

Abstract
This study examined NO- and non-NO-, non-prostanoid-dependent pathways of agonist-induced vasodilatation in streptozotocin (STZ)-induced diabetic rats and their age-matched controls at 1–2, 8–10 and 18–20 weeks after induction of diabetes. Using laser Doppler flowmetry, vasodilatory responses to acetylcholine (ACh; 0.1 mM) and morpholino-sydnonimine (SIN-1) were determined in the presence of Ringer solution, during inhibition of NO synthase (NOS) and cyclo-oxygenase (COX) with Nω-nitro-L-arginine (L-NNA; 1 mM) + indomethacin (10−5 M), and during inhibition of K+ channels, NOS and COX with tetraethylammonium (TEA; 10 mM) + L-NNA + indomethacin. Basal NOS activity and nerve conduction velocity were also determined. In age-matched controls, SIN-1-induced vasodilatation in the presence of TEA + L-NNA + indomethacin, basal NOS activity and the initial vasodilatory response to ACh during NOS and COX inhibition all decreased with maturation. In STZ-induced diabetics, SIN-1-induced vasodilatation in the presence of TEA + L-NNA + indomethacin was impaired immediately after induction of diabetes, but not at 18–20 weeks. NOS activity in STZ-induced diabetics displayed a transient 2-fold increase at 8–10 weeks, decreasing to age-matched control levels at 18–20 weeks. At 18–20 weeks of STZ-induced diabetes, ACh-induced vasodilatation during NOS and COX inhibition was prolonged due to increased K+ channel activity and experimental diabetic sensory neuropathy (EDN) had developed. Thus, in sciatic nerve microcirculation of STZ-induced diabetic rats: (1) diabetic impairment of vasodilatation in response to exogenous NO was transient; (2) non-NO-, non-prostanoid-dependent vasodilatation and K+ channel activity were augmented in STZ-induced diabetes; and (3) alterations in NO bioactivity were not related to the development of EDN.
Global endothelial dysfunction characterizes both human and experimental diabetes mellitus (Cosentino & Luscher, 1997). In streptozotocin (STZ)-induced diabetes, impaired acetylcholine (ACh)-induced vasodilatation, which is an index of endothelial dysfunction, has been found at all levels of the arterial bed. In the aorta, diabetic endothelial dysfunction has been attributed to defective nitric oxide (NO) synthesis or effect (for review, see Pieper, 1998), including quenching of NO by superoxide anions due to the increased oxidative stress found in diabetes (Hattori et al. 1991; Karasu, 2000). In smaller blood vessels, diabetic endothelial dysfunction may be due to impaired endothelium-dependent hyperpolarizing factor (EDHF) activity, since EDHF is the principal mediator of ACh-induced vasodilatation in the peripheral microvascular bed (Garland et al. 1995; Chen et al. 1997; Woodman et al. 2000). Acetylcholine-induced vasodilatation in rat sciatic nerve microcirculation is dependent upon three vasodilators: NO, a vasodilatory prostanoid, and a NOS- and COX-independent K+ channel-dependent factor, which may be EDHF (Thomsen et al. 2000). In this microcirculatory bed, endothelial dysfunction can result from an impairment of one or more of these vasodilators or their interactions.
Diabetes mellitus in humans is frequently complicated by distal symmetrical polyneuropathy affecting the nerves of the lower extremities. In the rat, experimental diabetic neuropathy (EDN) affects both motor and sensory nerves and is initially characterized by slower nerve conduction velocity levels compared to age-matched controls (Zochodne & Ho, 1994). Vascular as well as metabolic factors have been implicated in the pathogenesis of EDN. Vascular factors include increased peripheral resistance, which occurs immediately upon the onset of diabetic hyperglycaemia (Brands et al. 2000). In diabetic sciatic nerve, the increased vascular resistance is accompanied by reduced nerve blood flow (NBF) and tissue hypoxia (Tuck et al. 1984; Wright & Nukada, 1994). Support for the suggestion that vascular factors are essential to the pathogenesis of diabetic neuropathy came from the finding that treatment of established experimental diabetic neuropathy with supplemental oxygen improved nerve conduction (Low et al. 1984). Further support came from studies showing that systemic treatment with vasodilators (Hotta et al. 1995; Hohman et al. 2000) and inhibitors of vasoconstriction (Cameron et al. 1994; Kihara et al. 1999) improved nerve conduction while increasing nerve blood flow. However, treatment directed at correcting the metabolic imbalances found in STZ-induced diabetes, i.e. treatment with anti-oxidants, aldose reductase inhibitors or protein kinase C inhibitors, also ameliorates the impaired nerve conduction velocity and endoneurial blood flow of diabetes (Nagamatsu et al. 1995; Nakamura et al. 1998, 1999; Cameron et al. 1999; Pop-Busui et al. 2001), so the question arises whether EDN is primarily of vascular or metabolic origin.
Recently, endothelial dysfunction has been demonstrated in sciatic nerve microcirculation of STZ-induced diabetic rats (Maxfield et al. 1997; Terata et al. 1999). In the sciatic nerve, endothelial dysfunction develops before diabetic neuropathy (Coppey et al. 2000), suggesting that impaired vasoreactivity may also contribute to EDN. Indeed, treatment with anti-oxidants improves nerve conduction, vascular reactivity to acetylcholine and NBF in STZ-induced diabetic animals (Coppey et al. 2001a,b). In the present study, we examined the mechanisms of diabetic endothelial dysfunction in more detail by studying NO-related and non-NO-related pathways of agonist-induced vasodilatation. Specifically, we examined the influence of inhibition of nitric oxide synthase (NOS) and cyclo-oxygenase (COX) and K+ channel blockade on ACh- and SIN1-induced vasodilatation in the microcirculation of rat sciatic nerve. Basal NOS activity was determined as well. The above parameters were related to the development of EDN in STZ-induced diabetic rats with diabetes lasting up to 20 weeks, and to the normal electrophysiological development in their age-matched controls.
METHODS
Animals
This study was performed in full compliance with the guidelines set forth in the European Council's Convention for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes and was approved by the Danish National Ethics Committee. Seventy-seven male Sprague-Dawley rats were used, of which 41 were used for both the nerve conduction and in vivo haemodynamic studies, as well as the in vitro NOS assay. A further 24 rats were used for the nerve conduction study and 12 for the NOS assay. The animals were kept in plastic cages with even floors covered with wood shavings. They had free access to tap water and Altromin 1314 rat chow, and were acclimated to a 12 h:12 h light-dark cycle. After completion of the experimental protocols, all rats were killed with an overdose of pentobarbitone.
Experimental diabetes mellitus
Diabetes was induced at the age of 5 weeks (211 ± 13 g body weight) by an intraperitoneal injection of streptozotocin dissolved in citrate buffer (65 mg kg−1). Age-matched littermates were used as controls and injected with vehicle alone (pH 4.8). The presence of diabetes was assessed 7 days later using a GlucoTouch™ glucose meter (LifeScan Inc., Milpitas, CA, USA). Rats with whole blood glucose values of 18.0 mm or higher were accepted as diabetic; streptozotocin-injected rats with values under 18.0 mm were discarded. Before the experimental procedure, blood for glycosylated haemoglobin (HbA1C) and fasting plasma glucose analyses was drawn. HbA1C ranged from 3.41 to 4.30 % in age-matched control rats and from 5.03 to 12.29 % in streptozotocin-injected rats regardless of the duration of diabetes, except for one rat with an HbA1C value of 4.34 % after 18 weeks of diabetes. This rat was excluded from the study. Three groups of diabetic and age-matched control animals were examined 1–2, 8–10 or 18–20 weeks after injection. Mean values for HbA1C, fasting plasma glucose, initial and end weights for all six groups are shown in Table 1.
Table 1.
Glycosylated haemoglobin and fasting plasma glucose levels in age-matched controls and STZ-induced diabetic rats
| Age/duration of diabetes (weeks) | HbA1C (%) | Fasting plasma [glucose](mmol l−1) | Initial weight (g) | Final weight (g) | n |
|---|---|---|---|---|---|
| Age-matched controls | |||||
| 1–2 weeks | 3.58 ± 0.03 | 5.28 (CI:4.29–6.50) | 213 ± 2 | 315 ± 9 | 13 |
| 8–10 weeks | 3.92 ± 0.04* | 7.31 (CI:5.18–10.34) | 214 ± 5 | 466 ± 12 | 8 |
| 18–20 weeks | 3.84 ± 0.05* | 7.46*(CI:6.06–9.18) | 225 ± 4 | 535 ± 12 | 14 |
| STZ-induced diabetics | |||||
| 1–2 weeks | 6.69 ± 0.36†† | 8.69† (CI:6.99–10.81) | 203 ± 2 | 211 ± 6††† | 12 |
| 8–10 weeks | 9.93 ± 0.21††††‡‡ | 13.38† (CI:11.06–16.19) | 209 ± 3 | 246 ± 10††† | 19 |
| 18–20 weeks | 9.58 ± 0.44††‡ | 10.50 (CI:7.71–14.31) | 200 ± 4† | 313 ± 18†† | 6 |
Values are given as means ± S.E.M.,except for fasting plasma glucose values, which are given as means and 95% confidence intervals (CI). n is the number of animals.
P >0.01 vs. age-matched controls at 1–2 weeks after injection.
P < 0.01
P >10−4vs. STZ rats with 1–2 weeks of diabetes.
P >0.01
P < 10−4
P >10−7
P >10−15, age-matched controls vs. STZ-induced diabetics.
Analysis of fasting plasma glucose
Several days before the nerve conduction and in vivo haemodynamic studies were performed, the animals were anaesthetized with 70 % CO2-30 % O2 after an overnight fast. Blood was carefully taken from the retro-orbital venous plexus; samples were placed into heparinized Eppendorf microtubes, immediately cooled and centrifuged at 13 000 g for 20–30 s at 4 °C. The supernatant was snap frozen in liquid nitrogen and stored at -80 °C. Analysis was performed using an automated glucose analyser (YSI 2300, Yellow Springs Instruments, Yellow Springs, OH, USA).
Nerve conduction study
Motor and sensory nerve conduction determinations were made before nerve blood flow (NBF) measurements. Non-fasting animals were anaesthetized with intraperitoneal pentobarbitone (Mebumal, Nycomed, Roskilde, Denmark; initial dose 50 mg kg−1; supplemental doses 10–20 mg kg−1). Paired steel needle electrodes were inserted percutaneously with a distance of 1 cm between the reference electrode and the stimulating or recording electrode. Stimulating electrodes were inserted at the sciatic notch and at the knee. Recording electrodes were placed over the small muscles of the dorsum of the hind paw. Supramaximal stimulation current, defined as 110 % of the stimulation current which gave maximal response, was given in single, square-wave pulses with a duration of 0.2 ms. Motor nerve responses were amplified 100 times, and filtered at 10 Hz to 4 kHz. Motor nerve conduction velocity was calculated as the difference in latencies measured from the sciatic notch and from the knee divided by the difference in distance measured from each of these two points to the recording electrode on the hind paw. Motor nerve conduction velocities were measured twice in each hind limb and results averaged. Rectal temperatures were kept in the range of 37–38 °C using an infrared heating lamp.
For sensory nerve conduction determinations, the recording electrodes were placed at the base of the tail and the stimulating electrodes placed 8 cm distally. Stimulation current was 4 mA, based on pilot studies showing that stimulation with this amount of current produced maximal responses without producing artefacts due to current spread. Stimulation was given as a train of square-wave pulses (10 pulses per train, frequency 1 Hz, duration 0.2 ms) and the responses were averaged. Sensory nerve responses were amplified 1000 times and filtered at 30 Hz to 2 kHz. Sensory nerve conduction velocity was calculated as latency divided by distance between the stimulating and recording electrodes. Near-nerve temperatures in the tail were held constant at 37 °C with an infrared heating lamp. Both motor and sensory nerve responses were sampled at 83333 Hz, recorded and analysed off-line using Spike2 software program with a 1401+ interface (Cambridge Electronic Design, Cambridge, UK). Latencies were measured from the point of stimulation to the onset of the response and amplitudes were measured from peak to peak.
In vivo haemodynamic studies
Surgical procedures
Surgical procedures and experimental set-up have been described previously (Thomsen et al. 2000). Surgery was performed under pentobarbitone anaesthesia in animals having undergone nerve conduction measurements immediately preceding the surgical procedure. Otherwise, halothane anaesthesia was used (induction, 3 %; surgery, 1.8 %; Fluotec 3 vaporizer, Cyprane, UK). All animals were non-fasting. Both jugular veins were cannulated for the administration of drugs. The left carotid artery was cannulated for measurement of blood gases and arterial blood pressure. The rats were tracheotomized and artificially ventilated with 30 % O2:70 % N2O, ensuring arterial partial pressure of O2 >100 mmHg and arterial partial pressure of CO2 of 40 ± 3 mmHg. The left sciatic nerve was carefully exposed, the muscles of the thigh forming a well around it. After surgery, anaesthesia was maintained by an intravenous infusion of pentobarbitone (initial bolus 19 mg kg−1; infusion 23–29 mg kg−1 h−1). Suxamethonium (initial bolus 13 mg kg−1; supplemental doses 3 mg kg−1 (30 min)−1) was given through a peritoneal catheter. Adequate levels of anaesthesia were ensured by regularly verifying the absence of blood pressure responses to tail pinch throughout the experimental procedure.
Experimental set-up
Microcirculatory NBF responses were measured using laser Doppler flowmetry (LDF). Four identical LDF needle probes (Probe 411, Perimed, Jãrfãlla, Sweden; 140 μm separation between emitting and receiving fibres) measuring NBF down to a tissue depth of 0.5 mm were employed, connected to two PeriFlux 4001 monitors (Perimed). The probes were positioned over the sciatic nerve above the trifurcation, avoiding large epineurial blood vessels, with a minimum of 1 mm between neighbouring probes to ensure that areas of measurement did not overlap. The probes were kept in the same position throughout the experimental procedure. NBF and blood pressure were measured continuously with sampling rates of 10 and 100 Hz, respectively, with a time constant of 0.2 s. Spike2 software (Cambridge Electronic Design) was used for data acquisition and later off-line analysis.
Experimental procedures
The well was initially filled with Ringer solution (mm: NaCl 145, KCl 3.5, CaCl2 2, Hepes 5; pH adjusted to 7.4). After establishing baseline NBF, 3-morpholino-sydnonimine (SIN-1) was applied topically in increasing concentrations until the smallest concentration at which NBF increased by 25–50 % was achieved (range 0.1–0.75 mm). This concentration of SIN-1 was used throughout the experiment. NBF responses to SIN-1 and acetylcholine (ACh; 100 μM) were first measured in the presence of Ringer solution, then during inhibition of NOS and COX with Nω-nitro-l-arginine (l-NNA; 1 mm) + indomethacin (10−5m), and finally during combined blockade of K+ channels and inhibition of NOS and COX with tetraethylammonium (TEA; 10 mm) + l-NNA (1 mm) + indomethacin (10−5m). To reduce the variability of the NBF responses, the average of two consecutive stimulations with each vasodilator at each step was used. Each stimulation lasted 5 min, after which the well was washed out and the baseline re-established. Basal NBF responses to l-NNA (1 mm) + indomethacin (10−5m) and to TEA (10 mm) + l-NNA (1 mm) + indomethacin (10−5m) were also recorded after exposure to these inhibitors for 30 min. Systemic blood pressure was unaffected by the topical application of agonists and inhibitors.
Analysis of HbA1C
Blood for the analysis of HbA1C was drawn in connection with surgery. After cannulation of the left carotid artery and before washing the line with isotonic NaCl, a small sample of blood (0.05 ml) was drawn in Eppendorf microtubes containing 1 μl heparin (5000 i.u. ml−1), snap frozen in liquid nitrogen, and stored at -80 °C. Analysis of HbA1C was performed using a Roche Cobas spectrophotometer (Roche Diagnostic Systems, Branchburg, NJ, USA).
NOS assay
Biopsies of the undisturbed right sciatic nerve were excised after completion of the haemodynamic protocol. Rats not participating in the nerve blood flow study were anaesthetized with intraperitoneal injections of pentobarbitone (50 mg kg−1). The tissue specimens were snap frozen in liquid nitrogen and stored at -80 °C until the assays were performed.
The NOS assay was based on the conversion of l-[3H]-arginine to l-[3H]-citrulline. Specifically, tissue samples were homogenized in ice-cold 0.05 m Tris buffer (pH 7.4) containing 1.15 mm KCl, 1 mm EDTA, 5 mm glucose, 0.1 mmdl-dithiothreitol (DTT), 200 U ml−1 superoxide dismutase, 4.2 μM leupeptin, 2.9 μM pepstatin A, 50 nm trypsin inhibitor and 0.25 mm phenylmethylsulphonyl fluoride. The homogenate was centrifuged for 10 min at 2000 g at 2 °C and the protein content was determined according to the method of Groves et al. (1968). The NOS assay was performed using 50 mm Hepes buffer (pH 7.4) containing 1 mm EDTA, 1 mm DTT, the cofactors 1.25 mm CaCl2, 1 μM calmodulin, 15 μM tetrahydrobiopterin and 1 μM flavin adenine dinucleotide (FAD), and the substrate 100 μM arginine. Aliquots of supernatant from the tissue homogenate (40 μl) were added to the Hepes buffer (110 μl) to obtain final a protein concentration of 10–20 μg ml−1. Addition of the cofactor NADPH (final concentration 1 mm) to the buffer initiated the reaction, which ran for 60 min at 37 °C. The reaction was terminated by addition of 1 ml ice-cold Hepes buffer (pH 5.5) containing 10 mm EGTA (stop buffer). The total volume (1.15 ml) was applied to 0.5 ml Dowex AG50 WX-8 columns (Na+ form) that had been equilibrated with stop buffer (minus EGTA). l-[2,3-3H]-Citrulline was eluted twice with 0.5 ml stop buffer (minus EGTA), and radioactivity was determined by liquid scintillation counting.
Calculations and statistics
Nerve microvasular conductance vs. nerve blood flow
Since NBF varies proportionately with mean arterial blood pressure (Low & Tuck, 1984), NBF values were converted into nerve microvascular conductance (nMVC) values, using the equation:
where BP is mean arterial blood pressure. Since LDF measures NBF in arbitrary units of flux, nMVC was calculated as arbitrary units of flux per mmHg.
Components of ACh-induced vasodilatation
In rat sciatic nerve microcirculation, the initial increase in blood flow during acetylcholine-induced vasodilatation is NOS and COX independent and K+ channel dependent, while NO and a vasodilatory prostanoid mediate the following phase of sustained vasodilatation (Thomsen et al. 2000). NO- and prostanoid-dependent vasodilatation was taken as the NBF response to ACh during the last 60 s of the 5 min stimulation in the presence of Ringer solution. However, this applied only to nondiabetic rats, because K+ channel activity contributed significantly to the terminal ACh response in STZ-induced diabetics (see Results). Since the ACh response in the presence of l-NNA is not inhibited in rat sciatic nerve microcirculation (Thomsen et al. 2000), NO-mediated vasodilatation was assessed using the NO donor, SIN-1. Like ACh-induced vasodilatation, SIN-1-induced vasodilatation was also measured during the last 60 s to allow comparisons with the ACh responses.
Statistics
Ratios between nMVC responses and the immediately preceding nMVC baselines were determined for both vasodilators and inhibitors, and used for statistical calculations. The nMVC response ratios, NOS activity and fasting glucose values were log-transformed to ensure normal distribution of the residuals and, in these cases, the data are expressed as back-transformed means and confidence intervals. All other parameters had Gaussian distributions and these data are given as means ± s.e.m. Statistical analysis was carried out using analysis for linear trend, multiple linear regression, as well as one- and two-way analyses of variance (ANOVA). When significance was found using ANOVA, unpaired and Student's paired t tests were performed between and within groups, respectively. Bonferroni corrections were made in cases of multiple t testing. P < 0.05 was considered statistically significant.
Drugs
Fresh solutions of streptozotozin were made using citrate buffer and used immediately for the induction of diabetes. For the haemodynamic measurements, fresh solutions of all drugs were made using Ringer solution each day; fresh solutions of acetylcholine were made immediately preceding each application. Substances that were not readily dissolvable in Ringer solution were ultrasonicated for 5 min. ACh, SIN-1, l-NNA, indomethacin and TEA were all given topically.
ACh, SIN-1, l-NNA, guanosine-5-triphosphate (GTP), phenylmethylsulphonyl fluoride (PMSF), 1-methyl-3-isobutylxanthine (IBMX), theophylline, EDTA, STZ and citrate buffer solution were obtained from Sigma. Superoxide dismutase, leupeptin, pepstatin A, trypsin inhibitor, creatine kinase and creatine phosphate were obtained from Boehringer. TEA was obtained from ICN Biomedicals (Aurora, OH, USA). Indomethacin (Confortid; Alpharma, Copenhagen, Denmark), suxamethonium (50 g l−1) and pentobarbitone (50 g l−1) were obtained from the pharmacy of the National Hospital of Denmark.
RESULTS
HbA1C and fasting plasma glucose
In age-matched controls, HbA1C and fasting plasma glucose values increased with maturation (HbA1C: P = 1.433 × 10−5, plasma glucose: P = 2.81 × 10−4, both one-way ANOVA; Table 1). Both parameters were elevated in STZ-induced diabetic rats compared to age-matched controls (HbA1C: P < 2.20 × 10−16, plasma glucose: P = 1.128 × 10−6, both two-way ANOVA). However, fasting plasma glucose values of diabetic rats often fell within the range of those of age-matched controls, in contrast to HbA1C values, which were always elevated in diabetic rats (Fig. 1). This implies that the STZ-induced diabetic rats of this study were characterized to a larger degree by glucose intolerance than by fasting hyperglycaemia, even during prolonged diabetes. Therefore, HbA1C was taken as the more reliable indicator of the presence of diabetes mellitus.
Figure 1. Relationship between HbA1C and fasting plasma glucose.
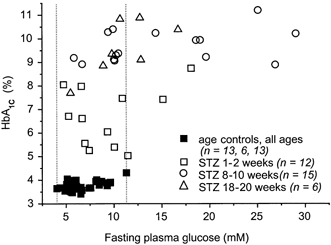
Fasting plasma glucose values of STZ-induced diabetic rats were often found within the range of those of age-matched controls (indicated by the dotted lines), while diabetic HbA1C values were always elevated. As this figure demonstrates, STZ-induced diabetes did not result in fasting hyperglycaemia in approximately 50 % of the animals, even in cases of prolonged diabetes. For this reason, HbA1C was taken as a more reliable indicator of the presence of diabetes.
In vivo haemodynamic study
Blood pressure
During the haemodynamic study, systemic blood pressure in each animal remained stable throughout the experimental procedure and was not influenced by the topical application to the nerve of vasodilators or inhibitors. Mean blood pressure was measured during each application of vasodilators and inhibitors and in the following, this series of mean blood pressure measurements is referred to as the blood pressure level (Table 2). Blood pressure levels in STZ-induced diabetics were not elevated in the 8–10 week (P = 0.2398, two-way ANOVA) and 18–20 week groups (P = 0.1973, two-way ANOVA) compared to the 1–2 week group. In control animals, blood pressure levels varied significantly between all age groups (P = 2.497 × 10−11, two-way ANOVA). Due to the variation among control animals, a significant difference was found between blood pressure levels of STZ-induced diabtetics and age-matched controls at 8–10 (P = 3.265 × 10−8, two-way ANOVA) and 18–20 weeks (P = 2.590 × 10−8, two-way ANOVA). In contrast, comparison of mean blood pressure measurements at any given time point revealed significant differences between diabetics and age-matched controls only twice. Nonetheless, since significant differences in blood pressure levels were found among the controls themselves, and between STZ-induced diabetics and age-matched controls, our data were examined for any dependence of agonist-induced vasodilatation upon blood pressure, but none was found.
Table 2.
Mean blood pressure in age-matched controls and STZ-induced diabetics during stimulation with SIN-1 and ACh
| Blood pressure at 1–2 weeks (mmHg) | Blood pressure at 8–10 weeks (mmHg) | Blood pressure at 18–20 weeks (mmHg) | ||||
|---|---|---|---|---|---|---|
| Experimental protocol | Control (n = 6) | Diabetic (n = 6) | Control (n = 6) | Diabetic (n = 8) | Control (n = 8) | Diabetic (n = 6) |
| Ringer solution | ||||||
| SIN-1 | 116.8 ± 5.0 | 111.9 ± 10.4 | 95.5 ± 8.3 | 108.8 ± 6.4 | 109.2 ± 6.7 | 114.2 ± 4.4 |
| ACh | 115.3 ± 5.3 | 109.7 ± 9.9 | 94.8 ± 7.8 | 109.1 ± 6.0 | 108.7 ± 5.0 | 115.8 ± 2.1 |
| l-NNA +indomethacin | ||||||
| SIN-1 | 114.4 ± 4.7 | 113.8 ± 8.8 | 96.4 ± 7.2 | 110.5 ± 6.4 | 102.0 ± 7.6 | 117.9 ± 4.0 |
| ACh | 113.6 ± 4.6 | 117.9 ± 9.5 | 95.8 ± 5.6 | 110.5 ± 7.2 | 100.9 ± 7.5 | 121.5 ± 3.7* |
| TEA + l-NNA +indomethacin | ||||||
| SIN-1 | 115.9 ± 5.1 | 120.4 ± 9.1 | 98.9 ± 6.0 | 118.5 ± 6.4* | 105.6 ± 6.9 | 122.0 ± 4.1 |
| ACh | 113.5 ± 4.3 | 116.7 ± 12.1 | 94.4 ± 5.1 | 110.9 ± 8.4 | 103.9 ± 8.3 | 121.9 ± 2.7 |
| Time-averaged blood pressure levels | ||||||
| 115.2 ± 0.6 | 115.2 ± 1.1 | 96.4 ± 0.6‡‡ | 111.5 ± 1.0** | 105.3 ± 0.9†‡ | 118.9 ± 1.1†** | |
Mean arterial blood pressure values in age-matched controls and STZ-induced diabetics were significantly different only twice throughout the experimental procedure, once in the 8–10 week group in the presence of TEA + L-NNA + indomethacin during stimulation with SIN-1, and once in the 18–20 week group in the presence of l-NNA + indomethacin during stimulation with ACh. Blood pressure levels, i.e.series of blood pressure values determined throughout the experimental procedure, are given here averaged over time. Among STZ-induced diabetics, mean blood pressure levels were not different between the 1–2 week and the 8–10 or the 18–20 week groups. Among age-matched controls, blood pressure levels varied significntly between all age groups. Data are given as means ± s.e.m.
P < 0.05
P >10−7 compared to age-matched controls.
P >10−4
P >10−13 compared to same treatment group at 1–2 weeks.
P < 0.005 compared to same treatment group at 8–10 weeks.
Basal nerve blood flow
In control rats, the reduction in basal nMVC produced by l-NNA + indomethacin was similar at all ages (P = 0.1186, analysis for linear trend; Fig. 2A). Addition of TEA to the l-NNA + indomethacin already present reduced nMVC yet further (Fig. 2B). The tendency for the effect of TEA to decrease with maturation was not significant (P = 0.2003, analysis for linear trend).
Figure 2. The effect of l-NNA + indomethacin ± TEA on basal nMVC.
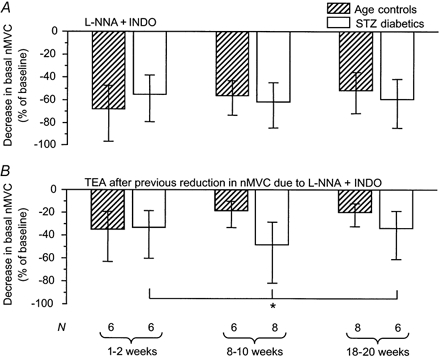
A, the decrease in basal nMVC upon application of l-NNA + indomethacin (1 mm + 10–5 m) was unchanged throughout maturation and was not influenced by the presence of diabetes. B, subsequent application of TEA (10 mm) caused a greater reduction of basal nMVC in STZ-induced diabetics than in age-matched controls (*P = 0.0339, two-way ANOVA), implying greater basal K+ channel activity in the diabetic state. The data are given as means and 95 % confidence intervals.
In STZ-induced diabetic rats, the reduction in nMVC due to l-NNA + indomethacin did not differ from that of age-matched controls (P = 0.8945, two-way ANOVA; Fig. 2A). In contrast, subsequent application of TEA reduced nMVC to a greater degree in STZ-induced diabetics than in age-matched controls (P = 0.0339, two-way ANOVA; Fig. 2B).
Sin-1-induced vasodilatation
In control animals, vasodilatory responses to SIN-1 in the presence of Ringer solution showed no variation with maturation (P = 0.2718, analysis for linear trend; Fig. 3A). As we have found previously, topical l-NNA + indomethacin potentiated SIN-1-induced vasodilatation 4- to 5-fold (Thomsen et al. 2000). In the presence of l-NNA + indomethacin ± TEA, the magnitude of SIN-1-induced vasodilatation declined with maturation (without TEA: P = 0.0134, with TEA: P = 0.0011, both analyses for linear trend; Fig. 3B and C).
Figure 3. SIN-1-induced vasodilatation.
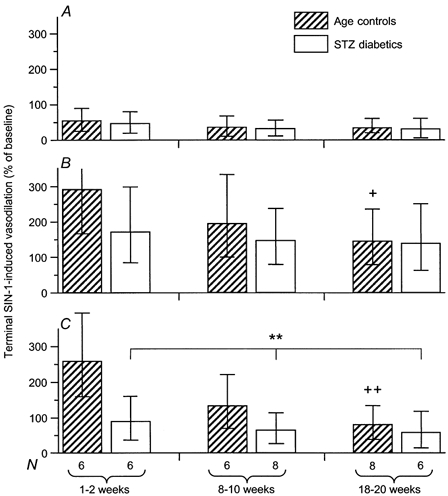
A, in the presence of Ringer solution. The vasodilatory response to SIN-1 was unaffected by diabetes and maturation. Blood pressure values in age-matched controls and STZ-induced diabetics were not significantly different at any age. B, in the presence of l-NNA + indomethacin (1 mm + 10−5m). The response to SIN-1 declined with maturation in age-matched controls (+P = 0.0134, analysis for linear trend), but not in STZ-induced diabetics. Blood pressure values in age-matched controls and STZ-induced diabetics were not significantly different at any age. C, in the presence of TEA + l-NNA + indomethacin (10 mm + 1 mm + 10−5m). A significant difference in the magnitude of the SIN-1 responses between age-matched controls and STZ-induced diabetics emerged after application of TEA subsequent to l-NNA + indomethacin. Under these conditions, the SIN-1 response declined with maturation (++P = 0.0011, analysis for linear trend) in age-matched controls, but was equally depressed in all STZ-induced diabetic groups. The difference between control and diabetic responses to SIN-1 in the presence of TEA + l-NNA + indomethacin (**P = 0.001, two-way ANOVA) demonstrates impaired reactivity of the NO-cGMP pathway to exogeneous NO in STZ-induced diabetes. This impairment occurred immediately after induction of diabetes in the 1–2 week group, was transient, and was no longer apparent in the 18–20 week group. At this point in the experimental protocol, significant differences between age-matched control and STZ-induced diabetic blood pressure values were found, but only between the 8–10 week groups (controls: 98.9 ± 6.0 mmHg; diabetics: 118.5 ± 6.4 mmHg). The data in all three panels are given as means and 95 % confidence intervals.
In STZ-induced diabetic animals, SIN-1-induced vasodilatation in the presence of Ringer solution and in the presence of l-NNA + indomethacin did not differ from that of age-matched controls (Ringer solution: P = 0.5463, l-NNA + indomethacin: P = 0.1444, two-way ANOVA; Fig. 3A and B). Conversely, in the presence of TEA + l-NNA + indomethacin, SIN-1-induced vasodilatation was clearly impaired in STZ-induced diabetic animals (P = 0.0011 vs. age-matched controls, two-way ANOVA; Fig. 3C). In the presence of these inhibitors, SIN-1-induced vasodilatation was of the same magnitude in all three diabetic groups, while decreasing in an age-dependent manner in the corresponding control groups. Thus, this impaired diabetic response to SIN-1 was most pronounced in animals having the least duration of diabetes and gradually disappeared with diabetes duration.
ACh-induced vasodilatation during NOS and COX inhibition
In both control and STZ-induced diabetic rats, acetylcholine in the presence of l-NNA + indomethacin induced a transient vasodilatory peak, which declined exponentially to baseline in age-matched controls and in the 1–2 week group of STZ-induced diabetic rats. In age-matched controls, time to peak and duration of the ACh response during NOS and COX inhibition were unaltered with maturation (time to peak: P = 0.3507, duration: P = 0.8414, both one-way ANOVA; Fig. 4). However, initial ACh-induced vasodilatation, measured from application of ACh to time of peak, decreased with maturation (P = 0.0410, analysis for linear trend; Fig. 5). Confirming the findings of our previous study, initial ACh-induced vasodilatation in the presence of l-NNA + indomethacin was inhibited, although not abolished, by TEA (Thomsen et al. 2000). The inhibitory effect of TEA on this ACh response did not vary with maturation (P = 0.1751, analysis for linear trend; Fig. 7).
Figure 4. Acetylcholine-induced vasodilatation in the presence of l-NNA + indomethacin (1 mm + 10−5m).
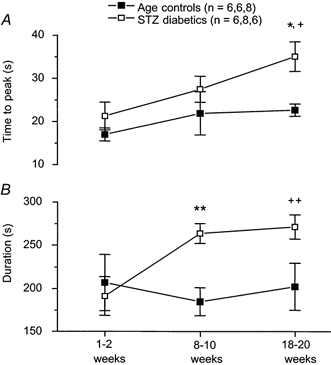
A, time-to-peak values in the presence of l-NNA + indomethacin increased with duration of diabetes (+P = 0.0112, analysis for linear trend) but not with maturation, and differed significantly from age-matched controls after 18–20 weeks of STZ-induced diabetes (*P = 0.0127). B, duration of ACh-induced vasodilatation in the presence of l-NNA + indomethacin increased with duration of diabetes (++P = 0.0076, analysis for linear trend), but not with maturation. The duration of this ACh response was significantly prolonged at 8–10 weeks of STZ-induced diabetes (**P = 0.0099). The data are given as means ± s.e.m.
Figure 5. Acetylcholine-induced vasodilatation in the presence of l-NNA + indomethacin (1 mm + 10−5m) in age-matched controls: the initial response.
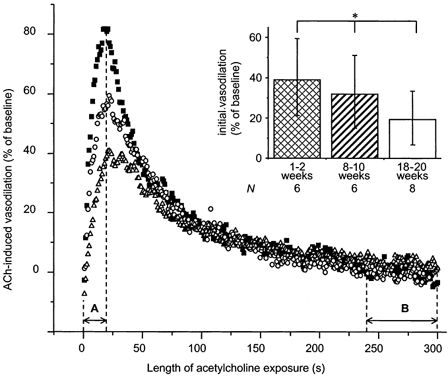
Each curve represents the averaged vasodilatory response to ACh of one age group, while each point represents a time average of 1 s. A, time to peak; B, the terminal 60 s interval of the 5 min exposure to ACh. Initial (inset) and terminal (Fig. 8B) responses are calculated as the area under the curve during A and B, respectively. Note that the ACh response during inhibition of NOS and COX was transient in spite of the continued presence of ACh. Importantly, the initial vasodilatory response declined with maturation in age-matched controls (inset, *P = 0.0410, analysis for linear trend). ▪, 1–2 week group; ○, 8–10 week group; and ▵, 18–20 week group.
Figure 7. The effect of TEA on initial ACh-induced vasodilatation during NOS and COX inhibition.
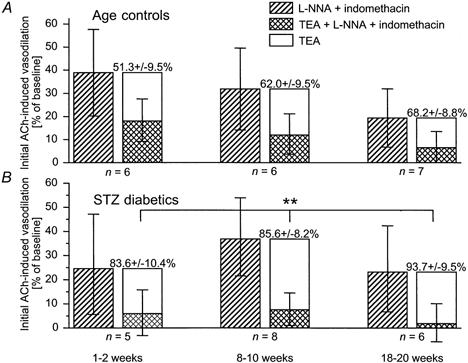
The effect of TEA is taken as the difference between ACh-induced vasodilatation in the presence of l-NNA + indomethacin (1 mm + 10−5m) and that in the presence of TEA + l-NNA + indomethacin (10 mm + 1 mm + 10−5m). The component of initial ACh-induced vasodilatation inhibited by 10 mm TEA was greater in STZ-induced diabetics (B) than in age-matched controls (A), demonstrating increased K+ channel activity during agonist-stimulated vasodilatation in STZ-induced diabetics. The effect of TEA is given as means ± s.e.m. and is shown by the stacked hollow columns. **P = 0.0012, STZ-induced diabetics vs. age-matched controls, two-way ANOVA.
In STZ-induced diabetics, time to peak and duration of the ACh response increased with diabetes duration (time to peak: P = 0.0112; duration: P = 0.0076, both analyses for linear trend; Fig. 4). These parameters were significantly prolonged compared to those of age-matched controls (time to peak: P = 0.0045; duration: P = 0.0181, both two-way ANOVA). Initial ACh-induced vasodilatation in the presence of l-NNA + indomethacin did not differ between diabetics and age-matched controls (P = 0.5882, two-way ANOVA). While this response clearly declined with maturation in controls, it did not do so in diabetics (P = 0.6851, analysis for linear trend; Fig. 6). As in controls, initial ACh-induced vasodilatation in the presence of l-NNA + indomethacin was inhibited by TEA and this inhibition was greater in STZ-induced diabetics compared to age-matched controls (P = 0.0012, two-way ANOVA, Fig. 7).
Figure 6. Acetylcholine-induced vasodilatation in the presence of l-NNA + indomethacin (1 mm + 10−5m) in STZ-induced diabetics: the initial response.
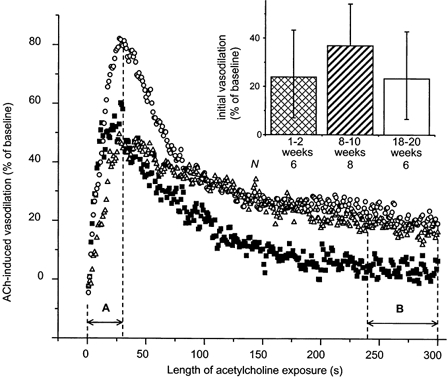
Each curve represents the averaged vasodilatory response to ACh of one age group, while each point represents a time average of 1 s. A, time to peak for the 8–10 week group (note that A varies, being less for the 1–2 week group and greater for the 18–20 week group); B, the terminal 60 s of the 5 min exposure to ACh. Initial (inset) and terminal (Fig. 8B) responses are calculated as the area under the curve during A and B, respectively. In contrast to age-matched controls, the initial response in STZ-induced diabetics did not decrease (P = 0.6851, analysis for linear trend), although the magnitudes of the initial responses in age-matched controls and STZ-induced diabetics did not differ (P = 0.5882). Note the sustained terminal ACh responses in the 8–10 and 18–20 week STZ-induced diabetic groups (compare to Fig. 5). ▪, 1–2 week group; ○, 8–10 week group; and ▵, 18–20 week group.
Terminal ACh-induced vasodilatation
In age-matched control rats, terminal ACh-induced vasodilatation in the presence of Ringer solution did not vary with maturation (P = 0.5392, one-way ANOVA; Fig. 8A). Consistent with terminal ACh responses in the presence of Ringer solution representing NO- and prostanoid-dependent vasodilatation, no terminal responses were found in the presence of l-NNA + indomethacin ± TEA in control animals (Fig. 8B and C).
Figure 8. Terminal vasodilatory responses to acetylcholine.
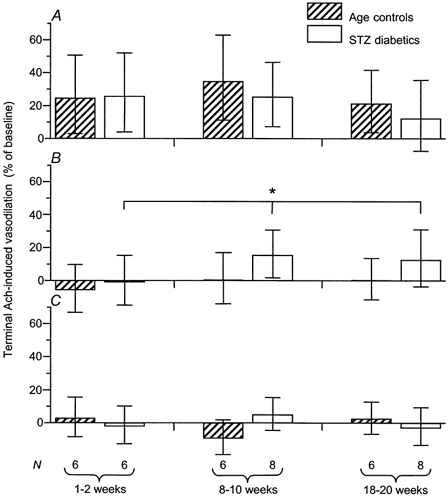
A, in the presence of Ringer solution. Terminal responses to acetylcholine did not decline significantly with maturation or with diabetes (controls: P = 0.5392, STZ-diabetics: P = 0.3927, both one-way ANOVA), nor did these responses differ significantly between age-matched controls and STZ-induced diabetics (P = 0.4173, two-way ANOVA). In controls, these responses represented NO- and prostanoid-dependent vasodilatation. In STZ-induced diabetic rats, the terminal responses in the older age groups were also dependent upon K+ channel activity, as shown in B and C. Blood pressure values in age-matched controls and STZ-induced diabetics were not significantly different at any age. B, in the presence of l-NNA + indomethacin (1 mm + 10−5m). Terminal responses to acetylcholine were abolished by inhibition of NOS and COX in controls, but not in STZ-induced diabetics with diabetes durations ≥ 8 weeks (*P = 0.0403, STZ vs. age-matched controls, two-way ANOVA). In the 18–20 week group, blood pressure values were significantly different between age-matched controls and STZ-induced diabetics at this point of the experimental protocol, being 105.5 ± 6.9 mmHg in controls and 121.5 ± 3.7 mmHg in diabetics. C, in the presence of TEA + l-NNA + indomethacin (10 mm + 1 mm + 10−5m). No terminal responses to ACh were found in either STZ-induced diabetics or controls after addition of TEA (P = 0.6324, STZ vs. age-matched controls, two-way ANOVA), demonstrating that the terminal ACh responses found in STZ-induced diabetics in the presence of l-NNA + indomethacin were dependent upon K+ channel activity. The data are given as means and 95 % confidence intervals. Blood pressure values in age-matched controls and STZ-induced diabetics were not significantly different at any age.
In STZ-induced diabetics, terminal ACh-induced vasodilatation in the presence of Ringer solution did not differ from that of age-matched controls (P = 0.4173, two-way ANOVA; Fig. 8A). In the presence of l-NNA + indomethacin, the prolonged duration of ACh-induced vasodilatation was reflected in significant terminal responses in rats with diabetes durations ≥ 8 weeks (P = 0.0403 vs. age-matched controls, two-way ANOVA; Fig. 8B). Subsequently, in the presence of TEA + l-NNA + indomethacin, no terminal responses were found in either STZ-induced diabetics or age-matched controls (P = 0.6324 vs. age-matched controls, two-way ANOVA; Fig. 8C). These findings indicate that K+ channel activity contributes to terminal ACh-induced vasodilatation in STZ-induced diabetic rats with durations of diabetes ≥ 8 weeks.
Summarizing the above findings, the same pattern of acetylcholine-induced vasodilatation in the presence of l-NNA + indomethacin was found in both controls and STZ-induced diabetics. An initial, transient, vasodilatory peak dependent upon K+ channel activity was seen, followed by a phase of no response in all control groups and by a phase of small, but sustained vasodilatation in STZ-induced diabetics with durations of diabetes of ≥ 8 weeks. In controls, the lack of response during this second phase was due to NOS and COX inhibition and was not influenced by K+ channel blockade, whereas in STZ-induced diabetics with diabetes lasting 8 weeks or more, the sustained vasodilatation of the second phase was entirely due to K+ channel activity.
NOS activity in sciatic nerve
Sciatic NOS activity in age-matched controls declined with maturation (P = 4.101 × 10−4, analysis for linear trend; Table 3). The decrease in NOS activity was significant at 18–20 weeks (P = 0.0015 vs. 1–2 weeks). In STZ-induced diabetics, sciatic NOS activity varied significantly (P = 3.996 × 10−4, one-way ANOVA; Table 3), increasing almost 2-fold after 8–10 weeks of diabetes, and declining again to age-matched control levels after a further 10 weeks. Diabetic NOS activity differed from that of age-matched controls (P = 0.0431, two-way ANOVA), although age-by-age comparisons revealed a difference only at 8–10 weeks.
Table 3.
Basal NOS activity in age-matched control and STZ-induced diabetic rats
| Duration of diabetes | Age-matched controls | STZ-induced diabetes |
|---|---|---|
| 1–2 weeks | 8.44 (CI:4.52–15.75) (n = 7) | 6.39 (CI:3.42–11.93) (n = 7) |
| 8–10 weeks | 4.84 (CI:2.41–9.73) (n = 6) | 11.57 (CI:7.46–17.95)*† (n = 12) |
| 18–20 weeks | 2.04 (CI:1.36–3.04)** (n = 14) | 3.60 (CI:1.90–6.82)(n = 7) |
Basal NOS activity in age-matched controls decreased with maturation (P = 4.101 × 10−5, analysis for linear trend). In STZ-induced diabetics, basal NOS activity was biphasic (P = 3.996 × 10−4, one-way ANOVA),first increasing to twice age-matched control levels at 8–10 weeks, then decreasing back to age-matched control levels at 18–20 weeks. The increase in NOS activity may be a compensatory mechanism to counteract the simultaneous impairment of NO–cGMP pathway responsiveness. The data are given as means and 95% confidence intervals (CI).
P < 0.05
P < 0.005 compared to 1–2 weeks in same group.
P < 0.05 compared to age-matched controls.
Nerve conduction velocity
Motor nerve conduction velocity
Motor nerve conduction velocity increased significantly with maturation in both controls and STZ-induced diabetic rats (controls: P = 4.414 × 10−6, diabetics: P = 1.341 × 10−5; both analyses for linear trend; Table 4). Although motor nerve conduction velocities of age-matched controls and those of STZ-induced diabetic rats differed significantly (P = 0.0066, two-way ANOVA), comparison of the two groups age-by-age did not reveal substantial differences.
Table 4.
Motor nerve conduction velocity and sensory action potentials in age-matched controls and STZ-induced diabetics
| Duration of diabetes | Age-matched controls | STZ-induced diabetes |
|---|---|---|
| A. Motor nerve conduction velocity (m s−1) | ||
| 1–2 weeks | 46.0 ± 2.0 (n = 12) | 42.4 ± 2.1 (n = 11) |
| 8–10 weeks | 60.8 ± 2.8 (n = 6) | 53.0 ± 2.1 (n = 11) |
| 18–20 weeks | 64.7 ± 2.2 (n = 10) | 59.5 ± 2.6 (n = 7) |
| B. Sensory action potential amplitude (nV) | ||
| 1–2 weeks | 20.3 ± 2.8 (n = 9) | 26.4 ± 2.1 (n = 8) |
| 8–10 weeks | 40.8 ± 3.7 (n = 6) | 45.6 ± 5.3 (n = 11) |
| 18–20 weeks | 83.7 ± 4.2 (n = 10) | 61.1 ± 8.3 (n = 7)* |
A, motor nerve conduction velocity increased with maturation in both controls (P = 4.414 ± 10−6, analysis for linear trend) and STZ-induced diabetics (P = 1.341 ± 10−5, analysis for linear trend). Although there was a significant difference between control and STZ-induced diabetic motor nerve conduction velocities (P = 0.0066, two-way ANOVA), age-by-age comparison of the two groups did not reveal significant differences. Data are given as means ± s.e.m. B, the amplitude of sensory action potentials also increased with maturation in age-matched controls (P = 7.16 ± 10−12, analysis for linear trend)and STZ-induced diabetics (P = 0.0022, analysis for linear trend). The two groups differed significantly from each other after 18–20 weeks of diabetes. Data are given as means ± s.e.m.
P < 0.05 compared to age-matched controls.
Sensory nerve conduction velocity
Like motor nerve conduction velocity, sensory nerve conduction velocity increased significantly with maturation in control and STZ-induced diabetic rats (controls: P = 1.347 × 10−13, STZ-induced diabetics: P = 2.097 × 10−4, both analyses for linear trend; Fig. 9). However, the increase in sensory nerve conduction velocity was not as great in STZ-induced diabetics as in age-matched controls (P = 4.357 × 10−5, two-way ANOVA). A significant deficit was found after 18–20 weeks of diabetes, amounting to -21 %. Taking age, weight, HbA1C and presence of diabetes into account, non-NO-, non-prostanoid-dependent ACh-induced vasodilatation was equally prognostic for the physiological development of sensory nerve function in controls and STZ-induced diabetics (P = 0.0143, coefficient ± standard error = 15.462 ± 6.037, adjusted r2 = 0.8644; multiple linear regression). Parameters related to NO, i.e. SIN-1-induced vasodilatation (with or without inhibitors) and NOS activity, were not related to sensory nerve conduction velocity in either age-matched controls or STZ-induced diabetics.
Figure 9. Sensory nerve conduction velocity during maturation and in STZ-induced diabetes.
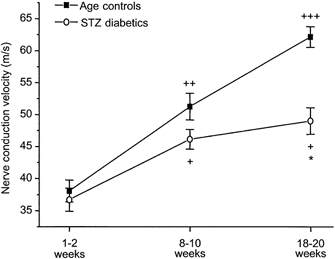
Sensory nerve conduction velocity increased with maturation in both age-matched control and STZ-induced diabetic rats, although this increase was attenuated in the diabetic animals. Significant difference in sensory nerve conduction velocities between STZ-induced diabetics and age-matched controls was found after 18–20 weeks of diabetes. Data are given as means ± s.e.m.+P < 0.01, ++P < 5 × 10−5 and +++P < 10−9vs. 1–2 weeks, same treatment. *P < 0.005, STZ-induced diabetics vs. age-matched controls, same age group.
The amplitudes of sensory action potentials also increased with maturation in both control and STZ-induced diabetic animals (controls: P = 7.16 × 10−12, STZ-induced diabetics: P = 0.0022; both analyses for linear trend; Table 4). The sensory action potential amplitudes of diabetic animals differed significantly from age-matched controls after 18–20 weeks of diabetes (P = 0.0378 vs. age-matched controls), where it amounted to 75 % of that of controls.
Discussion
This study examined NO-dependent and non-NO-, non-prostanoid-dependent vasodilatation in age-matched controls and STZ-induced diabetic rats, and examined diabetes-induced alterations of these variables in relationship to the development of experimental diabetic neuropathy. There were three major findings of this study: (1) ACh-induced vasodilatation during NOS and COX inhibition was prolonged in STZ-induced diabetes and, together with basal NBF, was dependent to a greater extent upon K+ channel activity in diabetics compared to age-matched controls; (2) SIN-1-induced vasodilatation during NOS, COX and K+ channel inhibition was impaired in STZ-induced diabetic animals immediately upon induction of diabetes; this impairment diminished with diabetes duration; and (3) when diabetic sensory neuropathy became manifest, both NOS activity and SIN-1-induced vasodilatation during NOS, COX and K+ channel inhibition were at age-matched control levels, indicating that neither NO synthesis nor the responsiveness of the NO-cGMP pathway was impaired at this stage of diabetes. In the following discussion, methodological considerations and the bearings of the above findings on NO-dependent, and non-NO-, non-prostanoid-dependent vasodilatation will be discussed.
Methodological considerations
STZ-induced diabetes can be accompanied by decreased, increased or unaltered systemic blood pressure (Brands & Hopkins, 1996; Angulo et al. 1998; Obrosova et al. 2000), while increased peripheral vascular resistance is a consistent finding (Angulo et al. 1998; Brands et al. 2000). Increased peripheral resistance is found immediately upon onset of diabetic hyperglycaemia (Brands et al. 2000), whereas endothelial function does not become impaired until ≥ 2 weeks after induction of diabetes (Pieper, 1999; Coppey et al. 2000). These studies indicate that endothelial dysfunction is present independently of the variable arterial blood pressure changes and increased peripheral resistance that characterize STZ-induced diabetes. However, differing blood pressure levels can influence the magnitude of the vasodilatory response to acetylcholine, since blood flow in the sciatic nerve is proportional to arterial blood pressure (Low & Tuck, 1984). Therefore, NBF responses were calculated as conductances to eliminate the potentiating effect of increased arterial blood pressure on blood flow.
Impaired acetylcholine-induced vasodilatation in the microcirculation could be due to reduced release or effect of NO or EDHF. In the present study, NOS activity and in vivo NBF responses to the NO donor SIN-1 were taken to reflect NO release and NO effect, respectively. To eliminate interactions between NO and other endothelium-dependent relaxing factors (EDRFs) (Bauersachs et al. 1996; Salvemini, 1997; Nishikawa et al. 2000), NBF responses to SIN-1 were determined in the presence of NOS, COX and K+ channel inhibition.
Biochemical studies suggest that SIN-1 may function as a peroxynitrite donor rather than a NO donor (Feelisch et al. 1989; Beckman & Koppenol, 1996). However, in arterial ring preparations using bovine intramammary arteries preconstricted with the thromboxane mimetic, U 46619, inhibition of adenylate and guanylate cyclases has diametrically opposing effects on the vasodilatation induced by SIN-1 compared to that induced by peroxynitrite (Trakranrungsie & Will, 2001). In particular, SIN-1-induced vasodilatation is entirely dependent upon activation of soluble guanylate cyclase (sGC; Plane et al. 1998), while inhibition of sGC does not affect vasodilatation induced by peroxynitrite (Trakranrungsie & Will, 2001). Thus, in biovascular preparations, SIN-1-induced vasodilatation is not mediated by peroxynitrite but by the NO-cGMP pathway.
NO-dependent vasodilatation
In a recent study using nondiabetic rats, maximal relaxation in response to NO donors using endothelium-denuded aortic rings was unaltered with age (Matz et al. 2000). Conversely, in the mesenteric arterial bed in which endothelial function had been removed by air bubble perfusion, the maximal response to sodium nitroprusside was significantly reduced at 12 vs. 2 months of age (Atkinson et al. 1994). Consistent with this microcirculatory study, we found a clear age-dependent reduction in SIN-1-induced vasodilatation during inhibition of known EDRFs in sciatic nerve microcirculation of control rats. As inhibition of NOS, COX and K+ channels leaves only the NO-cGMP pathway distal to NOS as a viable mediator of SIN-1-induced vasodilatation, these results indicate that in healthy animals, the response of the NO-cGMP pathway to NO decreases with maturation in sciatic nerve microcirculation.
In experimentally induced diabetic animals, unimpaired responses to NO and NO donors are found in conduit as well as resistance arteries compared to age-matched controls (for reviews, see Pieper, 1998; De Vriese et al. 2000b). Accordingly, in the present study, SIN-1-induced vasodilatation in the presence of Ringer solution did not differ between STZ-induced diabetic rats and their age-matched controls. In contrast, inhibition of NOS, COX and K+ channels resulted in impaired SIN-1 responses in the diabetic rats. The difference between STZ-induced diabetic and age-matched control responses to SIN-1 was most obvious after 1–2 weeks and was not evident after 18–20 weeks of diabetes. Thus, in sciatic nerve microcirculation, the responsiveness of the NO-cGMP pathway was impaired in early STZ-induced diabetes, but this impairment diminished gradually, disappearing with longer durations of diabetes.
Both the gradual age-dependent decline and the immediate diabetic impairment of SIN-1-induced vasodilatation in the presence of l-NNA + indomethacin + TEA could be related to superoxide anion production, NO quenching and peroxynitrite formation. Significant basal superoxide anion production has been found in aortas of nondiabetic rats 16–24 weeks of age (van der Loo et al. 2000), while increased levels of superoxide anion have been found in feed arterioles to the sciatic nerve after only 2 weeks of diabetes (Coppey et al. 2000). The reaction between superoxide anion and NO decreases NO bioavailability and forms peroxynitrite, which reduces the NO effect yet further by inhibiting the activity of NO's target molecule, soluble guanylate cyclase (Weber et al. 2001; Laber et al. 2002).
Basal NO synthesis
In nondiabetic rat aorta, basal endothelial NOS (eNOS) activity and eNOS mRNA decrease with age (Barton et al. 1997; Cernadas et al. 1998; Chou et al. 1998). Accordingly, we found an age-dependent reduction in basal constitutive, i.e. Ca2+-dependent, NOS activity that was significant in the 18–20 week group. This reduction in basal NOS activity occurred concurrently with the age-dependent decline of NO-cGMP pathway responsiveness, indicating that the importance of nitric oxide in maintaining baseline blood flow in the sciatic nerve microcirculation decreases with maturation.
After 2 months of STZ-induced diabetes, eNOS mRNA levels were increased in rat dorsal root ganglion (Zochodne et al. 2000). Constitutive NOS activity and mRNA were found to be elevated in rat heart muscle 4–6 weeks after induction of diabetes with STZ and to decrease again after diabetes duration of 20 weeks or more (Stockklauser-Farber et al. 2000). Consistent with these results, basal constitutive NOS activity in the sciatic nerve of 8–10 week STZ-induced diabetics increased almost 2-fold and was significantly greater than that of age-matched controls, returning to age-matched control levels again at 18–20 weeks. This increase in NOS activity occurred chronologically after the early diabetic inhibition of the NO-cGMP pathway, perhaps as a compensatory mechanism to maintain NO-dependent vasodilatation.
Terminal ACh-induced vasodilatation in age-matched controls
In ageing animals, decreased acetylcholine-induced vasodilatation is first found after 24–28 weeks of age in the aorta (Ibarra et al. 1995; Chen et al. 1997) and after 70–100 weeks in mesenteric arteries (Fujii et al. 1993; Matz et al. 2000). In good agreement with these studies, terminal acetylcholine-induced vasodilatation in the presence of Ringer solution was maintained in control rats throughout 25 weeks of age in the present study. However, as discussed above, both NO synthesis and NO-cGMP pathway responsiveness decreased in an age-dependent manner in these animals. Since terminal ACh responses in nondiabetic sciatic nerve microcirculation are mediated by NO and a vasodilatory prostanoid (Thomsen et al. 2000), our findings suggest that the importance of vasodilatory prostanoids in acetylcholine-induced vasodilatation increases as NO bioactivity diminishes with maturation. Similar findings have been reported in the microcirculation of rat skeletal muscle (Linderman & Boegehold, 1999).
ACh-induced, non-NO-, non-prostanoid-dependent vasodilatation
Previously, we have shown that ACh-induced, non-NO-, non-prostanoid-dependent vasodilatation in rat sciatic nerve microcirculation is transient and dependent upon K+ channel activity, as evidenced by partial inhibition by 10 mm TEA (Thomsen et al. 2000). These findings were confirmed by the present study, in which ACh-induced vasodilatation in the presence of l-NNA + indomethacin was also found to be transient and sensitive to TEA in both control and STZ-induced diabetic animals. The initial response to acetylcholine in the presence of l-NNA + indomethacin was found to decrease with age in control animals. This finding agrees with a recent study in ageing rats, in which ACh-induced hyperpolarization and vasodilatation during NOS and COX inhibition decreased in an age-dependent manner in mesenteric arteries (Goto et al. 2000).
In STZ-induced diabetes, impaired ACh-induced vasodilatation during NOS and COX inhibition has been found in the renal microcirculation in vivo and in mesenteric arteries in vitro (Fukao et al. 1997; De Vriese et al. 2000a). Conversely, in the present study, initial ACh-induced vasodilatation in the presence of l-NNA + indomethacin did not decrease, while the duration of this response was prolonged in the diabetic nerve microcirculation. Thus, we found that while non-NO-, non-prostanoid-dependent vasodilatation in rat sciatic nerve decreased with maturation in controls, it increased during STZ-induced diabetes. Similar findings in rat skeletal muscle arterioles have been reported, where vasodilatory responses to acetylcholine during NOS and COX inhibition were found to increase in magnitude with the duration of diabetes (Timar-Peregrin & Guy, 2001).
TEA at the concentration of 10 mm used in the present study inhibits K+ channels nonspecifically. The effect of TEA on basal microvascular conductance and non-NO-, non-prostanoid-dependent vasodilatation was greater in STZ-induced diabetics. TEA had a greater effect on basal microvascular conductance and inhibited ACh-induced vasodilatation during NOS and COX inhibition to a greater extent in STZ-induced diabetics than in age-matched controls. Thus, the activity of K+ channels in basal and agonist-stimulated vascular tone in the microcirculation of rat sciatic nerve was augmented in STZ-induced diabetes. Since the K+ channels involved were not determined, ACh-induced vasodilatation in the presence of l-NNA + indomethacin in this study may not only reflect EDHF activity (Edwards & Weston, 2001). In this context, it is interesting to note that 10 mm TEA did not abolish ACh-induced non-NO-, non-prostanoid vasodilatory response entirely, especially in the younger control animals, suggesting that non-NO-, non-prostanoid-dependent vasodilatation is not dependent upon K+ channel activity alone.
Interestingly, terminal ACh responses in the presence of Ringer solution were not impaired in STZ-induced diabetic animals of this study, in contrast to a previous study of sciatic nerve microcirculation employing a similar experimental set-up (Maxfield et al. 1997). However, in this earlier study, blood vessels of the sciatic nerve were preconstricted by suffusing the nerve with noradrenaline. Since α-adrenergic stimulation has been shown to inhibit K+ channel hyperpolarization and relaxation in mesenteric arterial rings (Dora & Garland, 2001), suffusion with noradrenaline may have suppressed K+ channel activity, resulting in the reduced NBF responses to acetylcholine found in STZ-induced diabetic animals of the earlier study.
Experimental diabetic neuropathy, NCI and NO-dependent vasodilatation
Both motor and sensory nerve conduction velocities and action potential amplitude increased with maturation in age-matched control and STZ-induced diabetic animals, as is reported in other studies (Zochodne & Ho, 1994). After 18–20 weeks of diabetes, sensory nerve conduction velocity and action potential amplitude displayed deficits of 21 and 25 %, respectively. Endothelial dysfunction in isolated feed arteries to the sciatic nerve has been shown to precede the development of diabetic motor neuropathy in early experimental diabetes (Coppey et al. 2000). The present study has demonstrated that while the responsiveness of the NO-cGMP pathway was impaired in early STZ-induced diabetes, this impairment was transient and had dissipated after 18–20 weeks of diabetes. Basal NOS activity was never diminished in STZ-induced diabetics compared to age-matched controls. These findings suggest that NO bioactivity was not impaired at the time diabetic sensory neuropathy became manifest. Nor did variables reflecting NO bioactivity, i.e. SIN-1-induced vasodilatation with and without inhibitors nor NOS activity, relate to the development of experimental diabetic neuropathy. However, non-NO-, non-prostanoid-dependent vasodilatation was prolonged after 18–20 weeks of diabetes and was related to the maturation of nerve function in both age-matched controls and STZ-induced diabetics. The above findings imply that neither NO-dependent nor non-NO-, non-prostanoid-dependent vasodilatation displayed deficits that related to the development of experimental diabetic sensory neuropathy.
Conclusion
The present study demonstated augmented K+ channel activity in STZ-induced diabetes during both basal regulation and agonist-induced stimulation of sciatic nerve microcirculation. This study also demonstrated that NO-cGMP pathway responsiveness, basal NOS activity and initial ACh-induced vasodilatation during NOS and COX inhibition decreased with maturation in nondiabetic controls. In contrast, the responsiveness of the NO-cGMP pathway in STZ-induced diabetic rats was immediately impaired after induction of diabetes. This impairment diminished with the duration of STZ-induced diabetes and disappeared after 18–20 weeks. Two measures, perhaps compensatory, came into play: (1) a 2-fold increase in NOS activity at 8–10 weeks of diabetes, which, however, returned to age-matched control levels at 18–20 weeks; and (2) prolonged duration of non-NO-, non-prostnoid-dependent vasodilatation after 8–10 weeks of STZ-induced diabetes. While non-NO-, non-prostnoid-dependent vasodilatation was related to the maturation of nerve function in both STZ-induced diabetics and age-matched controls, indices of NO bioactivity did not relate to either normal electrophysiological development or to the development of STZ-induced diabetic neuropathy. In conclusion, these findings demonstrate that the alterations induced by STZ-induced diabetes in NO bioactivity and non-NO-, non-prostanoid-dependent vasodilatation in the microcirculation of rat sciatic nerve do not contribute to the development of experimental diabetic sensory neuropathy.
Acknowledgments
The authors are indebted to Drs Christian Lehn Brand and Jeppe Sturis of NOVO-Nordisk for their generous collaboration in the HbA1C measurements, and to Marianne Boysen and Susanne Gronemann for their quick and expert technical assistance in performing them. We also wish to thank Dr Peter Dalgaard, Department of Biostatistics, University of Copenhagen for his willing and proficient help in statistical matters, and Lillian Grøndahl and Jens Romlund for their cheerful and absolutely indispensable assistance in the laboratory. This study was supported by the Danish Medical Research Council, NeuroScience PharmaBiotech, The Carlsberg Foundation, Brødrene Hartmann Foundation and the NOVO-Nordisk Foundation.
References
- Angulo J, Rodriguez-Manas L, Peiro C, Neira M, Marin J, Sanchez-Ferrer CF. Impairment of nitric oxide-mediated relaxations in anaesthetized autoperfused streptozotocin-induced diabetic rats. Naunyn-Schmiedeberg's Archives of Pharmacology. 1998;358:529–537. doi: 10.1007/pl00005289. [DOI] [PubMed] [Google Scholar]
- Atkinson J, Tatchum-Talom R, Capdeville-Atkinson C. Reduction of endothelial function with age in the mesenteric arterial bed of the normotensive rat. British Journal of Pharmacology. 1994;111:1184–1188. doi: 10.1111/j.1476-5381.1994.tb14870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton M, Cosentino F, Brandes RP, Moreau P, Shaw S, Luscher TF. Anatomic heterogeneity of vascular aging: role of nitric oxide and endothelin. Hypertension. 1997;30:817–824. doi: 10.1161/01.hyp.30.4.817. [DOI] [PubMed] [Google Scholar]
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. American Journal of Physiology. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Brands MW, Fitzgerald SM, Hewitt WH, Hailman AE. Decreased cardiac output at the onset of diabetes: renal mechanisms and peripheral vasoconstriction. American Journal of Physiology - Endocrinology and Metabolism. 2000;278:E917–924. doi: 10.1152/ajpendo.2000.278.5.E917. [DOI] [PubMed] [Google Scholar]
- Brands MW, Hopkins TE. Poor glycemic control induces hypertension in diabetes mellitus. Hypertension. 1996;27:735–739. doi: 10.1161/01.hyp.27.3.735. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA, Jack AM, Basso MD, Hohman TC. Protein kinase C effects on nerve function, perfusion, Na(+), K(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia. 1999;42:1120–1130. doi: 10.1007/s001250051280. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Dines KC, Cotter MA. The potential contribution of endothelin-1 to neurovascular abnormalities in streptozotocin-diabetic rats. Diabetologia. 1994;37:1209–1215. doi: 10.1007/BF00399794. [DOI] [PubMed] [Google Scholar]
- Cernadas MR, Sanchez DM, Garcia-Duran M, Gonzalez-Fernandez F, Millas I, Monton M, Rodrigo J, Rico L, Fernandez P, de Frutos T, Rodriguez-Feo JA, Guerra J, Caramelo C, Casado S, Lopez F. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circulation Research. 1998;83:279–286. doi: 10.1161/01.res.83.3.279. [DOI] [PubMed] [Google Scholar]
- Chen H, Chen CC, Jen CJ. Effects of age and hypertension on endothelium-dependent vasodilating responses. Chinese Journal of Physiology. 1997;40:157–164. [PubMed] [Google Scholar]
- Chou TC, Yen MH, Li CY, Ding YA. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension. 1998;31:643–648. doi: 10.1161/01.hyp.31.2.643. [DOI] [PubMed] [Google Scholar]
- Coppey LJ, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Slowing of motor nerve conduction velocity in streptozotocin-induced diabetic rats is preceded by impaired vasodilation in arterioles that overlie the sciatic nerve. International Journal of Experimental Diabetes Research. 2000;1:131–143. doi: 10.1155/EDR.2000.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Salvemini D, Yorek MA. Effect of M40403 treatment of diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. British Journal of Pharmacology. 2001a;134:21–29. doi: 10.1038/sj.bjp.0704216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001b;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Luscher TF. Endothelial function in coronary artery disease. Cardiologia. 1997;42:1221–1227. [PubMed] [Google Scholar]
- De Vriese AS, Van de Vorde J, Blom HJ, Vanhoutte PM, Verbeke M, Lameire NH. The impaired renal vasodilator response attributed to endothelium-derived hyperpolarizing factor in streptozotocin-induced diabetic rats is restored by 5-methyltetrahydrofolate. Diabetologia. 2000a;43:1116–1125. doi: 10.1007/s001250051502. [DOI] [PubMed] [Google Scholar]
- De Vriese AS, Verbeuren TJ, Van de Vorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. British Journal of Pharmacology. 2000b;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA, Garland CJ. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. American Journal of Physiology - Heart and Circulatory Physiology. 2001;280:H2424–2429. doi: 10.1152/ajpheart.2001.280.6.H2424. [DOI] [PubMed] [Google Scholar]
- Edwards G, Weston AH. EDHF - are there gaps in the pathway? Journal of Physiology. 2001;531:299. doi: 10.1111/j.1469-7793.2001.0299i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Ohmori S, Tominaga M, Abe I, Takata Y, Ohya Y, Kobayashi K, Fujishima M. Age-related changes in endothelium-dependent hyperpolarization in the rat mesenteric artery. American Journal of Physiology. 1993;265:H509–516. doi: 10.1152/ajpheart.1993.265.2.H509. [DOI] [PubMed] [Google Scholar]
- Fukao M, Hattori Y, Kanno M, Sakuma I, Kitabatake A. Alterations in endothelium-dependent hyperpolarization and relaxation in mesenteric arteries from streptozotocin-induced diabetic rats. British Journal of Pharmacology. 1997;121:1383–1391. doi: 10.1038/sj.bjp.0701258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends in Pharmacological Sciences. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- Goto K, Fujii K, Onaka U, Abe I, Fujishima M. Angiotensin-converting enzyme inhibitor prevents age-related endothelial dysfunction. Hypertension. 2000;36:581–587. doi: 10.1161/01.hyp.36.4.581. [DOI] [PubMed] [Google Scholar]
- Groves WE, Davis FC, Jr, Sells BH. Spectrophotometric determination of microgram quantities of protein without nucleic acid interference. Analytical Biochemistry. 1968;22:195–210. doi: 10.1016/0003-2697(68)90307-2. [DOI] [PubMed] [Google Scholar]
- Hattori Y, Kawasaki H, Abe K, Kanno M. Superoxide dismutase recovers altered endothelium-dependent relaxation in diabetic rat aorta. American Journal of Physiology. 1991;261:H1086–1094. doi: 10.1152/ajpheart.1991.261.4.H1086. [DOI] [PubMed] [Google Scholar]
- Hohman TC, Cotter MA, Cameron NE. ATP-sensitive K(+) channel effects on nerve function, Na(+), K(+) ATPase, and glutathione in diabetic rats. European Journal of Pharmacology. 2000;397:335–341. doi: 10.1016/s0014-2999(00)00227-2. [DOI] [PubMed] [Google Scholar]
- Hotta N, Koh N, Sakakibara F, Nakamura J, Hamada Y, Wakao T, Hara T, Mori K, Naruse K, Nakashima W, Fukasawa H, Kakuta H. Prevention of abnormalities in motor nerve conduction and nerve blood-flow by a prostacyclin analog, beraprost sodium, in streptozotocin-induced diabetic rats. Prostaglandins. 1995;49:339–349. doi: 10.1016/0090-6980(95)00066-j. [DOI] [PubMed] [Google Scholar]
- Ibarra M, Meneses A, Ransanz V, Castillo C, Hong E. Changes in endothelium-dependent vascular responses associated with spontaneous hypertension and age in rats. Archives of Medical Research. 1995;26:S177–183. Spec. No. [PubMed] [Google Scholar]
- Karasu C. Time course of changes in endothelium-dependent and -independent relaxation of chronically diabetic aorta: role of reactive oxygen species. European Journal of Pharmacology. 2000;392:163–173. doi: 10.1016/s0014-2999(00)00140-0. [DOI] [PubMed] [Google Scholar]
- Kihara M, Mitsui MK, Mitsui Y, Okuda K, Nakasaka Y, Takahashi M, Schmelzer JD. Altered vasoreactivity to angiotensin II in experimental diabetic neuropathy: role of nitric oxide. Muscle and Nerve. 1999;22:920–925. doi: 10.1002/(sici)1097-4598(199907)22:7<920::aid-mus16>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Laber U, Kober T, Schmitz V, Schrammel A, Meyer W, Mayer B, Weber M, Kojda G. Effect of hypercholesterolemia on expression and function of vascular soluble guanylyl cyclase. Circulation. 2002;105:855–860. doi: 10.1161/hc0702.103975. [DOI] [PubMed] [Google Scholar]
- Linderman JR, Boegehold MA. Growth-related changes in the influence of nitric oxide on arteriolar tone. American Journal of Physiology. 1999;277:H1570–1578. doi: 10.1152/ajpheart.1999.277.4.H1570. [DOI] [PubMed] [Google Scholar]
- Low PA, Tuck RR. Effects of changes of blood pressure, respiratory acidosis and hypoxia on blood flow in the sciatic nerve of the rat. Journal of Physiology. 1984;347:513–524. doi: 10.1113/jphysiol.1984.sp015079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low PA, Tuck RR, Dyck PJ, Schmelzer JD, Yao JK. Prevention of some electrophysiologic and biochemical abnormalities with oxygen supplementation in experimental diabetic neuropathy. Proceedings of the National Academy of Sciences of the USA. 1984;81:6894–6898. doi: 10.1073/pnas.81.21.6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz RL, de Sotomayor MA, Schott C, Stoclet JC, Andriantsitohaina R. Vascular bed heterogeneity in age-related endothelial dysfunction with respect to NO and eicosanoids. British Journal of Pharmacology. 2000;131:303–311. doi: 10.1038/sj.bjp.0703568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield EK, Cameron NE, Cotter MA. Effects of diabetes on reactivity of sciatic vasa nervorum in rats. Journal of Diabetes and its Complications. 1997;11:47–55. doi: 10.1016/s1056-8727(96)00112-2. [DOI] [PubMed] [Google Scholar]
- Nagamatsu M, Nickander KK, Schmelzer JD, Raya A, Wittrock DA, Tritschler H, Low PA. Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care. 1995;18:1160–1167. doi: 10.2337/diacare.18.8.1160. [DOI] [PubMed] [Google Scholar]
- Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, Naruse K, Kasuya Y, Mizubayashi R, Miwa K, Yasuda Y, Kamiya H, Ienaga K, Sakakibara F, Koh N, Hotta N. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48:2090–2095. doi: 10.2337/diabetes.48.10.2090. [DOI] [PubMed] [Google Scholar]
- Nakamura J, Koh N, Sakakibara F, Hamada Y, Hara T, Sasaki H, Chaya S, Komori T, Nakashima E, Naruse K, Kato K, Takeuchi N, Kasuya Y, Hotta N. Polyol pathway hyperactivity is closely related to carnitine deficiency in the pathogenesis of diabetic neuropathy of streptozotocin-diabetic rats. Journal of Pharmacology and Experimental Therapeutics. 1998;287:897–902. [PubMed] [Google Scholar]
- Nishikawa Y, Stepp DW, Chilian WM. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. American Journal of Physiology - Heart and Circulatory Physiology. 2000;279:H459–465. doi: 10.1152/ajpheart.2000.279.2.H459. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Van Huysen C, Fathallah L, Cao X, Stevens MJ, Greene DA. Evaluation of alpha(1)-adrenoceptor antagonist on diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB Journal. 2000;14:1548–1558. doi: 10.1096/fj.14.11.1548. [DOI] [PubMed] [Google Scholar]
- Pieper GM. Review of alterations in endothelial nitric oxide production in diabetes: protective role of arginine on endothelial dysfunction. Hypertension. 1998;31:1047–1060. doi: 10.1161/01.hyp.31.5.1047. [DOI] [PubMed] [Google Scholar]
- Pieper GM. Enhanced, unaltered and impaired nitric oxide-mediated endothelium-dependent relaxation in experimental diabetes mellitus: importance of disease duration. Diabetologia. 1999;42:204–213. doi: 10.1007/s001250051140. [DOI] [PubMed] [Google Scholar]
- Plane F, Wiley KE, Jeremy JY, Cohen RA, Garland CJ. Evidence that different mechanisms underlie smooth muscle relaxation to nitric oxide and nitric oxide donors in the rabbit isolated carotid artery. British Journal of Pharmacology. 1998;123:1351–1358. doi: 10.1038/sj.bjp.0701746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop-Busui R, Sullivan KA, Van Huysen C, Bayer L, Cao X, Towns R, Stevens MJ. Depletion of taurine in experimental diabetic neuropathy: implications for nerve metabolic, vascular, and functional deficits. Experimental Neurology. 2001;168:259–272. doi: 10.1006/exnr.2000.7591. [DOI] [PubMed] [Google Scholar]
- Salvemini D. Regulation of cyclooxygenase enzymes by nitric oxide. Cellular and Molecular Life Sciences. 1997;53:576–582. doi: 10.1007/s000180050074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockklauser-Farber K, Ballhausen T, Laufer A, Rosen P. Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochimica et Biophysica Acta. 2000;1535:10–20. doi: 10.1016/s0925-4439(00)00078-8. [DOI] [PubMed] [Google Scholar]
- Terata K, Coppey LJ, Davidson EP, Dunlap JA, Gutterman DD, Yorek MA. Acetylcholine-induced arteriolar dilation is reduced in streptozotocin-induced diabetic rats with motor nerve dysfunction. British Journal of Pharmacology. 1999;128:837–843. doi: 10.1038/sj.bjp.0702856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen K, Rubin I, Lauritzen M. In vivo mechanisms of acetylcholine-induced vasodilation in rat sciatic nerve. American Journal of Physiology - Heart and Circulatory Physiology. 2000;279:H1044–1054. doi: 10.1152/ajpheart.2000.279.3.H1044. [DOI] [PubMed] [Google Scholar]
- Timar-Peregrin A, Guy RG. Recovery of microvascular responses during streptozotocin-induced diabetes. European Journal of Pharmacology. 2001;414:63–70. doi: 10.1016/s0014-2999(01)00758-0. [DOI] [PubMed] [Google Scholar]
- Trakranrungsie N, Will JA. Comparative vasodilation of peroxynitrite and 3-morpholinosydnonimine. Life Sciences. 2001;69:2349–2359. doi: 10.1016/s0024-3205(01)01320-0. [DOI] [PubMed] [Google Scholar]
- Tuck RR, Schmelzer JD, Low PA. Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy. Brain. 1984;107:935–950. doi: 10.1093/brain/107.3.935. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Luscher TF. Enhanced peroxynitrite formation is associated with vascular aging. Journal of Experimental Medicine. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Lauer N, Mulsch A, Kojda G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radical Biology and Medicine. 2001;31:1360–1367. doi: 10.1016/s0891-5849(01)00706-7. [DOI] [PubMed] [Google Scholar]
- Woodman OL, Wongsawatkul O, Sobey CG. Contribution of nitric oxide, cyclic GMP and K+ channels to acetylcholine-induced dilatation of rat conduit and resistance arteries. Clinical and Experimental Pharmacology and Physiology. 2000;27:34–40. doi: 10.1046/j.1440-1681.2000.03199.x. [DOI] [PubMed] [Google Scholar]
- Wright RA, Nukada H. Vascular and metabolic factors in the pathogenesis of experimental diabetic neuropathy in mature rats. Brain. 1994;117:1395–1407. doi: 10.1093/brain/117.6.1395. [DOI] [PubMed] [Google Scholar]
- Zochodne DW, Ho LT. The influence of sulindac on experimental streptozotocin-induced diabetic neuropathy. Canadian Journal of Neurological Sciences. 1994;21:194–202. doi: 10.1017/s0317167100041160. [DOI] [PubMed] [Google Scholar]
- Zochodne DW, Verge VM, Cheng C, Hoke A, Jolley C, Thomsen K, Rubin I, Lauritzen M. Nitric oxide synthase activity and expression in experimental diabetic neuropathy. Journal of Neuropathology and Experimental Neurology. 2000;59:798–807. doi: 10.1093/jnen/59.9.798. [DOI] [PubMed] [Google Scholar]