy P, Hong J, Steinhardt RA. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. Journal of Biological Chemistry. 1996;271:22358–22367. doi: 10.1074/jbc.271.37.22358. [DOI] [PubMed] [Google Scholar]
- Inoue R. Effect of external Cd2+ and other divalent cations on carbachol-activated non-selective cation channels in guinea-pig ileum. Journal of Physiology. 1991;442:447–463. doi: 10.1113/jphysiol.1991.sp018802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circulation Research. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- Jabr RI, Toland H, Gelband CH, Wang XX, Hume JR. Prominent role of intracellular Ca2+ release in hypoxic vasoconstriction of canine pulmonary artery. British Journal of Pharmacology. 1997;122:21–30. doi: 10.1038/sj.bjp.0701326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiak R, Wilson SM, Montague S, Hume JR. Heterogeneity of calcium stores and elementary release events in canine pulmonary arterial smooth muscle cells. American Journal of Physiology - Cell Physiology. 2001;280:C22–33. doi: 10.1152/ajpcell.2001.280.1.C22. [DOI] [PubMed] [Google Scholar]
- Keizer J, De Young G. Effect of voltage-gated plasma membrane Ca2+ fluxes on IP3-linked Ca2+ oscillations. Cell Calcium. 1993;14:397–410. doi: 10.1016/0143-4160(93)90044-7. [DOI] [PubMed] [Google Scholar]
- Keizer J, Li YX, Stojilkovic S, Rinzel J. InsP3-induced Ca2+ excitability of the endoplasmic reticulum. Molecular Biology of the Cell. 1995;6:945–951. doi: 10.1091/mbc.6.8.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura J, Miyamae S, Noma A. Identification of sodium-calcium exchange current in single ventricular cells of guinea-pig. Journal of Physiology. 1987;384:199–222. doi: 10.1113/jphysiol.1987.sp016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama H, Kitamura K, Itoh T, Inoue R. Physiological features of visceral smooth muscle cells, with special reference to receptors and ion channels. Physiological Reviews. 1998;78:811–920. doi: 10.1152/physrev.1998.78.3.811. [DOI] [PubMed] [Google Scholar]
- Lewis RS. Store-operated calcium channels. Advances in Second Messenger Phosphoprotein Research. 1999;33:279–307. doi: 10.1016/s1040-7952(99)80014-7. [DOI] [PubMed] [Google Scholar]
- Ma HT, Patterson RL, Van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JXJ. Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2001;280:L870–880. doi: 10.1152/ajplung.2001.280.5.L870. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- McKay RR, Szymeczek-Seay CL, Lievremont JP, Bird GS, Zitt C, Jungling E, Luckhoff A, Putney JW. Cloning and expression of the human transient receptor potential 4 (TRP4) gene: localization and functional expression of human TRP4 and TRP3. Biochemical Journal. 2000;351:735–746. [PMC free article] [PubMed] [Google Scholar]
- Minke B, Cook B. TRP channel proteins and signal transduction. Physiological Reviews. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- Missiaen L, Declerck I, Droogmans G, Plessers L, De Smedt H, Raeymaekers L, Casteels R. Agonist-dependent Ca2+ and Mn2+ entry dependent on state of filling of Ca2+ stores in aortic smooth muscle cells of the rat. Journal of Physiology. 1990;427:171–186. doi: 10.1113/jphysiol.1990.sp018166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LC, Gurney AM. Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circulation Research. 2001;89:923–929. doi: 10.1161/hh2201.100315. [DOI] [PubMed] [Google Scholar]
- Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. Journal of Biological Chemistry. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW, Bird GS. The signal for capacitative calcium entry. Cell. 1993;75:199–201. doi: 10.1016/0092-8674(93)80061-i. [DOI] [PubMed] [Google Scholar]
- Putney JW, McKay RR. Capacitative calcium entry channels. Bioessays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- Robertson TP, Aaronson PI, Ward JP. Hypoxic vasoconstriction and intracellular Ca2+ in pulmonary arteries: evidence for PKC-independent Ca2+ sensitization. American Journal of Physiology. 1995;268:H301–307. doi: 10.1152/ajpheart.1995.268.1.H301. [DOI] [PubMed] [Google Scholar]
- Robertson TP, Hague D, Aaronson PI, Ward JP. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. Journal of Physiology. 2000;525:669–680. doi: 10.1111/j.1469-7793.2000.t01-1-00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD, Lee RJ, Oliver JM, Keizer J. Effect of Ca2+ influx on intracellular free Ca2+ responses in antigen- stimulated RBL-2H3 cells. American Journal of Physiology. 1996;270:C939–952. doi: 10.1152/ajpcell.1996.270.3.C939. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [published erratum appears in Nature (1994) 372(6508), 812] [DOI] [PubMed] [Google Scholar]
- Stojilkovic SS, Kukuljan M, Iida T, Rojas E, Catt KJ. Integration of cytoplasmic calcium and membrane potential oscillations maintains calcium signaling in pituitary gonadotrophs. Proceedings of the National Academy of Sciences of the USA. 1992;89:4081–4085. doi: 10.1073/pnas.89.9.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepakova ES, Csutora P, Hunton DL, Marchase RB, Cohen RA, Bolotina VM. Calcium influx factor (CIF) directly activates store-operated cation channels in vascular smooth muscle cells. Journal of Biological Chemistry. 2000;275:26158–26163. doi: 10.1074/jbc.M004666200. [DOI] [PubMed] [Google Scholar]
- Utz J, Eckert R, Trautwein W. Changes of intracellular calcium concentrations by phenylephrine in renal arterial smooth muscle cells. Pflügers Archiv. 1999;438:725–731. doi: 10.1007/s004249900091. [DOI] [PubMed] [Google Scholar]
- Walker RL, Hume JR, Horowitz B. Differential expression and alternative splicing of TRP channel genes in smooth muscles. American Journal of Physiology - Cell Physiology. 2001;280:C1184–1192. doi: 10.1152/ajpcell.2001.280.5.C1184. [DOI] [PubMed] [Google Scholar]
- Waurick R, Knapp J, Van Aken H, Boknik P, Neumann J, Schmitz W. Effect of 2,3-butanedione monoxime on force of contraction and protein phosphorylation in bovine smooth muscle. Naunyn-Schmiedeberg's Archives of Pharmacology. 1999;359:484–492. doi: 10.1007/pl00005380. [DOI] [PubMed] [Google Scholar]
- Welsh DG, Nelson MT, Eckman DM, Brayden JE. Swelling-activated cation channels mediate depolarization of rat cerebrovascular smooth muscle by hyposmolarity and intravascular pressure. Journal of Physiology. 2000;527:139–148. doi: 10.1111/j.1469-7793.2000.t01-1-00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SM, Johnston L, Nicholson N, Janiak R, Hume JR. Intracellular Ca2+ store depletion activates capacitative Ca2+ entry in canine pulmonary and renal artery smooth muscle cells. Biophysical Journal. 2001;80:351A. [Google Scholar]