

Abstract
Axons in WldS mutant mice are protected from Wallerian degeneration by overexpression of a chimeric Ube4b/Nmnat (Wld) gene. Expression of Wld protein was independent of age in these mice. However we identified two distinct neuromuscular synaptic responses to axotomy. In young adult Wlds mice, axotomy induced progressive, asynchronous synapse withdrawal from motor endplates, strongly resembling neonatal synapse elimination. Thus, five days after axotomy, 50–90 % of endplates were still partially or fully occupied and expressed endplate potentials (EPPs). By 10 days, fewer than 20 % of endplates still showed evidence of synaptic activity. Recordings from partially occupied junctions indicated a progressive decrease in quantal content in inverse proportion to endplate occupancy. In Wlds mice aged > 7 months, axons were still protected from axotomy but synapses degenerated rapidly, in wild-type fashion: within three days less than 5 % of endplates contained vestiges of nerve terminals. The axotomy-induced synaptic withdrawal phenotype decayed with a time constant of ∼30 days. Regenerated synapses in mature Wlds mice recapitulated the juvenile phenotype. Within 4–6 days of axotomy 30–50 % of regenerated nerve terminals still occupied motor endplates. Age-dependent synapse withdrawal was also seen in transgenic mice expressing the Wld gene. Co-expression of Wld protein and cyan fluorescent protein (CFP) in axons and neuromuscular synapses did not interfere with the protection from axotomy conferred by the Wld gene. Thus, Wld expression unmasks age-dependent, compartmentally organised programmes of synapse withdrawal and degeneration.
Following lesion of a peripheral nerve, distal axons and synaptic terminals normally degenerate via a cascade of molecular and cellular responses collectively termed Wallerian degeneration (WD). In rodents, neuromuscular transmission rapidly fails, the axonal and terminal cytoskeleton is disrupted, and fragmentation of nerve terminals is followed by phagocytosis in terminal Schwann cells. The axonal and nerve terminal debris is removed by Schwann cells and invading macrophages within 48 h (Birks et al. 1960; Miledi & Slater, 1970; Winlow & Usherwood, 1975). However, in the Wlds mutant mouse axotomy-induced degeneration is profoundly delayed (Lunn et al. 1989; Mack et al. 2001). Experiments utilising this mutant suggest that axon degeneration occurs by a mechanism distinct from those regulating cell body degeneration (Deckwerth & Johnson, 1994; Buckmaster et al. 1995). Independent analysis of synapse-specific proteases suggests that synapses also contain their own independent degenerative mechanisms (Mattson et al. 1998a, 1998b). Taken together, such findings militate towards a view that degeneration mechanisms are compartmentalised within neurones (Gillingwater & Ribchester, 2001).
By contrast, synapses are normally remodelled by a different mechanism in neonatal muscles or in reinnervated adult muscles. Competition between motor axons converging on the same motor endplate results in ‘synapse elimination’, a process which involves progressive withdrawal of synaptic boutons from polyneuronally innervated motor endplates, until those supplied by only one of the original axon collateral branches remain (Brown et al. 1976; Keller-Peck et al. 2001). A similar process occurs in reinnervated adult muscles after nerve injury and regeneration (see for example, Barry & Ribchester, 1995; Costanzo et al. 2000). The structural and physiological features of synaptic degeneration are normally quite different from those of synapses undergoing elimination, even in Wlds mice (Brown et al. 1976; Korneliussen & Jansen, 1976; Betz et al. 1979; Bixby, 1981; Colman et al. 1997; Parson et al. 1997; Ribchester, 2001; but see Rosenthal & Taraskevich, 1977).
Genetic analysis has shown that the Wlds mutation is inherited as a single autosomal dominant characteristic, by a gene located on the distal end of chromosome 4 (Perry et al. 1990b; Lyon et al. 1993). The Wlds mutation comprises an 85 kb tandem triplication (Coleman et al. 1998). This triplication contains the exons of three genes; Ube4b (the mammalian homologue of ubiquitin fusion degradation protein 2 (Ufd2)), nicotinamide mononucleotide adenylyl transferase (Nmnat) and a novel member of the cellular retinoid-binding protein family (Rbp7; see Conforti et al. 2000). The sequence for the N-terminal 70 amino acids of Ube4b and the complete sequence of Nmnat span the proximal and distal boundaries of the repeat unit, forming a chimeric gene with an open reading frame coding for a 43 kDa fusion protein. Transgenic mice expressing the Ube4b/Nmnat chimeric gene product (Wld) also show the Wlds phenotype, showing that this gene is both necessary and sufficient to produce slow axonal and synaptic degeneration (Mack et al. 2001).
Remarkably, however, the protection conferred by the mutant gene affects axons and synapses differently. Whereas distal axons persist after axotomy for up to three weeks in Wlds mice, the motor nerve terminals persist for only 4–10 days (Perry et al. 1990a; Brown et al. 1992; Tsao et al. 1994; Ribchester et al. 1995; Gillingwater & Ribchester, 2001). Moreover, there are conflicting reports concerning the neuroprotective effectiveness of the Wlds gene as these mice age. Some studies have suggested that the Wlds phenotype is lost, so that by six months of age, mutant mice exhibit almost normal rates of axon degeneration (Perry et al. 1992; Ribchester et al. 1995). By contrast, Crawford et al. (1995) reported that both axons and synapses were equally well-protected from axotomy-induced degeneration in Wlds mice of all ages up to 16 months. Thus, an initial objective of the present study was to resolve the discrepancy between the studies of Ribchester et al. (1995) and Crawford et al. (1995) using a combined genetic, biochemical, morphological and electrophysiological approach.
Our analysis shows that Wld gene expression is wholly independent of age, whether driven by the endogenous promoter or by a heterologous β-actin promoter. We also demonstrate that age has no effect on the biochemical or morphological preservation of Wld axons, but that preservation of axotomised synaptic terminals is clearly transformed with age in both character and kinetics. In young Wlds mice (< 4 months), or their transgenic equivalents, synaptic terminals degenerate asynchronously, apparently withdrawing boutons progressively from motor endplates. By contrast, in mice older than ∼7 months, most distal axons are preserved but synaptic terminals undergo a more rapid, synchronous degeneration, similar to the axotomy reaction in wild-type mice. Furthermore, the maturity of the synapse rather than age of the motor neurone appears to regulate this age-dependent phenotype.
Taken together, our observations suggest that newly-established, mononeuronally innervated junctions retain the molecular mechanisms required to execute retraction of supernumerary synapses, beyond the period when they are normally eliminated as part of post-natal development. Therefore, mice expressing the Wld protein will play a pivotal role in the understanding of cellular and molecular mechanisms of synapse elimination.
Preliminary data from this study were presented to meetings of the Physiological Society (Ribchester et al. 1999; Gillingwater et al. 2000; Ribchester et al. 2002).
METHODS
Mice
Natural mutant Wlds mice of ∼1–2 months were obtained from Harlan Olac Laboratories (Bicester, UK). Some of these mice were used within ∼1 week of arrival, whilst others were maintained within animal care facilities in Edinburgh until they reached the older ages (4, 7 and 12 months) required for the age-dependency experiments. The 4836 line of Wld transgenic mice expressing the Ube4b/Nmnat chimeric gene were generated in Cologne by pronuclear injection of a construct of the chimeric gene coupled to the β-actin promoter, as described previously (see Mack et al. 2001). Thy1-CFP mice were obtained from Jackson Laboratories (Bar Harbour, ME, USA) and crossbred with Wlds mice according to standard breeding techniques. CFP-expressing mice were identified from the fluorescence of axons in ear-punch biopsies. Wld homozygotes were confirmed post-mortem, by pulse-field gel electrophoresis of DNA isolated from their spleens (Mi et al. 2002).
Surgery
Mice were anaesthetized either by inhalation of halothane (2 % in 1:1 N2O/O2; Edinburgh) or via i.p. injection of Ketanest (100 mg kg−1) and Rompun (5 mg kg−1; Cologne). For experiments using flexor digitorum brevis (FDB) or lumbrical muscles, either the sciatic or tibial nerve was exposed and a 1–2 mm section was removed, denervating the majority of muscles in the hind foot. For experiments using transverses abdominus (TA), intercostal nerves were similarly exposed and lesioned. Mice were then kept in standard animal house conditions for 1–10 days before being killed by stunning and dislocation of cervical vertebrae. All surgical procedures were carried out in strict accordance with local ethical committee guidelines (Stadt Koön Veterinaäamt, Licence K13, 11/00; Cologne) and with the licensed authority of the UK Government Home Office (PPL 60/2434; Edinburgh).
Electrophysiology
Intracellular recordings were made between 1 and 10 days after surgery. Isolated FDB nerve/muscle preparations from WldS mice were pinned out in a Sylgard-lined bath and perfused with normal mammalian physiological saline (mm: NaCl 120; KCl 5; CaCl2 2; MgCl2 1; NaH2PO4 0.4; NaHCO3 23.8; d-glucose 5.6) bubbled to equilibrium with a 5 % CO2/95 % O2 mixture. Transgenic mouse FDB muscles were isolated in Cologne, bathed in Hepes-buffered saline and taken by courier to Edinburgh, where they were then transferred to bicarbonate-saline (above) for electrophysiological recording the same day, as described previously (Mack et al. 2001). Muscle contractions were reduced/eliminated by bathing the muscles in 2.5 μM μ-conotoxin GIIIB (Scientific Marketing Associates, Barnet, UK) for 30–45 min. Thirty fibres were impaled at random, per muscle, using a glass electrode filled with 5 m sodium acetate solution. Activity was recorded using an Axoclamp-2B amplifier (Axon Instruments, Inc., Union City, CA, USA) and stored and analysed on a PC using WinWCP v3.0.8 software (developed and distributed by Dr John Dempster, Strathclyde University, UK).
Electron microscopy
For ultrastructural studies, FDB muscles were fixed in ice-cold 0.1 m phosphate buffer containing 4 % paraformaldehyde/2.5 % glutaraldehyde for 4 h. Preparations were then washed in 0.1 m phosphate buffer before postfixing in a 1 % osmium tetroxide solution for 45 min and dehydration through an ascending series of ethanol solutions. Dehydrated muscles were embedded in Durcupan resin before sectioning at 75–90 nm and collection on formvar-coated grids (Agar Scientific, Stansted, UK). Grids were then stained with uranyl acetate and lead citrate in an LKB ‘Ultrostainer’ before viewing in a Philips CM12 TEM. Electron microscope (EM) negatives taken between 2000× and 60000× were scanned at 600 dpi using a Linoscan 1200 (Heidelberg, Germany) equipped with a transparency adaptor, before importing into Adobe Photoshop for analysis and presentation.
Axon counts
Lesioned tibial nerves from 2-month- and 7-month-old Wlds mice (n = 2 in each case) were fixed in ice-cold 0.1 m phosphate buffer containing 4 % paraformaldehyde/2.5 % glutaraldehyde for 4 h. The first 2 mm of the proximal and distal nerve stumps were discarded, and the remaining tissue was transferred into resin as described above. 1 μm cross-sections were cut and collected on glass slides before staining with toluidine blue (1 % in distilled H2O; Sigma). Sections were examined using a 40× water immersion objective (Zeiss) attached to a standard light microscope. Images were captured using Openlab (Improvision Software, Coventry, UK) before being transferred to Adobe Photoshop for analysis. The total number of myelinated axon profiles was recorded in six cross-sections from both the proximal and distal nerve stumps and the counts for each stump were averaged. To be included in the count, axons had to exhibit normal myelin sheaths and a uniform axoplasm.
Neuromuscular junction (NMJ) staining
FDB, lumbrical or TA preparations for immunocytochemistry were fixed in 0.1 m PBS containing 4 % paraformaldehyde for 30–40 min before labelling acetylcholine receptors by incubating for 20 min in α-bungarotoxin (BTX) conjugated to tetramethylrhodamine isothiocyanate (TRITC-α-bungarotoxin; 5 mg ml−1, Molecular Probes, Inc., Eugene, OR, USA). Muscles were blocked in 4 % bovine serum albumin (BSA) and 0.5 % Triton X in 0.1 m PBS for 30 min before incubation in primary antibodies directed against 165 kDa neurofilament proteins (2H3) and the synaptic vesicle protein SV2 (both 1:200 dilution; from the Developmental Studies Hybridoma Bank, IA, USA) overnight. After washing for 30 min in blocking solution (see above), muscles were incubated for 4 h in a 1:200 dilution of sheep anti-mouse antibody conjugated to the fluorescent label FITC (Diagnostics Scotland, Edinburgh, UK). Muscles were then whole-mounted in Vectashield (Burlingame, CA, USA) on slides for subsequent imaging.
TA nerve-muscle preparations for vital labelling and visualised recording were stained by repetitive stimulation (20 Hz, 10 V, 10 min) in the presence of 4 mm FM1-43 (Molecular Probes, Inc.) before washing for 15 min in oxygenated physiological saline.
Fluorescence imaging and analysis
Muscle preparations were imaged on either a standard fluorescence microscope (Micro Instruments M2B) or using a laser scanning confocal microscope (Biorad Radiance 2000, Hemel Hempstead, UK). Individual neuromuscular junction images were obtained using either a 40× (0.8 NA) or 60× (1.0 NA) water immersion objective. TRITC-α-BTX-labelled preparations were imaged using 543 nm excitation and 590 nm emission optics; FM1-43-stained preparations utilised a 400–440 nm excitation filter and 515 nm emission filter and FITC-labelled preparations utilised 488 nm excitation and 520 nm emission optics. For confocal microscopy, 488 nm and 543 nm laser lines were used for excitation and confocal Z-series were merged using Lasersharp (Biorad) software. All images were then assembled using Adobe Photoshop.
Western blotting
Wld protein expression was analysed by homogenising mouse brains in two volumes of 20 mm Hepes (pH 7.5), 0.2 m CaCl2, 0.2 m MgSO4, 1 ml (20 g tissue)−1 protease inhibitor cocktail (Sigma) and 1 mg ml−1 DNase (Sigma). Neurofilament preservation was assessed in distal sciatic nerve stumps homogenised in 20 volumes of this buffer. Proteins were separated using standard SDS-PAGE and semi-dry blotted onto nitrocellulose. Loading and transfer were checked using Ponceau S (Sigma) and Coomassie Blue. Overnight incubation in primary antibodies at 4 °C was followed by incubation in horseradish peroxidase-coupled secondary antibody (1 h at room temperature; goat anti-mouse 1:3000, goat anti-rabbit 1:5000; Dianova, Hamburg, Germany) and detection using enhanced chemiluminescence (Amersham Pharmacia, UK). Chimeric protein expression was quantified using affinity-purified N70 antibody and β-tubulin 2.1 (Sigma) control. Neurofilament protein degradation was analysed using phosphate-independent monoclonal N52 (1:2000; Sigma) against heavy neurofilament protein.
RESULTS
Age-independence of axon protection and Wld gene expression
We examined the levels of expression of the Wld gene in Wlds mice of different ages. Western blots of homogenised brain tissue in mice aged 2, 4, 7 and 12 months demonstrated that Wld protein expression did not diminish significantly with age (Fig. 1A). We also analysed the preservation of severed distal axons in 4 days-axotomised tibial nerve preparations from 2-month- and 7-month-old Wlds mice, based on Western blots and on counts of myelinated axon profiles in semi-thin cross-sections from proximal and distal nerve stumps (Fig. 1B and C). The data show there were no major qualitative differences in neurofilament preservation in the distal portions of lesioned tibial nerves and morphological analysis revealed virtually complete (93–100 %) retention of myelinated profiles in the distal stump. These data therefore indicate that both Wld gene expression and distal axon preservation are largely independent of age in Wlds mice.
Figure 1. Age-independent protection of axons and Wld gene expression in Wlds mice.
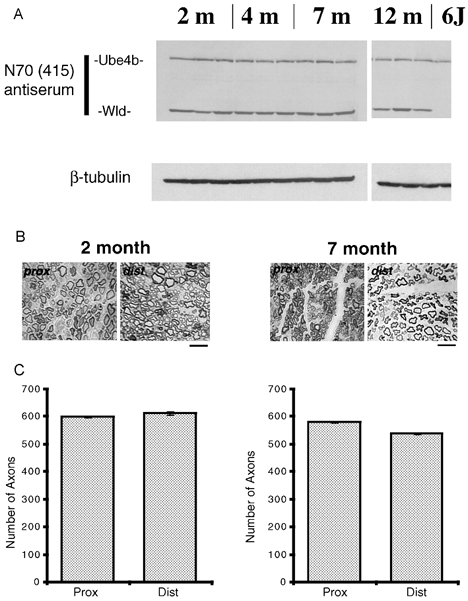
A, Western blot showing retained Ube4b, Wld and β-tubulin (loading control) expression in Wlds mice of different ages compared to a 6J mouse control. B, transverse sections through proximal and distal tibial nerve stumps, 4 days post axotomy in 2- and 7-month-old Wlds mice showing qualitative preservation of disconnected axons and no signs of axon loss or degeneration. Scale bars = 5 μm. C, quantitative analysis of distal axon preservation in 2- and 7-month-old Wlds mice tibial nerves 4 days post axotomy. There was no significant difference in the numbers of axon profiles between proximal and distal nerve stumps at either age.
Progressive loss of synaptic terminals in juvenile Wlds mice
Immunocytochemical staining of neurofilament (NF) and SV2 and TRITC-α-BTX labelling of axotomised NMJs in 2-month-old Wlds mice made at 3–7 days post-axotomy revealed prolonged retention of pre-terminal axons and motor nerve terminals (Fig. 2A). However, in addition to fully occupied endplates, we observed partially occupied and vacant motor endplates in all axotomised preparations from these young Wlds mice (Fig. 2B-D). Sometimes endplates were contacted by a single remaining synaptic bouton occupying less than 5 % of the endplate. These resembled ‘retraction bulbs’, previously identified in the final stages of synapse elimination from motor endplates (cf. Riley, 1977; Gorio et al. 1983; Balice-Gordon et al. 1993). Similar examples of fully occupied, partially occupied and vacant NMJs were detected in 2 month preparations examined ultrastructurally (Fig. 2E-G). Degenerating mitochondrial profiles were observed at < 3 % of terminals, but these preparations were otherwise free of classical degenerative markers: synaptic vesicle density and distribution appeared normal, terminal plasma membranes appeared intact and there was no evidence of phagocytosis by terminal Schwann cells. Two distinct features of many of the junctions were, firstly, accumulation of neurofilaments within the centre of synaptic boutons and secondly, instances of ‘giant’ synaptic vesicles (∼75–125 nm in diameter) were observed. Intracellular recordings demonstrated comparable retention of synaptic transmission over the same period, although evidence for a progressive decline in synaptic efficacy was detected at some junctions, where EPP amplitudes had a high coefficient of variation and ‘failures’, indicating a reduced quantal content (Fig. 2H-J). ‘Giant’ miniature endplate potentials (MEPPs) were occasionally observed in axotomised preparations (Fig. 2H).
Figure 2. Axotomized nerve terminals retract from endplates in young adult Wlds mice.
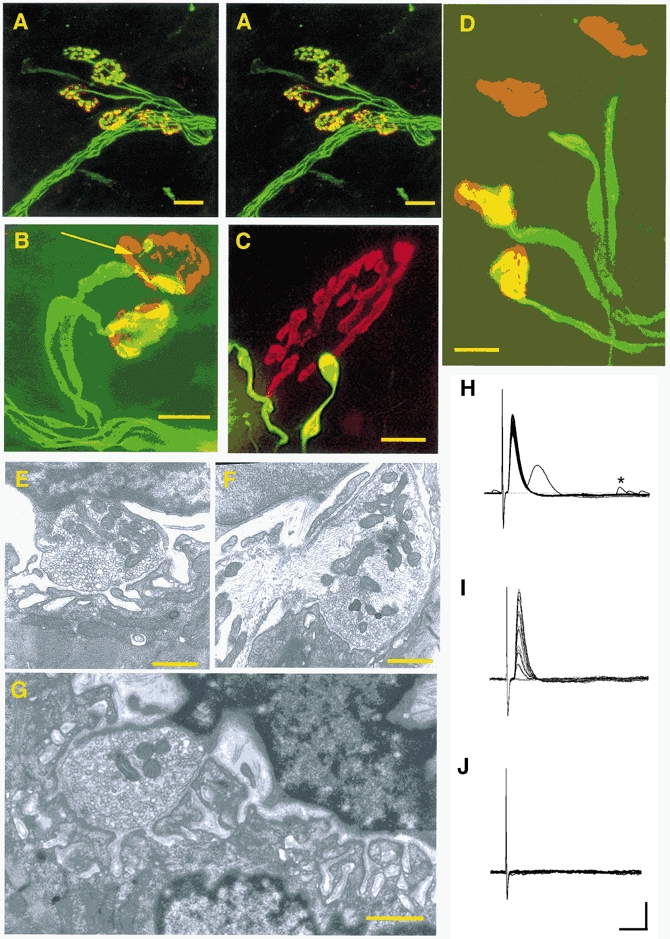
A, confocal stereo pair showing synapses protected from degeneration in an FDB muscle from a 2-month-old WldS mouse, 3 days post axotomy. Axons and motor nerve terminals were immunocytochemically labelled with NF and SV2 (FITC) and acetylcholine receptors were labelled with TRITC conjugated α-BTX. Scale bars = 20 μm. B, immunocytochemically labelled (NF, SV2, α-BTX) NMJs from a lumbrical muscle, 6 days post axotomy. Note the complete retention of the lower nerve terminal, but the partial occupancy of the upper endplate. The arrow indicates a very thin axon collateral terminating in a ‘retraction bulb’ swelling. Scale bar = 20 μm. C, immunocytochemically labelled (NF, SV2, α-BTX) NMJ from a TA muscle, 4 days post axotomy. The remaining axon collateral terminates in a ‘retraction bulb’ swelling, contacting < 5 % of the endplate. Scale bar = 10 μm. D, immunocytochemically labelled (NF, SV2, α-BTX) NMJs from a lumbrical muscle, 6 days post axotomy. The two endplates on the left are fully occupied whilst the two endplates on the right are vacant, with nearby axonal terminations. Scale bar = 20 μm. E, electron micrograph of a nerve terminal bouton from an FDB muscle, 3 days post axotomy. The nerve terminal is directly opposite the postsynaptic specialisations, is capped by a terminal Schwann cell, and has intact mitochondria, synaptic vesicles and membranes. Scale bar = 0.75 μm. F, electron micrograph of a nerve terminal bouton from an FDB muscle, 4 days post axotomy. The terminal shown has retained good synaptic ultrastructure, but neurofilaments are accumulated in the centre of the bouton. Scale bar = 0.75 μm. G, electron micrograph of a partially occupied 2 month Wlds NMJ, 3 days post axotomy. The remaining synaptic bouton neighbours a region of unoccupied postsynaptic specialisation which is covered by the nucleus of a terminal Schwann cell. Scale bar = 0.75 μm, H-J) Intracellular recordings from 2 month Wlds FDB muscle fibres at 5 days post axotomy. Examples of robust transmission (H), weak transmission (with varying EPP amplitude and ‘failures’, I) and loss of transmission (J) are shown. An example of a ‘giant’ MEPP is also shown (∼5 mV; denoted by *, H). Scale bars = 5 mV (vertical); 10 ms (horizontal).
The time course of withdrawal of Wlds nerve terminals in response to axotomy, as measured both morphologically and electrophysiologically, is shown in Fig. 3A and B. The functional analysis consistently gave lower estimates of the numbers of remaining synapses than those given by morphological measurements. However, both analyses showed that the time course of synapse loss appeared well fitted by sigmoidal functions. Thus, > 80 % of synapses were retained 3 days after axotomy, but by 5 days this level had dropped to ∼60 % of terminals, subsequently decreasing to ∼30–50 % by 7 days post axotomy. Figure 3C and D shows the incidence of morphological and functional correlates of nerve terminal withdrawal (partial occupancy of endplates; high coefficient of variation of EPP amplitudes and random failures in response to nerve stimulation). Levels of partial occupancy peaked at 5–7 days post axotomy, at ∼50 %, whilst the increase in the number of fibres showing failures followed a similar time course, but only reached a maximum level of ∼15 %. Withdrawal of synaptic boutons from endplates was asynchronous and independent of endplate size. Figure 4A shows the distribution of endplate occupancies plotted against the endplate area at 4, 6 and 8 days after axotomy in TA muscles. There was no correlation between endplate area and fractional occupancy at any time between 3 and 10 days. Thus, the onset of synapse withdrawal seemed to occur quite randomly, but to proceed at a constant rate once initiated.
Figure 3. Time course of synapse withdrawal in 2-month-old Wlds mice.
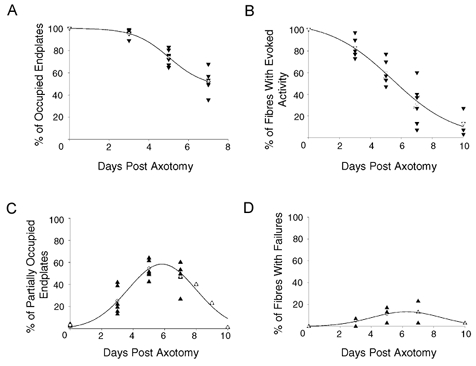
A, morphological data showing the time course of nerve terminal withdrawal in 2-month-old Wlds mice FDB muscles following axotomy. B, electrophysiological measurements of the time course of nerve terminal loss in 2-month-old Wlds mice FDB muscles following axotomy. C, morphological analysis of the incidence of endplate partial occupancy in 2-month-old Wlds mice FDB muscles following axotomy. D, electrophysiological analysis of the incidence of failures in response to nerve stimulation in 2-month-old Wlds mice FDB muscle fibres following axotomy. For all panels, ▴ and ▾ represent data points from individual muscles and ○ indicates the mean value calculated at each time point.
Figure 4. Effect of endplate size and occupancy on synaptic withdrawal.
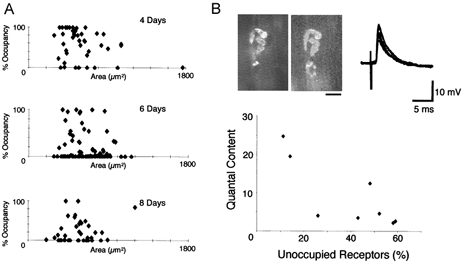
A, endplate size did not influence the withdrawal of Wlds synapses at 4, 6 and 8 days post axotomy. There was no significant correlation between the size of an endplate and the state of withdrawal of its corresponding nerve terminal. B, FM1-43 (left) and α-BTX (right) images, intracellular recordings and summary graph of electrophysiological recordings from visualised partially occupied NMJs on 2-month-old Wlds FDB muscles 4 days after axotomy. The NMJ shown has ∼50 % terminal occupancy of the endplate, yet still provides evidence of robust synaptic transmission. However, quantal analysis using the variance method, indicated a significantly reduced quantal content (m =∼5), compared to control muscles. The graph shows the relationship between quantal content and fractional vacancy from eight fibres, indicating that depression of transmitter release preceded structural withdrawal. Scale bar = 20 μm.
The discrepancies between morphological and physiological measurements could have been the result of impaired nerve conduction, or to deterioration in the physiological mechanisms coupling excitation to transmitter release. In a small sample of endplates we made direct recordings of synaptic physiology and morphology (Fig. 4B), using FM1-43 to label partially occupied neuromuscular junctions (Ribchester et al. 1994; Costanzo et al. 1999). Quantal analysis suggested that depression of transmitter release preceded structural withdrawal. Thus, endplates that were still more than 50 % occupied and had quantal contents at or below the lower bound of the normal range (Wood & Slater, 1997). Whether this decrease in quantal content occurred by a change in the quantal parameters n or p or both (cf. Kopp et al. 2000) remains open.
The most parsimonious explanation of the progressive appearance of partially occupied or vacant endplates, when taken together with the preservation of synaptic ultrastructure and the gradual decline in synaptic transmission, is that synaptic terminals in the young Wlds mice progressively withdrew from motor endplates following axotomy. Thus, the progression from full occupancy, through partial occupancy, to vacancy of the motor endplates (Fig. 2A-D); the absence of conventional signs of degeneration (Fig. 2E-G); and the pattern of strong and weak synaptic transmission (Fig. 2H-J), when taken together with the quantitative analysis, offer compelling similarities to stages of synapse elimination that occur at wild-type neuromuscular junctions during normal postnatal development. The difference here was that this progressive synaptic withdrawal was triggered in a juvenile (or young adult) mutant mouse, and the stimulus was axotomy, rather than competition between synaptic terminals (see Ribchester, 2001). Synapse elimination occurs with a normal time course in neonatal Wlds mice (Parson et al. 1997), and the time course of synapse withdrawal in response to axotomy in the juvenile/young adult Wlds mice is remarkably similar.
Rapid degeneration of Wlds synaptic terminals in mature mice
Ultrastructural, immunocytochemical and electrophysiological analyses of the responses to axotomy in older Wlds mice suggested that neuromuscular synapses reacted in a qualitatively different fashion. Ultrastructural examination revealed that some axotomised nerve terminals in 4 month preparations showed a curious morphology: they sometimes contained swollen and distorted mitochondria but intact synaptic vesicles. Interestingly, some of the boutons containing these mitochondrial abnormalities were adjacent to other boutons, from the same motor nerve terminal, that contained no signs of degeneration of organelles (Fig. 5A). In 7 month Wlds preparations, at 2 days post axotomy, most identifiable terminals showed classical degenerative signs including swollen and disrupted mitochondria, reduced synaptic vesicle densities, intra-terminal membrane whorls, fragmented terminal membranes and terminal Schwann cell phagocytosis (Fig. 5B and C). Thus, the appearance of 4 month terminals was intermediate between that of axotomised 2 month and 7 month terminals. These observations were corroborated by immunocytochemically stained preparations observed at the light microscope level. Very few instances of partially occupied endplates were seen in immunocytochemical preparations: most endplates were either fully occupied or vacant, and many of the occupied endplates had an abnormal, bloated appearance (see Fig. 6). Immunocytochemical analysis of the prevalence of innervated endplates with time after axotomy in mice aged 4–7 months is shown in Fig. 5D. In 4-month-old animals, the time course of synapse loss was well fitted by exponential curves (in contrast with 2-month-old animals, Fig. 3). Only ∼35 % of endplates were occupied 3 days after axotomy. In 7 month mice, fewer than 5 % remained by 3 days, and a similar rapid loss of synaptic terminals followed axotomy in Wlds mice aged 12 months. The number of endplates that were partially occupied 3–7 days after axotomy also declined exponentially with age (Fig. 5F), which further suggested that the synapses were removed synchronously rather than progressively in the older mice.
Figure 5. Degeneration of synaptic terminals in fully mature Wlds mice.
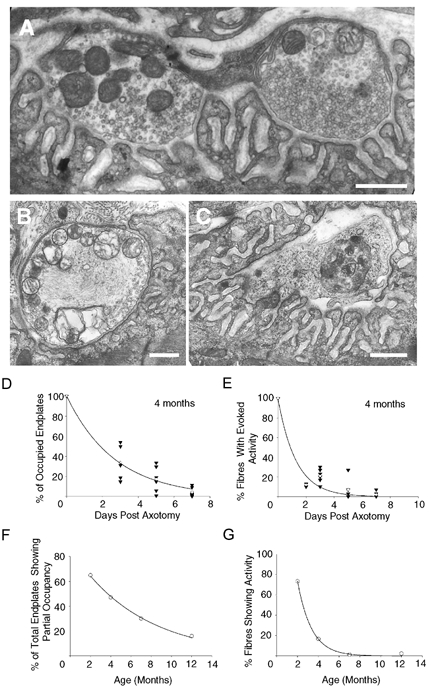
A, electron micrograph of two neighbouring nerve terminal boutons from a partially occupied endplate in a 4-month-old Wlds FDB muscle, 3 days post axotomy. The bouton on the right has retained terminal ultrastructure, but the bouton on the left contains disrupted mitochondria. Scale bar = 0.5 μm. B, electron micrograph of an individual nerve terminal bouton from a 7-month-old Wlds FDB muscle, 2 days post axotomy. Note the swollen and disrupted mitochondria, paucity of synaptic vesicles and membrane disruption. Scale bar = 0.5 μm. C, electron micrograph of a terminal Schwann cell phagocytosing a grossly fragmented nerve terminal in situ from a 7-month-old Wlds FDB muscle, 2 days post axotomy. Scale bar = 0.5 μm. D, immunocytochemical data, showing the incidence of retained terminals following axotomy in 4-month-old Wlds preparations, indicating an increase in the rate of terminal loss compared to 2 month preparations (see Fig. 3). E, graph plotted from electrophysiological data, showing the incidence of fibres exhibiting evoked activity following axotomy in 4 month Wlds preparations. The data support the morphological findings of an increase in the rate of terminal loss compared to 2 month preparations (see above). F, graph from immunocytochemical data showing a decrease in the incidence of partial occupancy with age in Wlds mice. G, graph showing the decline in the incidence of fibres showing synaptic activity with age. Each point shows the percentage of fibres with MEPPs or EPPs 3 days after axotomy in Wlds mice of different ages.
Figure 6. Synaptic protection in Wlds mice depends on synaptic maturity and not the age of the animal.
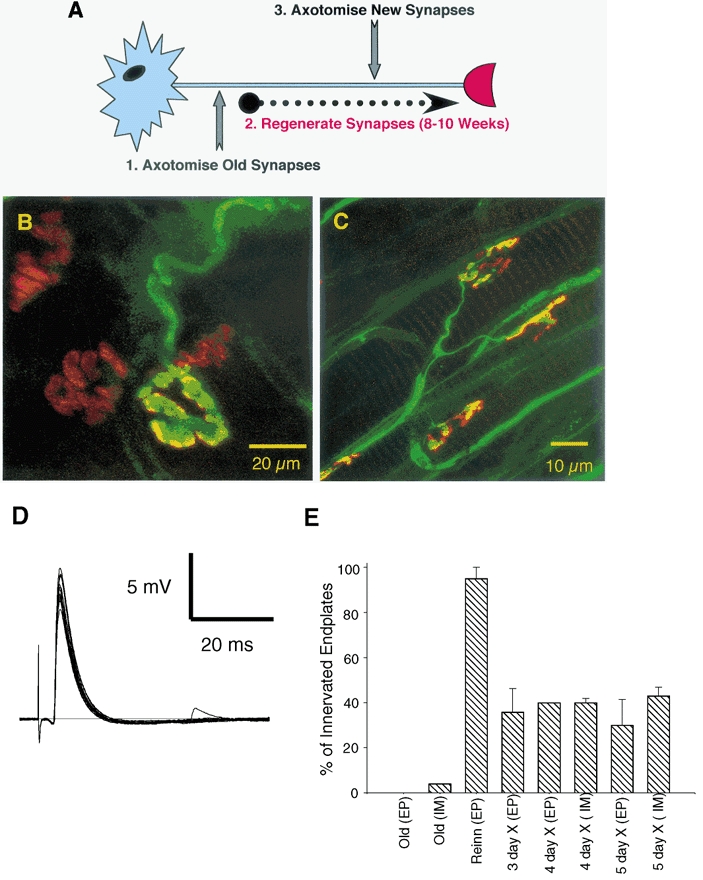
A, schematic representation of the experimental protocol used to assess the role of synaptic maturity versus chronological age on nerve terminal preservation following axotomy. The Wlds mice used were > 7 months old, therefore permitting a test of the responses to axotomy at immature (i.e. 2–4-week-old), regenerated synapses, in mature mice. B, confocal micrograph of an immunocytochemically labelled 7 month Wlds FDB muscle, 3 days post axotomy. One of the few remaining terminals from this preparation is shown, with a characteristic bloated appearance, surrounded by three vacated endplates. C, confocal micrograph of a regenerated synapse from a 14 month Wlds FDB muscle, 5 days post axotomy. Axons and motor nerve terminals were labelled immunocytochemically (NF and SV2; FITC) and acetylcholine receptors were labelled with TRITC-conjugated α-BTX. Note that all endplates shown are occupied and that the uppermost instance shows evidence of partial occupancy. D, electrophysiological recording from a 14 month Wlds FDB muscle fibre with regenerated synaptic connections, 5 days post axotomy, showing robust synaptic transmission. E, graph showing the percentage of fibres with synaptic innervation, as assessed by electrophysiological (EP) and immunocytochemical (IM) techniques, in old (non-regenerated) Wlds FDB muscle fibres at 3 days post axotomy; in reinnervated muscle fibres 8 weeks after the first lesion but before a second lesion; and in reinnervated endplates at 3–5 days after the second lesion. Note the rapid and almost complete loss of innervation following axotomy at the axotomised ‘old’ synapses (compare with Figure 4) and the retention of > 30 % of regenerated synaptic connections for up to 5 days post axotomy.
Electrophysiological analysis further supported these interpretations. Thus, synaptic transmission in axotomised 4 month mice was usually either robust, or it was absent and the incidence of junctions exhibiting weak synaptic transmission (including random failures) was low. Only ∼20 % of fibres showed evoked EPPs 3 days after axotomy in 4-month-old Wlds mice. By 7 months the corresponding figure was < 5 % (Fig. 5E). The proportion of fibres showing any spontaneous or evoked endplate activity also declined exponentially with a time constant of ∼30 days (Fig. 5G).
Thus, the axotomy-induced synaptic response in Wlds mice changes systematically, from one of withdrawal to one of degeneration, as these mice mature.
Recapitulation of synaptic withdrawal at reinnervated Wlds muscles
Because the expression of the Wld gene showed no age dependence, it was interesting to ask whether the transformation in the axotomy reaction of synaptic terminals with age was a function of age per se or whether it was a function of the maturational state of the synaptic terminals. To address this, we compared synaptic preservation of immature synapses at regenerated neuromuscular junctions in old Wlds mice (Fig. 6A). First, in mice aged 7–12 months, the sciatic nerve was crushed on one side (conditioning lesion). Regenerated axons were allowed to reinnervate the FDB muscle. Eight to ten weeks later, 95 % of fibres had regained synaptic activity from new synapses formed by the regenerated axons. Next, the repaired synapses (which could not have been more than 8 weeks old) were lesioned again, this time by cutting the tibial nerve. The incidence of innervated FDB muscle fibres following the second lesion was then scored electrophysiologically and immunocytochemically.
As expected, the first lesion produced rapid loss of synaptic transmission and morphology in these old mice; no fibres showed signs of synaptic activity at 3 days post axotomy and fewer than 2 % of endplates were occupied by nerve terminals (Fig. 6B). By contrast, synapses were consistently preserved after section of the regenerated axons (Fig. 6C). Intracellular recordings showed that 3–5 days after axotomy, 33.0 ± 15.3 % of endplates still showed spontaneous MEPPs and/or responded with EPPs to nerve stimulation (Fig. 6D). Furthermore, immunocytochemically labelled preparations showed that 4–5 days after axotomy 41.5 ± 2.02 % of endplates were contacted by overlying nerve terminal (Fig. 6E). These data therefore indicate that ‘new’ (i.e. regenerated) synapses in old Wlds mice are better protected from degeneration than the mature synapses innervating muscles without a prior, conditioning lesion applied to the nerve.
Age dependence of synaptic protection in Wld transgenic mice
Despite the uniform expression of Wld protein in the brain we considered the possibility that some subtle aspect of regulation of Wld gene expression by its endogenous promoter in motoneurones could result in an age-dependent synaptic phenotype. To address this, we examined the age dependence of synapse loss in two transgenic lines of Wld mice, lines 4836 and 4830 which also show the Wld phenotype (Mack et al. 2001). In both these lines the expression of the Wld gene is controlled by the β-actin promoter (Fig. 7A), but the 4836 line expresses the Wld protein more strongly than the 4830 line. The strength of the phenotype varies accordingly, as measured by the rate of axon loss after nerve injury. Heterozygotes of either strain also show weak expression of Wld protein. As in Wlds mice, Wld protein expression in the 4836 and 4830 transgenic lines was also independent of age (data not shown). Axon preservation, measured by retention of neurofilament heavy chains (Fig. 7B), or myelinated axon counts (data not shown) were also independent of age. In homozygous 4836-line transgenic mice, morphological and electrophysiological examination of FDB muscles indicated that most synapses were still present 5 days after axotomy, even in 4-month-old animals (Fig. 7C-E). However, intracellular recordings from hemizygous 4836 and homozygous 4830 muscles showed the same age dependence in synaptic response to axotomy as seen in Wlds mice. Thus the incidence of fibres showing synaptic responses 2–3 days after axotomy also declined exponentially with age with a time constant of ∼30 days, as in natural mutant Wlds mice (compare Fig. 7F with Fig. 5G).
Figure 7. Age-dependent synaptic protection in Wld transgenic mice.
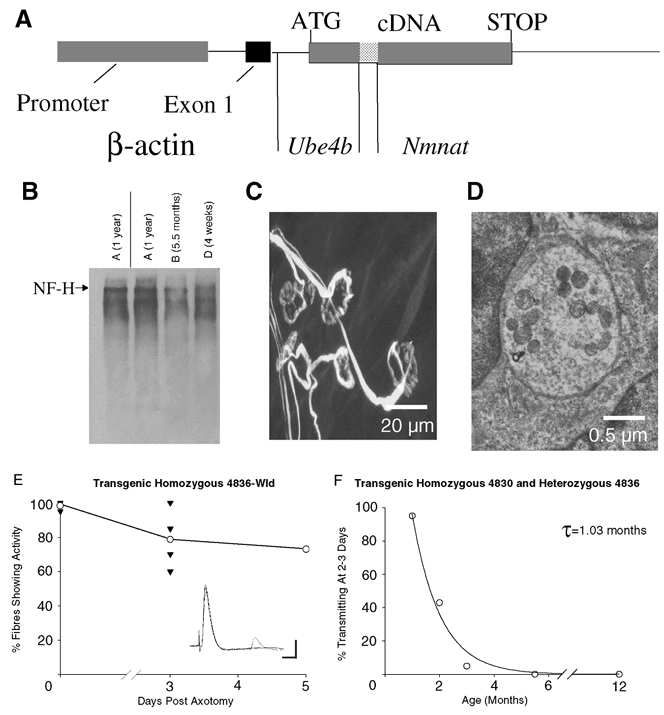
A, the transgene construct for Ube4b/Nmnat (Wld) transgenic mice. The chimeric cDNA was expressed with non-coding exon 1 of β-actin under the control of a human β-actin promoter and terminated with the SV40 polyadenylation signal. B, Western blot showing age-independent preservation of heavy chain neurofilament proteins (NF-H) in the distal stump of 4836 Wld transgenic mice sciatic nerve, 3 days post axotomy. C, confocal micrograph of immunocytochemically labelled (NF, SV2 and α-BTX) persistent synapses, 5 days post axotomy in a 4836 Wld transgenic lumbrical muscle. All of the endplates shown are either fully occupied or are missing only a small proportion of their nerve terminal. Examples of endplates with less than 50 % occupancy as well as evidence for retraction bulb formation were also found (data not shown). D, electron micrograph of a retained synaptic bouton from a 4836 homozygous Wld transgenic mouse FDB muscle, 5 days post axotomy, showing intact membranes and synaptic vesicles. E, time course of the loss of innervation following axotomy in 4836 Wld transgenic mice. The inset illustrates an electrophysiological recording showing robust synaptic transmission from a 2-month-old 4836 Wld transgenic FDB muscle fibre, 5 days post axotomy. Scale bars = 5 mV (vertical); 10 ms (horizontal). F, age dependence of synapse loss in 4830 and 4836 heterozygous Wld transgenic mice. The incidence of fibres showing synaptic responses 2–3 days after axotomy declined exponentially with age with a time constant (τ) of ∼30 days, similar to the natural mutant (compare with Fig. 3).
Protection of axons and synapses expressing fluorescent protein by the Wld gene
Recently mice have become available that express fluorescent protein in their axons and synapses under control of a thy1 promoter (Feng et al. 2000). This raises the possibility that axon and synaptic protection by the Wld gene might be visualised readily in living preparations. We therefore crossbred Wlds mice with thy1-CFP mice. The tibial nerve in CFP-expressing mice that were homozygous in the F2 generation for Wld was sectioned and FDB and lumbrical muscle preparations were examined electrophysiologically and using confocal microscopy, respectively. The results showed that fluorescent protein expression did not interfere with the protection of axons and synapses conferred by the Wld gene in young mice. Figure 8 shows that four days after axotomy, many axons remained intact, functional and endogenously fluorescent through expression of CFP and neuromuscular synapses were either fully or partly occupied by axotomised motor nerve terminals. Intracellular recordings from two muscles showed that at 4 days after axotomy 13 out of 14 fibres were innervated, and in the other muscle examined 1 day later, 19 out of 25 muscle fibres expressed either MEPPs, evoked EPPs or both. This degree of axonal and synaptic protection was similar to that in Wlds mice not expressing CFP after the same period of axotomy (compare with Fig. 2 and Fig. 3). These preliminary findings suggest that it should be possible in the future to visualise axotomy-induced synapse withdrawal in real time.
Figure 8. Persistence of neuromuscular junctions following axotomy in thy1-CFP/Wld-homozygous mice.
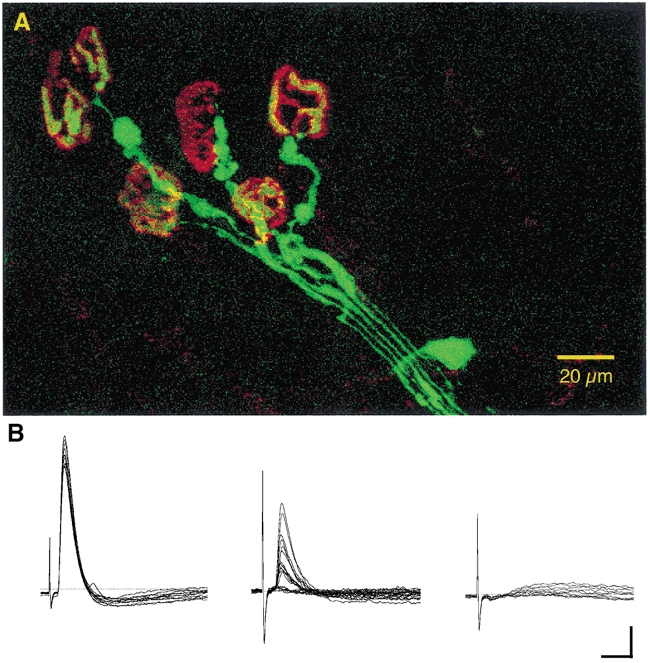
A, confocal projection image of endogenous CFP fluorescence of a group of axons and terminals supplying motor endplates, counterstained with TRITC-α-bungarotoxin, in an isolated, unfixed lumbrical muscle, 4 days after axotomy. CFP fluorescence has been pseudo-coloured green, TRITC fluorescence red. B, electrophysiological recordings of robust (left) and weak (middle: low quantal content, failures) synaptic responses recorded from FDB muscle fibres in the same axotomised foot. The record on the right shows that, as in normal Wld mice, a few fibres fail to respond, corresponding to unoccupied endplates. Scale bars = 5μm (vertical); 10 ms (horizontal).
Discussion
The main finding of the present study is that lesions of peripheral nerve induce one of at least two independent modes of synaptic degeneration in Wld-expressing mice, depending on the maturity of the synapses that are axotomised.
Synaptic terminals are progressively withdrawn from axotomised endplates in young Wlds mice. In more mature mice, this phenotype is absent, although axons are still protected from degeneration by the Wld gene. The pattern of synapse withdrawal in the young (juvenile) mice bears a compelling resemblance to synapse elimination - a phenomenon of immature or regenerating neuromuscular junctions which rationalises the innervation pattern of mammalian skeletal muscle fibres (see Ribchester, 2001 for review). This process is also distinct from Wallerian degeneration because, in most studies, none of the classical signs of Wallerian degeneration are seen at junctions undergoing synapse elimination (Miledi & Slater, 1970; Winlow & Usherwood, 1975; Korneliussen & Jansen, 1976; Bixby, 1981; but see Rosenthal & Taraskevich, 1977). Synapse elimination also occurs in reinnervated muscle (Hoffman 1953; McArdle, 1975; Brown & Ironton, 1978; Rich & Lichtman, 1989; Barry & Ribchester, 1995; Costanzo et al. 2000). In the present study we also found evidence for reinstatement of a synaptic withdrawal response to axotomy in mature Wld-expressing mice.
The main similarities between synapse elimination and axotomy-induced synapse withdrawal in Wlds are the partial occupancy of endplates and formation of retraction bulbs (Fig. 2) and a decline in synaptic efficacy (quantal content) that appears to precede loss of presynaptic terminals (Fig. 2 and Fig. 3; see Riley, 1977; Gorio et al. 1983; Balice-Gordon et al. 1993; Balice-Gordon & Lichtman, 1993; Colman et al. 1997; Kopp et al. 2000). An additional conspicuous feature of the terminals undergoing axotomy-induced withdrawal was accumulation of neurofilaments (see also Watson et al. 1993; Ribchester et al. 1995). It has previously been suggested that neurofilaments are removed from nerve terminals in advance of synapse elimination (Roden et al. 1991). However, EM reconstructions of retraction bulbs in neonatal muscle show clear evidence of neurofilament accumulation (D. L. Bishop & J. W. Lichtman, personal communication). Interestingly, the earliest study of motor nerve terminal degeneration at axotomised rabbit neuromuscular junctions (Tello, 1907) shows evidence of partial occupancy of endplates and retraction of synaptic terminals, but an ultrastructural study by Bixby (1981) showed that axotomy of wild-type rabbit neuromuscular junctions produced similar changes to those in rats and mice. It would be interesting to describe in more detail the pattern of synapse withdrawal in Wld mice using repeated or continuous visualisation methods (Keller-Peck et al. 2001; Lichtman & Fraser, 2001) in order to appraise further the similarities with or differences from synapse elimination. For example, such methods could be used to observe patterns of retraction of Wlds synaptic boutons at different endplates within the same motor unit (cf. Keller-Peck et al. 2001). This may also help to resolve whether synapse withdrawal is initiated locally at endplates, or stochastically as a result of differences in the rate of trafficking of maintenance molecules into different collateral branches of a motor axon, a mechanism sometimes referred to as ‘intrinsic withdrawal’ (Brown et al. 1976; Thompson & Jansen, 1977; cf. Betz et al. 1980). The phenomenon of axon retraction in the absence of competing inputs at the same endplate has been rather neglected since its endorsement by Fladby & Jansen (1987). Further studies of axotomy-induced retraction of synapses in Wld-expressing mice could throw further light on this issue.
We also demonstrated that age has no effect on Wld gene expression or on the biochemical or morphological preservation of Wld axons, but that preservation of axotomised synaptic terminals is clearly transformed with age in both character and kinetics, with a time constant of ∼1 month. Synaptic terminals in mice of ∼4 months and older were lost more rapidly and the degeneration of synapses began to show all the hallmarks of ‘classical’ degeneration. Thus, the present findings help to resolve a discrepancy in the earlier reports concerning the age-dependent loss of the Wlds phenotype. On the one hand, our findings support those of Crawford et al. (1995), by showing that axonal loss was independent of age, and therefore suggest that the loss of nerve conduction with age (Perry et al. 1992; Tsao et al. 1994) must have some other explanation. Crawford et al. (1995) used a cholinesterase stain combined with immunostaining for ubiquitin hydrolase to assay the preservation of terminals following axotomy in mice aged up to 16 months. On the other hand, our present analysis clearly shows - using a combination of electrophysiology, vital staining with FM1-43, immunocytochemistry and electron microscopy - that preservation of axotomised Wlds nerve terminals is strongly age dependent. Moreover, when we compared the level of protection conferred upon existing synapses in old Wlds mice and regenerated synapses, it became clear that it is the maturity of the synapse rather than age of the motor neurone (or the mouse) which is important. Whether this is a consequence of the biochemical state of the regenerated terminal and local regulation of the response to axotomy, or to recapitulation of patterns of gene expression in motor neurone nuclei remains unknown (for review see Herdegen & Leah, 1998). Another possibility that has recently come to light is that synapses in different muscles may react quite differently to nerve injury and in an age-dependent fashion over a similar time-scale to that we have reported here (Pun et al. 2002). The loss of a synaptic withdrawal response revealed in WldS mice as they mature may also have a bearing on previous findings that synaptic boutons can be induced to withdraw by selectively blocking activity at some normal adult mouse muscle motor endplates, but not others (see Fig. 6 in Balice-Gordon & Lichtman, 1994). Perhaps the age of the mice should be an important consideration in the design of such experiments.
The present findings provide further support for the theory that neurones are compartmentalised with respect to the mechanisms they contain for bringing about degeneration. They extend the notion of ‘dynamic polarisation’ of the neurone, to degenerative mechanisms as well as those responsible for sub-cellular differentiation (Changeux, 2001; Gillingwater & Ribchester, 2001). Specifically the response to axotomy of young, old and regenerated Wlds synapses suggests that newly-established, mononeuronally innervated junctions retain the molecular mechanisms required to execute retraction of supernumerary synapses, beyond the period when they are normally eliminated as part of postnatal development (Parson et al. 1997; Ribchester, 2001). An extension of this hypothesis is that neonatal synapse elimination, and by extension axonal branch elimination, may be triggered by a ‘physiological axotomy’, perhaps arising from small axonal constrictions in an axon collateral or from selective trafficking of an as yet unidentified synaptic maintenance factor produced by the cell body (Gillingwater & Ribchester, 2001; see also Raff et al. 2002). In addition, the finding that Wlds axons are protected from degeneration at all ages, but synapses are not, suggests that both mechanisms of synaptic degeneration (withdrawal in young Wld mice and Wallerian-like degeneration in older mice) occur independently of axonal degeneration. Thus, the present study of young and old Wlds mice (or their transgenic equivalents) has revealed a continuum between at least two types of synaptic degeneration, both distinct from Wallerian degeneration (Fig. 9).
Figure 9. Schematic representation of the compartmental model of neurodegeneration.
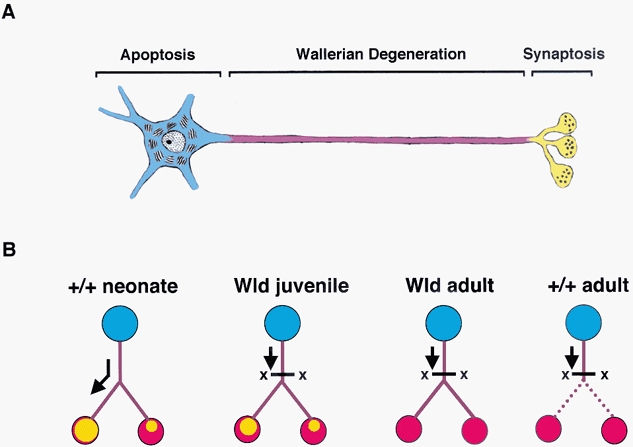
A, schematic representation of the compartmental model of neurodegeneration indicating the different degenerative compartments present in neurones (blue = cell body; purple = axon; yellow = synaptic terminals; drawing based on Nicholls et al. 1992, reproduced with permission from Gillingwater & Ribchester, 2001). B, schematic representation, based on the findings of the present study, of alternative modes of synaptic degeneration. We envisage a continuum extending from developmental synapse elimination (left) to Wallerian degeneration (right). Between these limits, are the typical responses to axotomy in young Wld mice (middle left) and more mature Wld mice (right). Axotomy induces withdrawal, resembling synapse elimination in young Wld mice. In older Wld mice the synaptic response to axotomy more closely resembles wild-type, although axons are still protected by the Wld gene. Arrows on the left diagram (synapse elimination) imply one hypothetical link: perhaps selective physiological trafficking of maintenance factors during early postnatal development (or after reinnervation) results in the same withdrawal response as that induced by surgical axotomy in young Wld mice.
How does Wld gene initially protect axons and synapses from degeneration? The Wld protein is localised to cell nuclei (Mack et al. 2001), and this means that the protection of axonal and synaptic protection must be an indirect consequence of the expression of the gene. Our cross-breeding of the Wld gene into mice expressing fluorescent protein, as well as offering unrivalled future opportunities for further analysis of the mechanisms of synaptic degeneration, suggests that in general the mutant gene does not interact with other genes that are uniquely expressed in axons and synapses. An important clue may lie with the incorporation of the N-terminal 70 amino acids of Ube4b, a ubiquitination cofactor, in the Wld gene. This raises interesting questions about the possible role of ubiquitination in neuromuscular synaptic plasticity and the synaptic response to axotomy. It is noteworthy that Drosophila mutants in which ubiquitin ligases or deubiquitinating proteases are up- or downregulated show systematic changes in neuromuscular synaptic form and function (Wan et al. 2000; DiAntonio et al. 2001). Speculation that ubiquitination might regulate mammalian synaptic form and function (Chang & Balice-Gordon, 2000) receives circumstantial support from the present findings. Thus, it will be important to find ways to critically evaluate the role of protein ubiquitination in synapse withdrawal and degeneration, whether induced by axotomy in Wlds mice, or occurring during normal synapse elimination. The way ahead may lie with the manufacture of other genes that more directly protect synapses from the normal consequences of nerve lesions.
Acknowledgments
We thank Diana Wagner (Cologne) for transgenic animal care and breeding, Weiqian Mi (Cologne) for N70 antiserum and Adrian Thomson (Edinburgh) for assistance with confocal microscopy. This work was supported by grants from the Wellcome Trust, MRC and the Federal Ministry of Education and Research, Germany (FKZ: 01 KS 9502).
References
- Balice-Gordon RJ, Chua CK, Nelson CC, Lichtman JW. Gradual loss of synaptic cartels precedes axon withdrawal at developing neuromuscular junctions. Neuron. 1993;11:801–815. doi: 10.1016/0896-6273(93)90110-d. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. Journal of Neuroscience. 1993;13:834–855. doi: 10.1523/JNEUROSCI.13-02-00834.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. Long-term synapse loss induced by focal blockade of postsynaptic receptors. Nature. 1994;372:519–524. doi: 10.1038/372519a0. [DOI] [PubMed] [Google Scholar]
- Barry JA, Ribchester RR. Persistent polyneuronal innervation in partially denervated rat muscle after reinnervation and recovery from prolonged nerve conduction block. Journal of Neuroscience. 1995;15:6327–6339. doi: 10.1523/JNEUROSCI.15-10-06327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Caldwell JH, Ribchester RR. The size of motor units during post-natal development of rat lumbrical muscle. Journal of Physiology. 1979;297:463–478. doi: 10.1113/jphysiol.1979.sp013051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Caldwell JH, Ribchester RR. The effects of partial denervation at birth on the development of muscle fibres and motor units in rat lumbrical muscle. Journal of Physiology. 1980;303:265–279. doi: 10.1113/jphysiol.1980.sp013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks R, Katz B, Miledi R. Physiological and structural changes at the amphibian myoneural junction, in the course of nerve degeneration. Journal of Physiology. 1960;150:145–168. doi: 10.1113/jphysiol.1960.sp006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bixby JL. Ultrastructural observations on synapse elimination in neonatal rabbit skeletal muscle. Journal of Neurocytology. 1981;10:81–100. doi: 10.1007/BF01181746. [DOI] [PubMed] [Google Scholar]
- Brown MC, Ironton R. Sprouting and regression of neuromuscular synapses in partially denervated mammalian muscles. Journal of Physiology. 1978;278:325–348. doi: 10.1113/jphysiol.1978.sp012307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Jansen JKS, Van Essen DC. Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. Journal of Physiology. 1976;261:387–422. doi: 10.1113/jphysiol.1976.sp011565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Lunn ER, Perry VH. Consequences of slow Wallerian degeneration for regenerating motor and sensory axons. Journal of Neurobiology. 1992;23:521–536. doi: 10.1002/neu.480230507. [DOI] [PubMed] [Google Scholar]
- Buckmaster EA, Perry VH, Brown MC. The rate of Wallerian degeneration in cultured neurons from wild type and C57Bl/Wlds mice depends on time in culture and may be extended in the presence of elevated K+ levels. European Journal of Neuroscience. 1995;7:1596–1602. doi: 10.1111/j.1460-9568.1995.tb01155.x. [DOI] [PubMed] [Google Scholar]
- Chang Q, Balice-Gordon RJ. Highwire, rpm-1, and futsch: Balancing synaptic growth and stability. Neuron. 2000;26:287–290. doi: 10.1016/s0896-6273(00)81161-7. [DOI] [PubMed] [Google Scholar]
- Changeux JP. Cajal on neurons, molecules, and consciousness. Annals of the New York Academy of Sciences. 2001;929:147–151. doi: 10.1111/j.1749-6632.2001.tb05713.x. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proceedings of the National Academy of Sciences of the USA. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman H, Nabekura J, Lichtman JW. Alterations in synaptic strength preceding synaptic withdrawal. Science. 1997;275:356–361. doi: 10.1126/science.275.5298.356. [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TGA, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wlds) mouse. Proceedings of the National Academy of Sciences of the USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo EM, Barry JA, Ribchester RR. Co-regulation of synaptic efficacy at stable polyneuronally innervated neuromuscular junctions in reinnervated rat muscle. Journal of Physiology. 1999;521:365–374. doi: 10.1111/j.1469-7793.1999.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo EM, Barry JA, Ribchester RR. Competition at silent synapses in reinnervated skeletal muscle. Nature Neuroscience. 2000;3:694–700. doi: 10.1038/76649. [DOI] [PubMed] [Google Scholar]
- Crawford TO, Hsieh S-T, Schryer BL, Glass JD. Prolonged axonal survival in transected nerves of C57Bl/Ola mice is independent of age. Journal of Neurocytology. 1995;24:333–340. doi: 10.1007/BF01189060. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM. Neurites can remain viable after the destruction of the neuronal soma by programmed cell death. Developmental Biology. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS. Ubiquitin-dependent mechanisms regulate synaptic growth and function. Nature. 2001;412:449–452. doi: 10.1038/35086595. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Gross J, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fladby T, Jansen JKS. Postnatal loss of synaptic terminals in the partially denervated mouse soleus muscle. Acta Physiologica Scandinavica. 1987;129:239–246. doi: 10.1111/j.1748-1716.1987.tb08064.x. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Koutsikou S, Barry JA, Ribchester RR. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wlds mutant mice. Journal of Physiology. 2000;523.P:53P. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the Wlds mutant mouse. Journal of Physiology. 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorio A, Carmignoto G, Finesso M, Polato P, Nunzi MG. Muscle reinnervation - II. Sprouting, synapse formation and repression. Neuroscience. 1983;8:403–415. doi: 10.1016/0306-4522(83)90188-4. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: Control of gene expression by jun, fos and krox, and creb/atf proteins. Brain Research Reviews. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- Hoffman H. The persistence of hyperneurotized end-plates in mammalian muscles. Journal of Comparative Neurology. 1953;99:331–345. doi: 10.1002/cne.900990207. [DOI] [PubMed] [Google Scholar]
- Keller-Peck CR, Walsh MK, Gan W-B, Feng G, Sanes JR, Lichtman JW. Asynchronous synapse elimination in neonatal motor units: Studies using GFP transgenic mice. Neuron. 2001;31:381–394. doi: 10.1016/s0896-6273(01)00383-x. [DOI] [PubMed] [Google Scholar]
- Kopp DM, Perkel DJ, Balice-Gordon RJ. Disparity in neurotransmitter release probability among competing inputs during neuromuscular synapse elimination. Journal of Neuroscience. 2000;20:8771–8779. doi: 10.1523/JNEUROSCI.20-23-08771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen H, Jansen JKS. Morphological aspects of the elimination of polyneuronal innervation of skeletal muscle fibres in newborn rats. Journal of Neurocytology. 1976;5:591–604. doi: 10.1007/BF01175572. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Fraser SE. The neuronal naturalist: watching neurons in their native habitat. Nature Neuroscience. 2001;4:1215–1220. doi: 10.1038/nn754. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does hinder regeneration in peripheral nerve. European Journal of Neuroscience. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH. A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4. Proceedings of the National Academy of Sciences of the USA. 1993;90:9717–9720. doi: 10.1073/pnas.90.20.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack TGA, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Shama Fernando F, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nature Neuroscience. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Experimental Neurology. 1998a;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Partin J, Begley JG. Amyloid β-peptide induces apoptosis-related events in synapses and dendrites. Brain Research. 1998b;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- McArdle JJ. Complex end-plate potentials at the regenerating neuromuscular junction of the rat. Experimental Neurology. 1975;49:629–638. doi: 10.1016/0014-4886(75)90048-5. [DOI] [PubMed] [Google Scholar]
- Mi W, Conforti L, Coleman MJ. Genotyping methods to detect a unique neuroprotective factor (Wlds) for axons. Journal of Neuroscience Methods. 2002;113:215–218. doi: 10.1016/s0165-0270(01)00501-5. [DOI] [PubMed] [Google Scholar]
- Miledi R, Slater CR. On the degeneration of rat neuromuscular junctions after nerve section. Journal of Physiology. 1970;207:507–528. doi: 10.1113/jphysiol.1970.sp009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls JG, Martin AR, Wallace BG. From Neuron to Brain. 3. Massachusetts USA: Sinauer Associates; 1992. p. 389. [Google Scholar]
- Parson SH, Mackintosh CL, Ribchester RR. Elimination of motor nerve terminals in neonatal mice expressing a gene for slow Wallerian degeneration (C57Bl/Wlds) European Journal of Neuroscience. 1997;9:1586–1592. doi: 10.1111/j.1460-9568.1997.tb01516.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER. Very slow retrograde and Wallerian degeneration in the CNS of C57Bl/Ola mice. European Journal of Neuroscience. 1990a;3:102–105. doi: 10.1111/j.1460-9568.1991.tb00815.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Tsao JW. The effectiveness of the gene which slows the rate of Wallerian degeneration in C57Bl/Ola mice declines with age. European Journal of Neuroscience. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S. Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. European Journal of Neuroscience. 1990b;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Pun S, Sigrist M, Santos AF, Ruegg MA, Sanes JR, Jessell TM, Arber S, Caroni P. An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron. 2002;34:357–370. doi: 10.1016/s0896-6273(02)00670-0. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-degeneration and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ribchester RR. Development and plasticity of neuromuscular connections. In: Kalverboer AF, Gramsbergen A, editors. Brain and Behaviour in Human Neural Development. London: Kluwer Academic Press; 2001. pp. 261–341. [Google Scholar]
- Ribchester RR, Coleman MP, Thomson D, Gillingwater TH, Mack TGA, Wagner D. Recapitulation of axotomy-induced synapse withdrawal at regenerated neuromuscular junctions in mature Wlds mice. European Journal of Physiology. 2002;443:183S. [Google Scholar]
- Ribchester RR, Mao F, Betz WJ. Optical measurements of activity-dependent membrane recycling in motor nerve terminals of mammalian skeletal muscle. Proceedings of the Royal Society. 1994;255:61–66. doi: 10.1098/rspb.1994.0009. [DOI] [PubMed] [Google Scholar]
- Ribchester RR, Pakiam JG, O'Carroll CM, Thomson D, Mattison RJ, Costanzo EM, Gillingwater TH, Barry JA. Impaired neuromuscular transmission preceding synapse withdrawal in axotomised adult Wlds mutant mouse skeletal muscle. Journal of Physiology. 1999;520.P:76P. [Google Scholar]
- Ribchester RR, Tsao JW, Barry JA, Asgari-Jirandeh N, Perry VH, Brown MC. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57Bl/Wlds) European Journal of Neuroscience. 1995;7:1641–1650. doi: 10.1111/j.1460-9568.1995.tb01159.x. [DOI] [PubMed] [Google Scholar]
- Rich MM, Lichtman JW. In vivo visualization of presynaptic and postsynaptic changes during synapse elimination in reinnervated mouse muscle. Journal of Neuroscience. 1989;9:1781–1805. doi: 10.1523/JNEUROSCI.09-05-01781.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley DA. Spontaneous elimination of nerve terminals from the endplates of developing skeletal myofibers. Brain Research. 1977;134:279–285. doi: 10.1016/0006-8993(77)91073-3. [DOI] [PubMed] [Google Scholar]
- Roden RL, Donahue SP, Schwartz GA, Wood JG, English AW. 200kD neurofilament protein and synapse elimination in the rat soleus. Synapse. 1991;9:239–243. doi: 10.1002/syn.890090402. [DOI] [PubMed] [Google Scholar]
- Rosenthal JL, Taraskevich PS. Reduction of multiaxonal innervation at the neuromuscular junction of the rat during development. Journal of Physiology. 1977;270:299–310. doi: 10.1113/jphysiol.1977.sp011953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tello JF. Degeneration et regeneration des plaques motrices après la section des nerfs. Travaux du Laboratoire de Recherches Biologiques de l'Université de Madrid. 1907;5:117–149. [Google Scholar]
- Thompson W, Jansen JKS. The extent of sprouting of remaining motor units in partly denervated immature and adult rat soleus muscle. Neuroscience. 1977;2:523–535. doi: 10.1016/0306-4522(77)90049-5. [DOI] [PubMed] [Google Scholar]
- Tsao JW, Brown MC, Carden MJ, McLean WG, Perry VH. Loss of the compound action potential: an electrophysiological, biochemical and morphological study of early events in axonal degeneration in the C57Bl/Ola mouse. European Journal of Neuroscience. 1994;6:516–524. doi: 10.1111/j.1460-9568.1994.tb00295.x. [DOI] [PubMed] [Google Scholar]
- Wan HI, Diantonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Watson DF, Glass JD, Griffin JW. Redistribution of cytoskeletal proteins in mammalian axons disconnected from their cell bodies. Journal of Neuroscience. 1993;13:4354–4360. doi: 10.1523/JNEUROSCI.13-10-04354.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winlow W, Usherwood PNR. Ultrastructural studies of normal and degenerating mouse neuromuscular junctions. Journal of Neurocytology. 1975;4:377–394. doi: 10.1007/BF01261371. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. Journal of Physiology. 1997;500:165–176. doi: 10.1113/jphysiol.1997.sp022007. [DOI] [PMC free article] [PubMed] [Google Scholar]