

Abstract
The regulation of calcium (Ca2+) sparks and transient calcium-sensitive K+ (KCa) currents by acute changes in sarcoplasmic reticulum (SR) Ca2+ load ([Ca2+]SR) was investigated in rat cerebral artery smooth muscle cells using laser-scanning confocal microscopy in combination with patch clamp electrophysiology. [Ca2+]SR was elevated by: (i) increasing the activity of the SR Ca2+-ATPase with an anti-phospholamban monoclonal antibody, or (ii) blocking Ca2+ release from the SR with tetracaine, a membrane-permeant, reversible ryanodine-sensitive Ca2+ release (RyR) channel blocker. Alternatively, [Ca2+]SR was progressively decreased over time with a low concentration of thapsigargin (20 nm), a SR Ca2+-ATPase blocker. An elevation in [Ca2+]SR increased Ca2+ spark and transient KCa current frequency, but did not alter the amplitude, decay or spatial spread of Ca2+ sparks or the coupling ratio or amplitude correlation between Ca2+ sparks and evoked transient KCa currents. Decreasing [Ca2+]SR reduced Ca2+ spark frequency, amplitude and spatial spread and this reduced transient KCa current frequency and amplitude. However, even when mean Ca2+ spark amplitude and spread decreased by up to 47 and 56 % of control, respectively, the coupling ratio or amplitude correlation between Ca2+ sparks and transient KCa currents was not affected. These data demonstrate that acute changes in [Ca2+]SR regulate Ca2+ sparks and transient KCa currents in arterial smooth muscle cells.
Intracellular calcium (Ca2+) regulates a diverse range of cellular functions, including contraction, secretion and gene transcription. However, recent studies have determined that the intracellular Ca2+ concentration ([Ca2+]i) of many cell types is not homogeneously distributed (for reviews see Clapham, 1995; Berridge, 1997). Several different types of intracellular Ca2+ elevations have been described to occur that differ in respect to temporal kinetics, spatial localization and physiological function (Bootman et al. 2001). One type of Ca2+ signalling modality that has been observed in cardiac (Cheng et al. 1993), skeletal (Tsugorka et al. 1995) and smooth muscle (Nelson et al. 1995) cells is termed a ‘Ca2+ spark’. Ca2+ sparks are highly localized, cytosolic Ca2+ transients that occur due to the opening of a number of ryanodine-sensitive Ca2+ release (RyR) channels in the sarcoplasmic reticulum (SR; Jaggar et al. 2000).
In smooth muscle cells, Ca2+ sparks exhibit a rise time of ≈20 ms, a half-time of decay of ≈50 ms and a spatial spread of ≈2-3 μm when imaged using a confocal microscope and fluorescent Ca2+ indicators such as fluo-3 (Nelson et al. 1995; Jaggar et al. 2000). Although Ca2+ sparks elevate intracellular Ca2+ in the immediate vicinity of the Ca2+ release site to micromolar concentrations (Perez et al. 2001), the impact of Ca2+ sparks on the global [Ca2+]i is low, due to their transient and localized properties (Nelson et al. 1995; Jaggar et al. 2000). In arterial smooth muscle cells, most Ca2+ sparks occur in close proximity to the plasma membrane and activate a number of large-conductance Ca2+-sensitive K+ (KCa) channels to evoke a transient outward K+ current (Nelson et al. 1995; Bolton & Gordienko, 1998; Perez et al. 1999), which has been termed a ‘spontaneous transient outward current’ or ‘STOC’ (Benham & Bolton, 1986). In arteries at physiological levels of pressure, inhibition of Ca2+ sparks or KCa channels leads to membrane depolarization, activation of voltage-dependent Ca2+ channels, an elevation in the arterial wall [Ca2+]i and constriction (Nelson et al. 1995; Jaggar, 2001). An elevation in intravascular pressure activates voltage-dependent Ca2+ channels in arterial smooth muscle cells leading to an increase in the global [Ca2+]i and activation of Ca2+ sparks (Jaggar, 2001). The resulting elevation in KCa channel activity opposes the pressure-induced constriction (Brayden & Nelson, 1992).
Several signalling elements regulate Ca2+ sparks in smooth muscle cells, including intracellular Ca2+ and protein kinases (Jaggar et al. 2000). Smooth muscle RyR channels that are incorporated into lipid bilayers are activated by cytosolic Ca2+ elevations (Herrmann-Frank et al. 1991; Xu et al. 1994), suggesting that an elevation in [Ca2+]i may regulate Ca2+ sparks via an interaction with activation sites located on the cytosolic face of the RyR channel. However, an increase in cytosolic [Ca2+]i or activation of the SR Ca2+-ATPase may increase SR Ca2+ load ([Ca2+]SR), which could also regulate RyR channels. Cardiac (Sitsapesan & Williams, 1994; Gyorke & Gyorke, 1998; Xu & Meissner, 1998; Ching et al. 2000) and skeletal (Herrmann-Frank & Lehmann-Horn, 1996; Tripathy & Meissner, 1996) muscle RyR channels incorporated into lipid bilayers are activated by an elevation in luminal Ca2+ concentration. Furthermore, an elevation in [Ca2+]SR activates Ca2+ sparks in cardiac myocytes (Santana et al. 1997; Satoh et al. 1997; Lukyanenko et al. 2001). Recent studies have also suggested that [Ca2+]SR may regulate Ca2+ sparks in smooth muscle cells. Genetic ablation of phospholamban, an endogenous inhibitor of the SR Ca2+-ATPase, leads to a chronic elevation in [Ca2+]SR and Ca2+ spark frequency in arterial smooth muscle cells, when compared to wild type controls (Wellman et al. 2001). In Bufo marinus stomach smooth muscle cells, following partial depletion of the [Ca2+]SR with caffeine, Ca2+ spark frequency and amplitude increase during [Ca2+]SR refilling (ZhuGe et al. 1999). Vasodilators also stimulate Ca2+ sparks, in part, by elevating [Ca2+]SR (Porter et al. 1998; Wellman et al. 2001). However, despite the important role that the [Ca2+]SR may play in intracellular Ca2+ signalling in smooth muscle, the regulation of Ca2+ sparks and transient KCa currents by [Ca2+]SR is poorly understood.
The goal of this study was to investigate the regulation of Ca2+ sparks and transient KCa currents by acute changes in [Ca2+]SR in arterial smooth muscle cells. [Ca2+]SR was elevated with a monoclonal antibody raised against phospholamban, or with tetracaine, a membrane-permeant reversible RyR channel blocker. Alternatively, [Ca2+]SR was progressively decreased with a low concentration of thapsigargin, a selective inhibitor of the SR Ca2+-ATPase. Data suggest that an elevation in [Ca2+]SR increases Ca2+ spark and transient KCa current frequency, but does not alter the amplitude, decay or spatial spread of Ca2+ sparks or the coupling ratio or amplitude correlation between Ca2+ sparks and evoked transient KCa currents. In contrast, decreasing [Ca2+]SR reduces Ca2+ spark frequency, amplitude and spatial spread, which decreases the frequency and amplitude of evoked transient KCa currents, although the coupling ratio and amplitude correlation between Ca2+ sparks and KCa channels does not change. These findings provide evidence that acute changes in [Ca2+]SR directly alter the frequency and amplitude properties of Ca2+ sparks and evoked transient KCa currents.
METHODS
Tissue preparation
Sprague-Dawley rats (≈250 g) of either sex were killed by peritoneal injection of a sodium pentobarbital overdose (150 mg kg−1), in accordance with the Animal Care and Use Committee policies and procedures at the University of Tennessee. The brain was removed and placed into ice-cold (4 °C), oxygenated (95 % O2-5 % CO2), physiological saline solution (PSS) containing (mm): 119 NaCl, 4.7 KCl, 24 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, 0.023 EDTA and 11 glucose (adjusted to pH 7.4 with NaOH). Posterior cerebral and cerebellar arteries (50-200 μm diameter) were removed, cleaned of basolateral connective tissue and maintained in ice-cold PSS. Where appropriate, the endothelium was removed by allowing an air bubble to remain in the lumen of the artery for 2 min, followed by a 30 s wash with H2O (Jaggar & Nelson, 2000). Individual smooth muscle cells were enzymatically dissociated from cerebral arteries using a procedure similar to that described in Jaggar (2001).
Confocal calcium imaging
Arterial segments (1-2 mm in length) or isolated smooth muscle cells were placed into Hepes-buffered PSS of composition (mm): 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes and 10 glucose (pH 7.4, NaOH) containing 10 μM fluo-4 AM and 0.05 % pluronic F-127 for 60 min or 15 min, respectively, at 22 °C. To allow indicator de-esterification, arteries or isolated cells were subsequently placed into Hepes-buffered PSS for 30 min at 22 °C. Smooth muscle cells were imaged using a Noran Oz laser scanning confocal microscope (Noran Instruments, Middleton, WI, USA) and a ×60 water immersion objective (NA = 1.2) attached to a Nikon TE300 microscope by illuminating with a krypton-argon laser at 488 nm. Planar images (56.3 μm × 52.8 μm) were recorded every 16.7 ms, i.e. 60 images s−1. To compare confocal Ca2+ imaging data with electrophysiological recordings performed in this study, Ca2+ sparks in smooth muscle cells of arterial segments were measured in an extracellular solution containing 30 mm K+, which depolarizes smooth muscle cells to ≈-40 mV, and is similar to the membrane potential of cerebral arteries pressurized to 60 mmHg (see Jaggar et al. 1998; Jaggar & Nelson, 2000; Jaggar, 2001 for similar procedure). The 30 mm K+ bath solution contained (mm): NaCl 110; KCl 30; Hepes 10; CaCl2 2; MgCl2 1; and glucose 10 (pH 7.4, NaOH). For imaging smooth muscle cells in arteries, at least two different representative areas (56.3 μm × 52.8 μm) of the same segment were each scanned for at least 10 s under each condition. The same area of artery was scanned only once to avoid any laser-induced changes in Ca2+ signalling, and the effects of drugs were measured in paired experiments. Where appropriate, diltiazem (50 μM) was applied for 15 min to ensure a complete and steady state decrease in Ca2+ sparks and global Ca2+ fluorescence prior to tetracaine application. Tetracaine was then added in the presence of diltiazem, and further Ca2+ imaging measurements were made 5 min later. Time-matched control experiments confirmed that diltiazem induced steady-state block of Ca2+ sparks prior to tetracaine application. Ca2+ spark frequency in smooth muscle cells was not different after 15 (0.32 ± 0.09 Hz) or 20 min (0.26 ± 0.09 Hz) in the continued presence of diltiazem (50 μM, P > 0.05, n= 5 arteries). In experiments where confocal microscopy was employed in combination with patch clamp electrophysiology, current and fluorescence measurements were synchronized using a light emitting diode positioned above the recording chamber that was triggered during acquisition. Each single smooth muscle cell was imaged for at least 10 s under each condition. Ca2+ sparks were detected in smooth muscle cells using custom analysis software written using IDL 5.0.2 (Research Systems Inc., Boulder, CO, USA) kindly provided by Drs M. T. Nelson and A. D. Bonev (University of Vermont, VT, USA). Automated and manual detection of Ca2+ sparks was performed by dividing an area 1.54 μm (7 pixels) × 1.54 μm (7 pixels) (i.e. 2.37 μm2) in each image (F) by a baseline (F0) which was determined by averaging 10 images without Ca2+ spark activity. The entire area of each image was analysed to detect Ca2+ sparks. A Ca2+ spark was defined as a localized increase in F/F0 that was greater than 1.2. In intact artery experiments, Ca2+ spark frequency (Hz) in each condition was calculated from at least two different areas of each artery wall. Ca2+ spark spatial spread was calculated as the full width at half-maximal amplitude (FWHM). Global Ca2+ fluorescence was calculated from the same images used for Ca2+ spark analysis and was the mean pixel value of 100 different images acquired over 10 s.
Ratiometric calcium measurements
Cerebral artery segments (1-2 mm in length) were incubated with the ratiometric fluorescent Ca2+ indicator fura-2 AM (2 μM) and 0.05 % pluronic F-127 for 20 min, followed by a 15 min wash. Experiments were performed using a 6 mm potassium Hepes-buffered PSS (composition described above). Arteries were alternately excited at 340 or 380 nm using a PC driven Hyperswitch (Ionoptix, Milton, MA, USA), and background corrected ratios were collected at every 0.2 s at 510 nm using a photomultiplier tube. [Ca2+]SR was determined by rapidly applying a high concentration of caffeine (20 mm), a RyR channel activator, wherein the amplitude of the [Ca2+]i transient would be related to the [Ca2+]SR. [Ca2+]i concentrations were calculated using the following equation (Grynkiewicz et al. 1985):
where R is the 340/380 nm ratio, Rmin and Rmax are the minimum and maximum ratios determined in Ca2+-free and saturating Ca2+ solutions, respectively, Sf2/Sb2 is the Ca2+-free/Ca2+-replete ratio of emissions at 380 nm excitation, and Kd is the dissociation constant for fura-2. Rmin, Rmax, Sf2 and Sb2 were determined at the end of each experiment and in separate experiments by increasing the Ca2+ permeability of smooth muscle cells with ionomycin (10 μM), and perfusing cells with a high Ca2+ (50 mm) or Ca2+-free (10 mm EGTA) solution. The in situ apparent dissociation constant (Kd) for fura-2 used in this study was 282 nm (Knot & Nelson, 1998).
Patch clamp electrophysiology
Isolated cells were allowed to adhere to a glass coverslip in the bottom of a chamber for 10 min prior to experimentation. K+ currents were measured using either the conventional whole-cell or perforated patch configuration (Horn & Marty, 1988) of the patch clamp technique (Hamill et al. 1981) with an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA). Bath solution was 6 mm K+ Hepes-buffered PSS (composition described above). For perforated patch experiments, the pipette solution contained (mm): 110 potassium aspartate, 30 KCl, 10 NaCl, 1 MgCl2, 10 Hepes, 0.05 EGTA (pH 7.2 with KOH). For conventional whole-cell experiments, the pipette (i.e. intracellular) solution contained (mm): 140 KCl, 1.9 MgCl2, 0.037 CaCl2, 0.1 EGTA, 10 Hepes, 1 Na2ATP (pH 7.2 with KOH); the calculated free Ca2+ and free Mg2+ concentrations of this solution are 100 nm and 1 mm, respectively (WEBMAXC, Stanford University, CA, USA). The conventional whole-cell configuration was used in experiments with anti-phospholamban antibody. Elsewhere in this study, the perforated patch configuration was used. All patch clamp experiments were performed with a holding potential of −40 mV. Membrane currents were recorded with a sample rate of 2.5 kHz and filtered at 1 kHz. Transient KCa current analysis was performed off-line using a custom analysis program provided by Drs M. T. Nelson and A. D. Bonev (University of Vermont). A transient KCa current was defined as the simultaneous opening of three or more KCa channels. In the presence of Ca2+ spark blockers, the simultaneous opening of three single KCa channels was not observed at −40 mV (Nelson et al. 1995; Bonev et al. 1997; Porter et al. 1998). For experiments performed in the absence or presence of anti-phospholamban antibody, transient KCa current properties were compared between 5 and 10 min after whole-cell patch formation. Where appropriate, anti-phospholamban antibody was heat-inactivated by incubation at 94 °C for 15 min.
Statistical analysis
Values are expressed as means ± standard error of the mean. Student's t test or Student-Newman-Keuls tests were used for comparing paired or multiple data sets, respectively. First-order polynomial linear fits were used to calculate statistical correlation between the amplitude of Ca2+ sparks and evoked transient KCa currents (Origin, OriginLab Corp., Northampton, MA, USA). ANCOVA of first-order polynomial best fits were used to compare amplitude correlation data sets (Prism, GraphPad Software, Inc., San Diego, CA, USA). P < 0.05 was considered significant.
Chemicals
Unless stated otherwise, all chemicals used in this study were obtained from Sigma Chemical Company (St Louis, MO, USA). Papain was purchased from Worthington Biochemical Co. (Lakewood, NJ, USA), fluo-4 AM and pluronic F-127 from Molecular Probes (Eugene, OR, USA), and anti-phospholamban mouse IgG1 clone A1 from Upstate Biotechnology (Lake Placid, NY, USA).
RESULTS
A monoclonal antibody targeted against phospholamban elevates transient KCa current frequency in cerebral artery smooth muscle cells
To investigate the regulation of transient KCa currents by an acute elevation in [Ca2+]SR, we used a commercially available monoclonal antibody (mouse IgG1) raised against phospholamban, an endogenous inhibitor of the SR Ca2+-ATPase. This antibody has previously been demonstrated to elevate [Ca2+]SR in cardiac myocytes (Suzuki & Wang, 1986; Sham et al. 1991; Lukyanenko et al. 2001). Transient KCa current properties were compared between 5 and 10 min after conventional whole-cell formation in cells in which antibody (4 μg ml−1) or heat-inactivated antibody (4 μg ml−1) was included in the pipette solution, or antibody was absent.
Mean transient KCa current frequency was ≈2.7-fold higher in cells in which anti-phospholamban antibody was included in the pipette solution (n= 11), when compared with cells in which heat-inactivated antibody was included (n= 6 cells), or antibody was absent (n= 11, Fig. 1). In contrast, the anti-phospholamban antibody did not change mean transient KCa current amplitude (Fig. 1). These data suggest that acute activation of the SR Ca2+-ATPase with an anti-phospholamban antibody increases the frequency, but does not alter the amplitude, of transient KCa currents in arterial smooth muscle cells.
Figure 1. A monoclonal antibody raised against phospholamban (anti-PLB antibody) elevates transient KCa current frequency, but not amplitude, in cerebral artery smooth muscle cells.
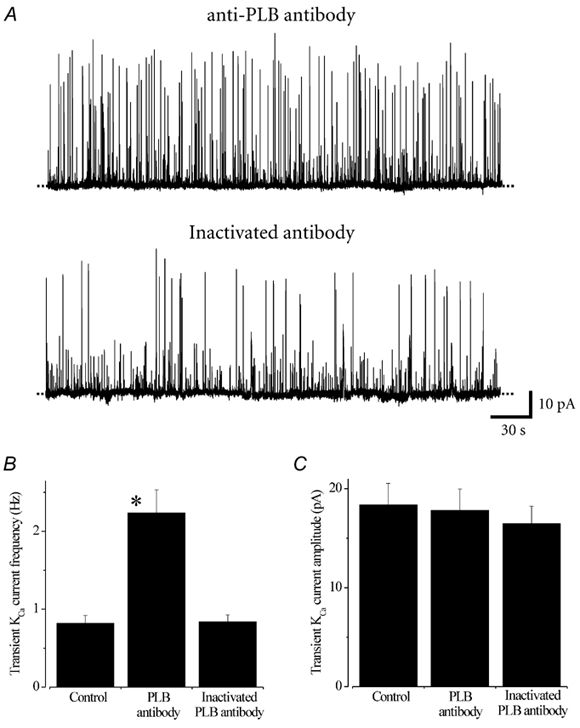
A, original traces illustrating typical transient KCa current activity at −40 mV in cells in which anti-PLB antibody (4 μg ml−1, upper panel) or heat-inactivated anti-PLB antibody (4 μg ml−1, lower panel) was included in the pipette solution. Average transient KCa current frequency (B) and amplitude (C) in cells in which no antibody (control), anti-PLB antibody, or heat-inactivated antibody was included in the pipette solution. *P < 0.05; Student's unpaired t test.
Tetracaine inhibits Ca2+ sparks in cerebral artery smooth muscle cells
We sought to measure the regulation of Ca2+ sparks and transient KCa currents before and after an elevation in [Ca2+]SR in the same cell. We hypothesized that RyR channel blockers would increase [Ca2+]SR by preventing Ca2+ release from the SR under conditions where the SR Ca2+-ATPase would continue to sequester Ca2+ from the cytosol. To examine this possibility we used tetracaine, a membrane-permeant, reversible blocker of cardiac (Gyorke et al. 1997; Lukyanenko et al. 2001) and skeletal (Xu et al. 1993) muscle RyR channels. To determine if tetracaine blocks RyR channels in smooth muscle cells, the regulation of Ca2+ sparks by tetracaine (50 μM) was measured in the smooth muscle cells of endothelium-denuded cerebral artery segments. To compare confocal Ca2+ imaging data with electrophysiological data performed in this study, Ca2+ sparks were measured in a bath solution containing 30 mm K+, which depolarizes smooth muscle cells from ≈-60 to −40 mV, a membrane potential similar to that of cerebral arteries pressurized to 60 mmHg (see Jaggar et al. 1998; Jaggar & Nelson, 2000; Jaggar, 2001 for similar procedure). To prevent any potential indirect regulation of Ca2+ sparks due to non-specific effects on voltage-dependent Ca2+ channels (Sugiyama & Muteki, 1994), tetracaine was applied in the continued presence of diltiazem (50 μM), a voltage-dependent Ca2+ channel blocker.
Diltiazem reduced mean global Ca2+ fluorescence in smooth muscle cells (F/F0) to 53 ± 3 % of control, and decreased mean Ca2+ spark frequency to ≈20 % of control, although mean Ca2+ spark amplitude (F/F0) did not change (n= 6 arteries, Fig. 2). In the same arteries in the continued presence of diltiazem, tetracaine reduced mean Ca2+ spark frequency to ≈3 % of control, but did not change mean Ca2+ spark amplitude (Fig. 2). Tetracaine did not alter mean global F/F0 (98 % ± 2 % of diltiazem), suggesting that Ca2+ sparks were not blocked via a reduction in global [Ca2+]i (Fig. 2). These data demonstrate that tetracaine, a RyR channel blocker, inhibits Ca2+ sparks and does not alter global [Ca2+]i in cerebral artery smooth muscle cells, when applied in the continued presence of diltiazem.
Figure 2. Tetracaine blocks Ca2+ sparks in smooth muscle cells of intact cerebral arteries.
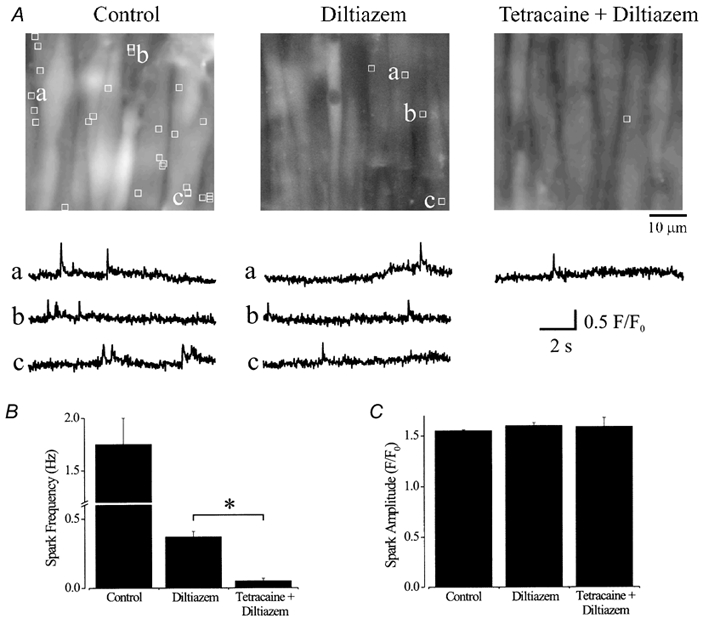
A, average fluorescence (100 of 600 images) over 10 s of three different 56.3 μm × 52.8 μm areas of the same cerebral artery in control (30 mm K+), diltiazem (50 μM) and tetracaine (50 μM) + diltiazem (50 μM). The locations of Ca2+ sparks that occurred during 10 s are indicated by white boxes (1.54 μm × 1.54 μm) and representative localized F/F0 changes with time are illustrated below respective images and labelled accordingly. In this artery over 10 s, 27 sparks occurred in control, five sparks in diltiazem and one spark in tetracaine + diltiazem. Average effects on Ca2+ spark frequency (B) and amplitude (C). *P < 0.05; Student-Newman-Keuls test.
Tetracaine elevates sarcoplasmic reticulum Ca2+ load in cerebral arteries
Blocking Ca2+ release from the SR with tetracaine should lead to an increase in [Ca2+]SR. To examine this hypothesis, the effect of tetracaine (50 μM) on the amplitude of caffeine (20 mm)-induced [Ca2+]i transients was investigated in endothelium-denuded cerebral artery segments using the ratiometric Ca2+ indicator fura-2. Experiments were performed in a bath solution containing 6 mm K+.
Diltiazem did not change mean arterial wall [Ca2+]i (control, 99 ± 5 nmvs. diltiazem, 100 ± 5 nm) or the amplitude of caffeine-induced [Ca2+]i transients (Fig. 3, n= 6 arteries). In the same arteries, although tetracaine did not change mean arterial wall [Ca2+]i (104 ± 4 nm), mean caffeine-induced [Ca2+]i transients increased ≈1.4-fold after a 7 min application. Five minutes after tetracaine washout, caffeine-induced [Ca2+]i transients returned to pre-tetracaine levels (Fig. 3). These data suggest that tetracaine, a RyR channel blocker, reversibly elevates [Ca2+]SR in cerebral artery smooth muscle cells, presumably by blocking Ca2+ release from the SR (Fig. 2).
Figure 3. Tetracaine elevates [Ca2+]SR in cerebral artery segments.
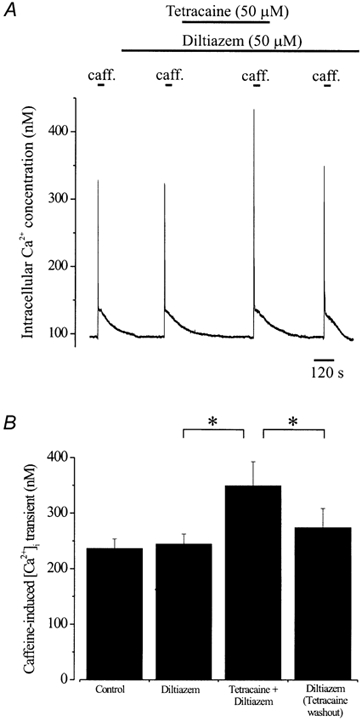
A, intracellular Ca2+ concentration and caffeine (20 mm)-induced [Ca2+]i transients in an endothelium-denuded cerebral artery segment. Diltiazem (50 μM) did not change arterial wall [Ca2+]i or caffeine-induced [Ca2+]i transients. Tetracaine (50 μM, 7 min application) did not change arterial wall [Ca2+]i, but significantly increased the amplitude of the caffeine-induced [Ca2+]i transient. Five minutes after washout of tetracaine, the caffeine-induced [Ca2+]i transient was of similar amplitude to control. B, average caffeine-induced [Ca2+]i transients in control, diltiazem, tetracaine + diltiazem, and 5 min after tetracaine washout + diltiazem. *P < 0.05; Student-Newman-Keuls test.
Regulation of transient KCa currents by tetracaine
If tetracaine inhibits Ca2+ sparks, it should also block transient KCa currents. Furthermore, if an increase in [Ca2+]SR activates Ca2+ sparks, transient KCa currents should be elevated immediately after washout of tetracaine, when compared with transient KCa currents prior to tetracaine application. To investigate this hypothesis, the regulation of transient KCa currents by tetracaine was measured in voltage-clamped (-40 mV) cerebral artery smooth muscle cells. Tetracaine was applied, and washed out, in the continued presence of Cd2+ (250 μM), a voltage-dependent Ca2+ channel blocker.
Cd2+ reduced mean transient KCa current frequency to ≈54 % of control, but did not change mean transient KCa current amplitude (Fig. 4, n= 6 cells). In the continued presence of Cd2+, tetracaine (50 μM, 7 min application) further reduced mean transient KCa current frequency, but did not alter mean amplitude. Washout of tetracaine was rapid and led to a significant increase in mean transient KCa current frequency. Between 1 and 3 min after tetracaine washout, mean transient KCa current frequency was ≈3.1-fold higher than that prior to tetracaine application, although mean transient KCa current amplitude was not significantly different (Fig. 4). Approximately 5 min after tetracaine washout, but in the continued presence of Cd2+, transient KCa current frequency returned to pre-tetracaine levels (Fig. 4).
Figure 4. Regulation of transient KCa currents by tetracaine.
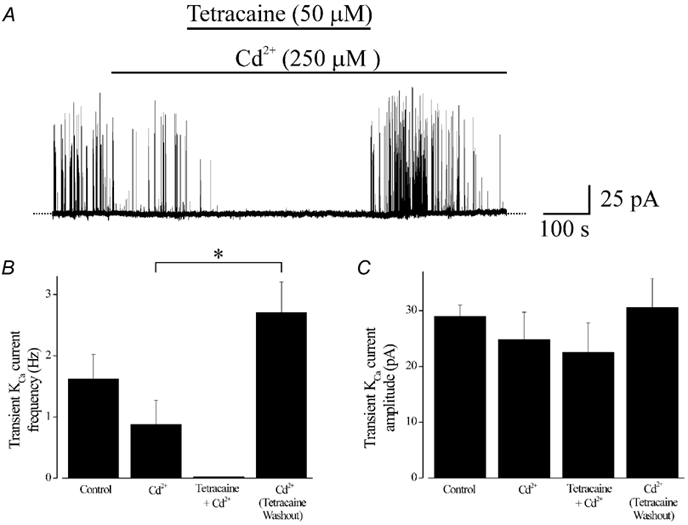
A, original record illustrating transient KCa currents in a cerebral artery smooth muscle cell voltage-clamped at −40 mV using the perforated-patch configuration. Cd2+ (250 μM) reduced transient KCa current frequency. Tetracaine (50 μM, 7 min application), applied in the continued presence of Cd2+, further reduced transient KCa currents. Immediately after tetracaine washout, transient KCa current frequency was significantly higher than before tetracaine application, although transient KCa current amplitude was not significantly different. Transient KCa current frequency returned to pre-tetracaine levels approximately 5 min after tetracaine washout. Average effects of Cd2+ (250 μM), tetracaine (50 μM) + Cd2+, and 1–3 min after tetracaine washout + Cd2+ on transient KCa current frequency (B) and amplitude (C). *P < 0.05; Student-Newman-Keuls test.
These data demonstrate that tetracaine blocks transient KCa currents. After washout of tetracaine, when [Ca2+]SR would have increased (Fig. 3), transient KCa current frequency was significantly higher than prior to tetracaine application, although transient KCa current amplitude was not different. Because tetracaine washout does not alter cytosolic [Ca2+]i (Fig. 3), these data suggest that an acute elevation in [Ca2+]SR increases transient KCa current frequency, but not amplitude. Furthermore, the elevation in transient KCa current frequency was short lived after tetracaine washout, suggesting that elevated Ca2+ release from the SR leads to a return of [Ca2+]SR and transient KCa current frequency to pre-tetracaine levels.
Tetracaine washout elevates Ca2+ spark frequency, but not amplitude
We sought to investigate the effect of an acute elevation in [Ca2+]SR on Ca2+ sparks and transient KCa currents. Essentially 100 % of Ca2+ sparks induce a transient KCa current in rat cerebral artery smooth muscle cells (Perez et al. 1999). Therefore, our finding that an elevation in [Ca2+]SR elevates transient KCa current frequency suggests that this occurs via an increase in Ca2+ spark frequency. However, an acute increase in [Ca2+]SR could conceivably elevate Ca2+ spark amplitude via activation of RyR channels (Herrmann-Frank & Lehmann-Horn, 1996; Tripathy & Meissner, 1996; Gyorke & Gyorke, 1998; Xu & Meissner, 1998; Ching et al. 2000) and by increasing the driving force for Ca2+ from the SR. A significant proportion of Ca2+ sparks do not evoke a transient KCa current in human cerebral artery (28 %, Wellman et al. 2002), feline oesophageal (27 %, Kirber et al. 2001) or Bufo marinus stomach (21 %, ZhuGe et al. 2000) smooth muscle cells. If Ca2+ spark:transient KCa current coupling is not 1:1, an increase in Ca2+ spark amplitude may augment this coupling ratio, which would also increase transient KCa current frequency. Therefore, we measured the effect of an elevation in [Ca2+]SR on Ca2+ spark and evoked transient KCa current properties, and the coupling ratio and amplitude relationship between these events. Simultaneous measurements of Ca2+ sparks and transient KCa currents were made in the same cells before tetracaine, and 90 s after tetracaine washout (50 μM, 7 min application), in the continued presence of diltiazem (50 μM).
Prior to tetracaine application, 100 % of detected Ca2+ sparks evoked a transient KCa current (n= 4 cells, n= 33 sparks) and 71.7 % of transient KCa currents were associated with a Ca2+ spark, suggesting that some sparks occurred outside the cytosolic volume detected by the confocal microscope (Fig. 5). The amplitude of a Ca2+ spark and that of the evoked transient KCa current were significantly correlated (P < 0.0001) (Fig. 6). In the same cells, tetracaine significantly reduced transient KCa current frequency, consistent with effects in non-fluo-4 loaded cells (Fig. 4). After tetracaine washout, when [Ca2+]SR would have increased, mean Ca2+ spark frequency was ≈2.7-fold higher than prior to tetracaine application (Fig. 5, n= 88 sparks). In contrast, mean Ca2+ spark amplitude (Fig. 5), half-time for decay (t1/2; control, 52 ± 9 vs. tetracaine washout, 57 ± 6 ms) and spatial spread (FWHM; control, 2.23 ± 0.15 vs. tetracaine washout, 1.95 ± 0.17 μm) were not different (P > 0.05 for each). Consistent with effects in non-fluo-4 loaded cells, mean transient KCa current amplitude (control, 23.8 ± 2.8 vs. tetracaine washout, 23.1 ± 4.6 pA) also did not change (P > 0.05). After tetracaine washout, 100 % of Ca2+ sparks evoked a transient KCa current and 68.2 % of transient KCa currents were associated with a Ca2+ spark, suggesting that an elevation in [Ca2+]SR did not change the coupling ratio between sparks and transient KCa currents. Furthermore, tetracaine washout did not significantly alter the amplitude correlation between Ca2+ sparks and evoked transient KCa currents (Fig. 6). Global F/F0 was not altered by washout of tetracaine (99 ± 6 % of pre-tetracaine), suggesting that Ca2+ sparks were activated via an elevation in [Ca2+]SR and not via an increase in cytosolic [Ca2+]i, and that the increase in Ca2+ spark frequency did not elevate global cytosolic [Ca2+]i. These data suggest that an elevation in [Ca2+]SR increases Ca2+ spark frequency, but does not alter Ca2+ spark amplitude, decay, spatial spread, or the coupling ratio or amplitude correlation between sparks and transient KCa currents.
Figure 5. Tetracaine washout elevates Ca2+ spark frequency.
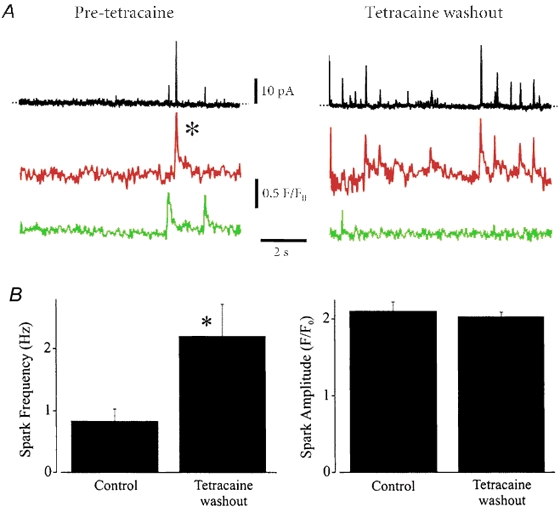
A, original simultaneous recordings of Ca2+ sparks and transient KCa currents in a voltage-clamped (-40 mV) cerebral artery smooth muscle cell. The black line illustrates whole cell K+ current. Red and green lines illustrate fluorescence changes (F/F0) measured in two different 1.54 μm × 1.54 μm (i.e. 2.37 μm2) areas of the cell in which Ca2+ sparks occurred. Traces indicate activity before tetracaine (50 μM) application (pre-tetracaine) and 90 s after tetracaine washout. Tetracaine was applied for 7 min. The experiment was performed in the continued presence of diltiazem (50 μM). B, summary of Ca2+ spark frequency and amplitude pre-tetracaine (n= 33 sparks, n= 4 cells) and after tetracaine washout (n= 88 sparks). *P < 0.05; Student's paired t test.
Figure 6. Tetracaine washout does not alter the amplitude correlation between Ca2+ sparks and transient KCa currents.
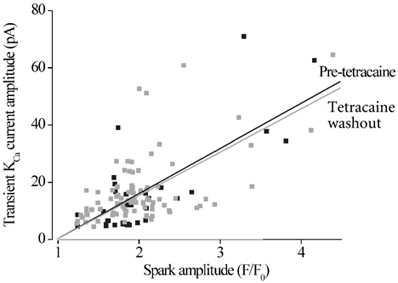
Scatter plot of Ca2+ spark and evoked transient KCa current amplitude at −40 mV before tetracaine application (pre-tetracaine, black), and after tetracaine washout (grey). The amplitudes of Ca2+ sparks and evoked transient KCa currents were significantly correlated, both for pre-tetracaine (P < 0.0001) and tetracaine washout data (P < 0.0001). Correlation co-efficients (r) were: control, 0.55; tetracaine washout, 0.58. The amplitude correlations before, and after, tetracaine were not significantly different from each other (P= 0.79).
Thapsigargin progressively decreases SR Ca2+ load in cerebral arteries
If an elevation in [Ca2+]SR increases Ca2+ spark frequency in arterial smooth muscle cells, then a decrease in [Ca2+]SR should decrease Ca2+ spark frequency. Furthermore, because the [Ca2+]SR establishes the driving force for Ca2+ from the SR, a decrease in [Ca2+]SR may also reduce Ca2+ spark amplitude. To investigate the effect of decreasing [Ca2+]SR on Ca2+ sparks and transient KCa currents, thapsigargin, a selective blocker of the SR Ca2+-ATPase, was used. In smooth muscle cells, thapsigargin has most commonly been applied at a high concentration (≈100 nm), to rapidly deplete the SR of Ca2+ (e.g. see Jaggar, 2001). To determine the effect of a low concentration of thapsigargin (20 nm) on [Ca2+]SR, caffeine-induced [Ca2+]i transients were measured in control, and 5, 10 and 15 min after application of thapsigargin (20 nm) in the same endothelium-denuded cerebral artery segments.
Thapsigargin (20 nm) induced a time-dependent decrease in caffeine-induced [Ca2+]i transients (Fig. 7, n= 9 arteries). Five, ten and fifteen minutes after addition of thapsigargin, caffeine-induced [Ca2+]i transients decreased to ≈76, ≈57 and ≈41 % of control, respectively. In contrast, 5, 10 and 15 min after thapsigargin application, steady-state arterial wall [Ca2+]i was 99 ± 2, 101 ± 2 and 102 ± 2 nm, respectively, which were not significantly different from control (P > 0.05 for each, Fig. 7). To ensure that [Ca2+]SR was reduced by thapsigargin, and not by repetitive addition of caffeine or insufficient recovery of [Ca2+]SR after each caffeine application, caffeine-induced [Ca2+]i transients were measured in six separate arteries in the absence of thapsigargin using the same time protocol. In the absence of thapsigargin, caffeine-induced [Ca2+]i transients were not significantly different at 0, 5, 10 or 15 min (P > 0.05 for each, Fig. 7). In these control arteries, arterial wall [Ca2+]i also did not change over the same time course (0 min, 99 ± 3 nm; 15 min, 96 ± 5 nm, P > 0.05). These data demonstrate that a low concentration of thapsigargin (20 nm) progressively decreases [Ca2+]SR in cerebral arteries, and that significant depletion of [Ca2+]SR does not alter arterial wall [Ca2+]i.
Figure 7. Thapsigargin reduces [Ca2+]SR in cerebral arteries.
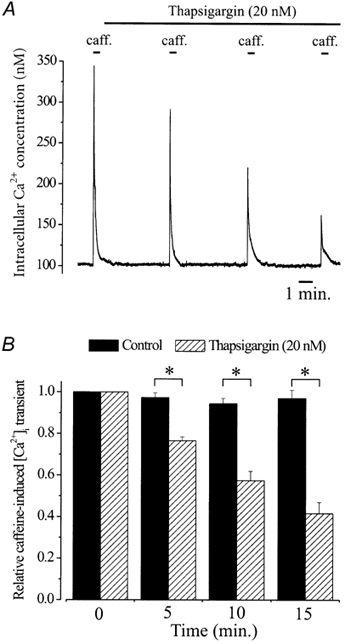
A, intracellular Ca2+ concentration and caffeine (20 mm)-induced [Ca2+]i transients in control and 5, 10 and 15 min after thapsigargin application (20 nm) in an endothelium-denuded cerebral artery segment. B, relative time-dependent change in caffeine-induced [Ca2+]i transients when compared with control in the absence (black bars) or presence of thapsigargin (20 nm, hatched bars). Mean caffeine-induced [Ca2+]i transients for each time point were (control vs. thapsigargin): 0 min, 240 ± 11 vs. 245 ± 14 nm; 5 min, 235 ± 8 vs. 184 ± 10 nm; 10 min, 229 ± 8 vs. 136 ± 7 nm; 15 min, 234 nm± 9 vs. 98 ± 8 nm. *P < 0.05 when compared using unpaired Student's t test.
Thapsigargin progressively decreases Ca2+ spark frequency, amplitude and spatial spread
We sought to examine the effect of progressively decreasing [Ca2+]SR on Ca2+ sparks and transient KCa currents. Simultaneous measurements of Ca2+ sparks and transient KCa currents were made in the same voltage-clamped (-40 mV) cerebral artery smooth muscle cells in control, and 5, 10 and 15 min after addition of thapsigargin (20 nm).
In control, 98.2 % of detected Ca2+ sparks evoked a transient KCa current (Fig. 8, Table 1, n= 55 sparks, n= 6 cells). The amplitudes of coupled Ca2+ sparks and evoked transient KCa currents were correlated (Fig. 8). In control, one Ca2+ spark out of a total of 56 did not evoke a transient KCa current. The amplitude (F/F0) of this uncoupled Ca2+ spark was 1.26, which was significantly smaller than the mean amplitude of coupled sparks (Table 1), and is only just larger than our designated threshold for a Ca2+ spark, which is an F/F0 greater than 1.2. In addition, the spatial spread of the uncoupled spark was smaller (0.96 μm) and the decay was faster (18.0 ms) than the mean value for coupled sparks (Table 1). In the same cells, thapsigargin (20 nm) induced a time-dependent decrease in mean Ca2+ spark frequency, amplitude and spatial spread, which reduced transient KCa current frequency and amplitude (Table 1). Five, ten or fifteen minutes after addition of thapsigargin, 100 % of Ca2+ sparks evoked a transient KCa current, suggesting that uncoupling of Ca2+ sparks from KCa channels did not occur, even when mean spark amplitude and spread were reduced to 47 and 56 % of control, respectively (Table 1). The amplitudes of Ca2+ sparks and transient KCa currents were correlated at 5, 10 or 15 min after thapsigargin (Fig. 8). Furthermore, the amplitude correlation for each time point after thapsigargin addition was not significantly different when compared with the amplitude correlation in control (Fig. 8). These data suggest that although a decrease in [Ca2+]SR reduces Ca2+ spark amplitude and spatial spread, the amplitude correlation between a spark and a transient KCa current is maintained (Fig. 8). Thapsigargin did not change global F/F0 (103 ± 5 % of control at 15 min, P > 0.05), suggesting that all modifications in Ca2+ sparks were due to a decrease in [Ca2+]SR and not due to alterations in cytosolic [Ca2+]i.
Figure 8. Regulation of Ca2+ sparks and transient KCa currents by thapsigargin.
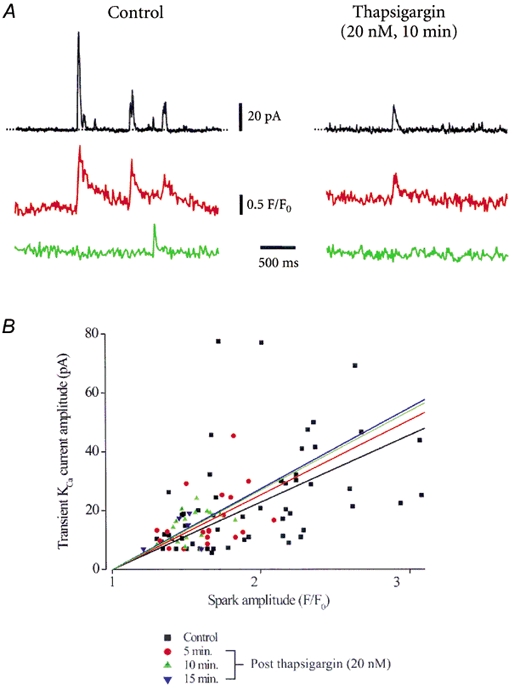
A, original simultaneous recordings of Ca2+ sparks (red and green lines) and whole-cell current (black lines) in the same voltage-clamped (-40 mV) cerebral artery smooth muscle cell in control and 10 min after thapsigargin (20 nm) application. B, the amplitudes of Ca2+ sparks and evoked transient KCa currents were correlated for control (P= 0.002), and data obtained 5 (P= 0.04), 10 (P < 0.0001), or 15 (P < 0.0001) min after thapsigargin. Linear correlation coefficients (r) were: control (black), 0.46; thapsigargin: 5 min (red), 0.41; 10 min (green), 0.38; 15 min (blue), 0.32. Amplitude correlations for each time point after thapsigargin application were not significantly different when compared with control (significance values: 5 min, P= 0.76; 10 min, P= 0.94; 15 min, P= 0.83).
Table 1.
Summary of the effects of thapsigargin on Ca2+ spark and transient KCa currents
| Post thapsigargin (20 nM)time (min) | ||||
|---|---|---|---|---|
| Control | 5 | 10 | 15 | |
| Ca2+ sparks | ||||
| Frequency (Hz) | 1.0 ± 0.23 (6 cells) | 0.43 ± 0.14 (6 cells)* | 0.32 ± 0.15 (6 cells)* | 0.16 ± 0.07 (6 cells)* |
| Amplitude (F/F0) | 1.97 ± 0.07 (55) | 1.65 ± 0.05 (19)* | 1.55 ± 0.03 (16)* | 1.46 ± 0.07 (5)* |
| Decay (t1/2, ms)† | 40.9 ± 4.4 (29) | 35.6 ± 2.5 (14) | 32.6 ± 3.1 (12) | 30.6 ± 2.8 (5) |
| Spread (FWHM, μm) | 2.11 ± 0.19 (18) | 2.07 ± 0.08 (11) | 1.21 ± 0.11 (11)* | 1.18 ± 0.21 (11)* |
| KCa transients | ||||
| Amplitude (pA) | 23.7 ± 2.4 (61) | 17.0 ± 2.3 (36)* | 15.0 ± 1.3 (29)* | 12.2 ± 2.6 (11)* |
| Decay (t1/2, ms)‡ | 16.6 ± 0.9 (61) | 15.8 ± 0.9 (36) | 15.2 ± 0.8 (29) | 14.8 ± 1.0 (11) |
| Rise time (10–90%,ms) | 11.8 ± 0.6 (61) | 11.5 ± 0.7 (36) | 11.7 ± 0.7 (29) | 10.7 ± 1.0 (11) |
All Ca2+ spark data refer to events associated with transient KCa currents. Only one Ca2+ spark was detected in this series of experiments that did not evoke a transient KCa current and this spark occurred in control conditions. Transient KCa current data also includes events for which an associated Ca2+ spark was not detected. Values of n shown in parentheses refer to number of sparks or transient KCa currents used for analysis, unless specified otherwise.
Ca2+ spark decays were fit with a second-order exponential.
Transient KCa current decays were fit with a first-order exponential. t1/2: time for decay to half-maximal amplitude. FWHM: full width at half maximal amplitude.
P < 0.05 when compared with control; Student-Newman-Keuls test.
To investigate if the repetitive confocal acquisition protocol was responsible for the observed changes in Ca2+ sparks and transient KCa currents, the same imaging procedure was performed in voltage-clamped (-40 mV) cerebral artery smooth muscle cells in the absence of thapsigargin. In six cerebral artery smooth muscle cells, Ca2+ spark frequency was not different at 0 min (first laser exposure, 0.76 ± 0.1 Hz) and 15 min (fourth laser exposure, 0.81 ± 0.1 Hz, P > 0.05 for each). Similarly, Ca2+ spark amplitude (F/F0; 0 min, 2.21 ± 0.16 n= 48; 15 min, 2.15 ± 0.11, n= 51) and transient KCa current amplitude (0 min, 18.8 ± 2.2 pA; 15 min, 17.6 ± 2.2 pA) were unchanged (P > 0.05 for each). These data indicate that the repetitive confocal Ca2+ imaging protocol did not induce changes in Ca2+ sparks and transient KCa currents. In summary, progressively decreasing [Ca2+]SR reduced Ca2+ spark frequency, amplitude and spatial spread, which reduced transient KCa current frequency and amplitude. However, even when Ca2+ spark amplitude and spread were reduced by 47 and 56 % of control, respectively, the coupling ratio or the amplitude correlation between Ca2+ sparks and transient KCa currents was not altered.
DISCUSSION
We demonstrate that an acute increase or decrease in [Ca2+]SR regulates Ca2+ sparks and evoked transient KCa currents in cerebral artery smooth muscle cells. An elevation in [Ca2+]SR increased Ca2+ spark and transient KCa current frequency, but did not change Ca2+ spark amplitude, spatial spread or decay, or the coupling ratio or amplitude correlation between sparks and transient KCa currents. Decreasing [Ca2+]SR reduced Ca2+ spark frequency, amplitude and spatial spread, and these effects reduced the frequency and amplitude of evoked transient KCa currents, although the coupling ratio or amplitude correlation between Ca2+ sparks and transient KCa currents was not affected.
An elevation in [Ca2+]SR increases Ca2+ spark frequency in arterial smooth muscle cells
[Ca2+]SR was elevated in cerebral artery smooth muscle cells by: (1) selectively increasing the activity of the SR Ca2+-ATPase, or (2) blocking RyR channels. To activate the SR Ca2+-ATPase we used a monoclonal antibody raised against phospholamban that elevates [Ca2+]SR in cardiac myocytes (Suzuki & Wang, 1986; Sham et al. 1991; Lukyanenko et al. 2001). The anti-phospholamban antibody increased mean transient KCa current frequency, but did not alter transient KCa current amplitude. [Ca2+]SR was also elevated with tetracaine, a reversible RyR channel blocker. Local anaesthetics block RyR channels by inducing a long closed state rather than by reducing the single channel conductance or the mean open times of the channel (Zahradnikova & Palade, 1993). Tetracaine was applied in the continued presence of a voltage-dependent Ca2+ channel blocker, to prevent any non-specific effects on voltage-dependent Ca2+ channels that would also regulate Ca2+ sparks (Jaggar et al. 1998; Jaggar, 2001). After tetracaine washout when [Ca2+]SR had increased, Ca2+ sparks and transient KCa currents were significantly more frequent than prior to tetracaine application, but Ca2+ spark amplitude, decay and spatial spread were not altered, nor were transient KCa current amplitude or the coupling ratio or amplitude correlation between Ca2+ sparks and transient KCa currents. [Ca2+]SR and transient KCa current frequency returned to control levels ≈5 min after tetracaine washout, presumably due to the elevation in Ca2+ release.
Ca2+ spark amplitude should depend on the number and activity of contributing RyR channels and the driving force for Ca2+ from the SR. However, although Ca2+ sparks in smooth muscle cells exhibit a wide range of amplitudes (see Fig. 6, Jaggar et al. 2000; ZhuGe et al. 2000), data suggest that an acute elevation in [Ca2+]SR did not change mean Ca2+ spark amplitude. Conceivably, this could be because resting [Ca2+]SR saturates Ca2+ flux through RyR channels. If this is the case, the SR Ca2+-ATPase maintains this saturating [Ca2+]SR at a cytosolic [Ca2+]i of ≈100 nm, and the wide range of Ca2+ spark amplitudes that occur at this saturating [Ca2+]SR may be due to different numbers and/or activities of RyR channels that contribute to sparks. The decline in mean Ca2+ spark amplitude and spatial spread that occurs when [Ca2+]SR is reduced could also be due to a decrease in the driving force for Ca2+ from the SR in addition to a reduction in the number or activity of contributing RyR channels. However, mechanisms that regulate Ca2+ spark amplitude may be more complex and may involve multiple processes, particularly since Ca2+ activation and inactivation sites may be located on the luminal (Ching et al. 2000) and cytosolic side (Herrmann-Frank et al. 1991; Xu et al. 1994; Herrmann-Frank & Lehmann-Horn, 1996; Tripathy & Meissner, 1996) of RyR channels. In addition, during a Ca2+ spark, the localized free Ca2+ concentration may partially deplete near the luminal side of RyR channels, and this may also influence Ca2+ spark amplitude.
Our finding that an acute increase in SR Ca2+ load elevates Ca2+ spark frequency but mean Ca2+ spark amplitude does not change is consistent with findings in arterial smooth muscle cells of a phospholamban-deficient mouse, wherein SR Ca2+ load is chronically elevated (Wellman et al. 2001). However, modifications in Ca2+ signalling modalities in smooth muscle cells regulate the transcription factor, CREB, the immediate-early gene, c-fos (Cartin et al. 2000) and the transcription factor, NFAT (Stevenson et al. 2001). Conceivably, long-term modifications in Ca2+ signalling modalities, particularly during development, could modify the expression levels of Ca2+ signalling proteins, such as RyR channels, and this could have led to the observation that a chronic elevation in SR Ca2+ load does not change Ca2+ spark amplitude. Forskolin, an activator of adenylyl cyclase, also elevates SR load and Ca2+ spark frequency in arterial smooth muscle cells, but does not change mean spark amplitude (Porter et al. 1998). However, cAMP-dependent protein kinase also activates RyR channels (Porter et al. 1998; Wellman et al. 2001), which could have masked a change in Ca2+ spark amplitude. Our data, which examined the regulation of Ca2+ sparks and transient KCa current by an acute elevation in SR Ca2+ load, also suggest that frequency is increased, but amplitude does not change. When comparing our data with that in previous studies, findings suggest that an acute, or chronic, elevation in [Ca2+]SR elevates Ca2+ spark frequency, but the amplitude and decay of Ca2+ sparks are unchanged.
A decrease in [Ca2+]SR reduces Ca2+ spark frequency, amplitude and spatial spread
Thapsigargin induced a progressive reduction in [Ca2+]SR and Ca2+ spark frequency, amplitude and spatial spread, and these changes decreased transient KCa current frequency and amplitude. Presumably, transient KCa current amplitude was reduced because: (1) a lower amplitude Ca2+ spark would be a less effective activator of KCa channels, and (2) a smaller spark spread would envelop a reduced sarcolemmal surface area and impact a lower number of KCa channels. The thapsigargin-induced decrease in Ca2+ spark amplitude (47 % of control) and spatial spread (56 % of control) did not induce uncoupling of Ca2+ sparks from KCa channels. In this study, out of a total of 216 sparks, only one uncoupled Ca2+ spark was detected, suggesting a coupling ratio of sparks to transient KCa currents of 99.5 %, which is similar to previous reports in rat cerebral artery smooth muscle cells (Perez et al. 1999). The amplitude (F/F0) and spatial spread of this uncoupled spark were significantly smaller than the mean value for coupled sparks (Table 1), which may partially explain why it did not evoke a transient KCa current. Mechanisms that lead to uncoupling are unclear, but may involve a reduced density (ZhuGe et al. 2000) or Ca2+ sensitivity (Bayguinov et al. 2001) of KCa channels, in addition to the properties of the Ca2+ release event. The variability in coupling ratio that occurs in different smooth muscle preparations (ZhuGe et al. 2000; Kirber et al. 2001; Wellman et al. 2002) may reflect species- and tissue-specific diversity in Ca2+ signalling between RyR channels and KCa channels.
In cardiac myocytes, Ca2+ sparks decay primarily due to diffusion, but some of the released Ca2+ is also sequestered into the SR by the SR Ca2+-ATPase (Gomez et al. 1996; Santana et al. 1997). In our experiments, partially blocking the SR Ca2+-ATPase reduced spark amplitude and spread, and slightly decreased half-time for decay (t1/2). If the SR Ca2+-ATPase sequestered a portion of the released Ca2+, thapsigargin should have prolonged decay (t1/2), which was not observed. In experiments with tetracaine where [Ca2+]SR was increased, spark spread and decay were unaltered. This finding suggests that decreasing the driving force for SR Ca2+ uptake also has no effect on spark decay. Thus, the SR Ca2+-ATPase does not appear to contribute directly to spark decay by sequestering the released Ca2+ into the SR. Data suggest that amplitude largely determines the decay of a spark, since spread and decay (t1/2) decreased when amplitude was reduced (Table 1). Possible explanations for a correlation between amplitude and spread/decay may be that spark diffusion is space limited or that sparks saturate cytosolic Ca2+ buffers within the immediate vicinity of the release site. Thus, lower amplitude sparks would decay more rapidly and would exhibit lower spread due to a higher ratio of cytosolic buffering molecules to Ca2+.
A decrease in [Ca2+]SR elevates sarcolemmal Ca2+ entry in a variety of cell types via activation of store-operated Ca2+ channels, which are also referred to as capacitative Ca2+ entry channels or Ca2+-release activated Ca2+ (CRAC) channels (Parekh & Penner, 1997). Although a store-operated Ca2+ entry pathway has been demonstrated in arterial smooth muscle (Xu & Beech, 2001), this observation is not universal (e.g. see Knot et al. 1998). In the present study, a decrease or an increase in [Ca2+]SR did not alter cytosolic [Ca2+]i (Fig. 3 and Fig. 7), suggesting that store-operated Ca2+ entry is not significant in rat cerebral artery smooth muscle cells, at least in the conditions used in our experiments.
Physiological relevance of modifications in [Ca2+]SR in arterial smooth muscle cells
An elevation in [Ca2+]SR that increases Ca2+ spark and transient KCa current frequency should lead to membrane hyperpolarization, a decrease voltage-dependent Ca2+ channel activity, a reduction in global [Ca2+]i, and dilation (Jaggar et al. 2000). An elevation in Ca2+ spark frequency may also increase the driving force for Ca2+ for sarcolemma extrusion mechanisms that are located within the vicinity of the release site, such as the Na+-Ca2+ exchanger or the Ca2+-ATPase. A similar mechanism, referred to as the ‘superficial buffer barrier hypothesis’ (van Breemen et al. 1995), suggests that Ca2+ entering smooth muscle cells is buffered by the SR and is discharged vectorially towards the sarcolemma without any effect on global [Ca2+]i. Elevating Ca2+ release from the SR may also enhance Ca2+-dependent inactivation of sarcolemma voltage-dependent Ca2+ channels, an additional mechanism that would further reduce Ca2+ entry. Thus, an increase in [Ca2+]SR may be intimately involved in directly regulating a number of negative feedback mechanisms via activation of Ca2+ sparks. Conversely, a decrease in [Ca2+]SR would reduce Ca2+ spark frequency, amplitude and spatial spread, which would have the opposite effect.
In summary, our data suggest that an elevation in [Ca2+]SR increases Ca2+ spark and transient KCa current frequency in cerebral artery smooth muscle cells, but Ca2+ spark amplitude, spatial spread or decay, or the coupling ratio or amplitude correlation between sparks and transient KCa currents are not altered. A decrease in [Ca2+]SR reduces Ca2+ spark frequency, amplitude and spatial spread, and the frequency and amplitude of evoked transient KCa currents, although the coupling ratio or amplitude correlation between sparks and transient KCa currents remains unchanged. These findings demonstrate that acute changes in [Ca2+]SR regulate Ca2+ sparks and transient KCa currents in cerebral artery smooth muscle cells.
Acknowledgments
We would like to thank Dr Charles W. Leffler and Xiaoyang Cheng for helpful comments on the manuscript. This study was supported by grants (to J.H.J.) from the National Institutes of Health (HL67061-01) and American Heart Association National Center (0130190N) and Southeast Affiliate (0060331B).
REFERENCES
- Bayguinov O, Hagen B, Kenyon JL, Sanders KM. Coupling strength between localized Ca2+ transients and K+ channels is regulated by protein kinase C. American Journal of Physiology - Cell Physiology. 2001;281:C1512–1523. doi: 10.1152/ajpcell.2001.281.5.C1512. [DOI] [PubMed] [Google Scholar]
- Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells of the rabbit. Journal of Physiology. 1986;381:385–406. doi: 10.1113/jphysiol.1986.sp016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Elementary and global aspects of calcium signalling. Journal of Physiology. 1997;499:290–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton TB, Gordienko DV. Confocal imaging of calcium release events in single smooth muscle cells. Acta Physiologica Scandinavica. 1998;164:567–575. doi: 10.1046/j.1365-201X.1998.00464.x. [DOI] [PubMed] [Google Scholar]
- Bonev AD, Jaggar JH, Rubart M, Nelson MT. Activators of protein kinase C decrease Ca2+ spark frequency in smooth muscle cells from cerebral arteries. American Journal of Physiology. 1997;273:C2090–2095. doi: 10.1152/ajpcell.1997.273.6.C2090. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Lipp P, Berridge MJ. The organisation and functions of local Ca2+ signals. Journal of Cell Science. 2001;114:2213–2222. doi: 10.1242/jcs.114.12.2213. [DOI] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Cartin L, Lounsbury KM, Nelson MT. Coupling of Ca2+ to CREB activation and gene expression in intact cerebral arteries from mouse: roles of ryanodine receptors and voltage-dependent Ca2+ channels. Circulation Research. 2000;86:760–767. doi: 10.1161/01.res.86.7.760. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Ching LL, Williams AJ, Sitsapesan R. Evidence for Ca2+ activation and inactivation sites on the luminal side of the cardiac ryanodine receptor complex. Circulation Research. 2000;87:201–206. doi: 10.1161/01.res.87.3.201. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Cheng H, Lederer WJ, Bers DM. Ca2+ diffusion and sarcoplasmic reticulum transport both contribute to [Ca2+]i decline during Ca2+ sparks in rat ventricular myocytes. Journal of Physiology. 1996;496:575–581. doi: 10.1113/jphysiol.1996.sp021708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophysical Journal. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke S, Lukyanenko V, Gyorke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. Journal of Physiology. 1997;500:297–309. doi: 10.1113/jphysiol.1997.sp022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Herrmann-Frank A, Darling E, Meissner G. Functional characterization of the Ca2+-gated Ca2+ release channel of vascular smooth muscle sarcoplasmic reticulum. Pflügers Archiv. 1991;418:353–359. doi: 10.1007/BF00550873. [DOI] [PubMed] [Google Scholar]
- Herrmann-Frank A, Lehmann-Horn F. Regulation of the purified Ca2+ release channel/ryanodine receptor complex of skeletal muscle sarcoplasmic reticulum by luminal calcium. Pflügers Archiv. 1996;432:155–157. doi: 10.1007/s004240050117. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. Journal of General Physiology. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. American Journal of Physiology - Cell Physiology. 2001;281:C439–448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Nelson MT. Differential regulation of Ca2+ sparks and Ca2+ waves by UTP in rat cerebral artery smooth muscle cells. American Journal of Physiology - Cell Physiology. 2000;279:C1528–1539. doi: 10.1152/ajpcell.2000.279.5.C1528. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. American Journal of Physiology - Cell Physiology. 2000;278:C235–256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Stevenson AS, Nelson MT. Voltage dependence of Ca2+ sparks in intact cerebral arteries. American Journal of Physiology. 1998;274:C1755–1761. doi: 10.1152/ajpcell.1998.274.6.C1755. [DOI] [PubMed] [Google Scholar]
- Kirber MT, Etter EF, Bellve KA, LifshitZ LM, Tuft RA, Fay FS, Walsh JV, Fogarty KE. Relationship of Ca2+ sparks to STOCs studied with 2D and 3D imaging in feline oesophageal smooth muscle cells. Journal of Physiology. 2001;531:315–327. doi: 10.1111/j.1469-7793.2001.0315i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. Journal of Physiology. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. Journal of Physiology. 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Viatchenko-Karpinski S, Smirnov A, Wiesner TF, Gyorke S. Dynamic regulation of sarcoplasmic reticulum Ca2+ content and release by luminal Ca2+-sensitive leak in rat ventricular myocytes. Biophysical Journal. 2001;81:785–798. doi: 10.1016/S0006-3495(01)75741-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Perez GJ, Bonev AD, Nelson MT. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. American Journal of Physiology - Cell Physiology. 2001;281:C1769–1775. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- Perez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. Journal of General Physiology. 1999;113:229–238. doi: 10.1085/jgp.113.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter VA, Bonev AD, Knot HJ, Heppner TJ, Stevenson AS, Kleppisch T, Lederer WJ, Nelson MT. Frequency modulation of Ca2+ sparks is involved in regulation of arterial diameter by cyclic nucleotides. American Journal of Physiology. 1998;274:C1346–1355. doi: 10.1152/ajpcell.1998.274.5.C1346. [DOI] [PubMed] [Google Scholar]
- Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. Journal of Physiology. 1997;503:21–29. doi: 10.1111/j.1469-7793.1997.021bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. American Journal of Physiology. 1997;272:H657–668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- Sham JS, Jones LR, Morad M. Phospholamban mediates the beta-adrenergic-enhanced Ca2+ uptake in mammalian ventricular myocytes. American Journal of Physiology. 1991;261:H1344–1349. doi: 10.1152/ajpheart.1991.261.4.H1344. [DOI] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca2+-release channel by luminal Ca2+ Journal of Membrane Biology. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- Stevenson AS, GomeZ MF, Hill-Eubanks DC, Nelson MT. NFAT4 movement in native smooth muscle. A role for differential Ca2+ signaling. Journal of Biological Chemistry. 2001;276:15018–15024. doi: 10.1074/jbc.M011684200. [DOI] [PubMed] [Google Scholar]
- Sugiyama K, Muteki T. Local anesthetics depress the calcium current of rat sensory neurons in culture. Anesthesiology. 1994;80:1369–1378. doi: 10.1097/00000542-199406000-00025. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Wang JH. Stimulation of bovine cardiac sarcoplasmic reticulum Ca2+ pump and blocking of phospholamban phosphorylation and dephosphorylation by a phospholamban monoclonal antibody. Journal of Biological Chemistry. 1986;261:7018–7023. [PubMed] [Google Scholar]
- Tripathy A, Meissner G. Sarcoplasmic reticulum lumenal Ca2+ has access to cytosolic activation and inactivation sites of skeletal muscle Ca2+ release channel. Biophysical Journal. 1996;70:2600–2615. doi: 10.1016/S0006-3495(96)79831-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugorka A, Rios E, Blatter LA. Imaging elementary events of calcium release in skeletal muscle cells. Science. 1995;269:1723–1726. doi: 10.1126/science.7569901. [DOI] [PubMed] [Google Scholar]
- van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends in Pharmacological Sciences. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- Wellman GC, Nathan DJ, Saundry CM, PereZ G, Bonev AD, Penar PL, Tranmer BI, Nelson MT. Ca2+ sparks and their function in human cerebral arteries. Stroke. 2002;33:802–808. doi: 10.1161/hs0302.104089. [DOI] [PubMed] [Google Scholar]
- Wellman GC, Santana LF, Bonev AD, Nelson MT. Role of phospholamban in the modulation of arterial Ca2+ sparks and Ca2+-activated K+ channels by cAMP. American Journal of Physiology - Cell Physiology. 2001;281:C1029–1037. doi: 10.1152/ajpcell.2001.281.3.C1029. [DOI] [PubMed] [Google Scholar]
- Xu L, Jones R, Meissner G. Effects of local anesthetics on single channel behavior of skeletal muscle calcium release channel. Journal of General Physiology. 1993;101:207–233. doi: 10.1085/jgp.101.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Lai FA, Cohn A, Etter E, Guerrero A, Fay FS, Meissner G. Evidence for a Ca2+-gated ryanodine-sensitive Ca2+ release channel in visceral smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1994;91:3294–3298. doi: 10.1073/pnas.91.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Meissner G. Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum lumenal Ca2+ Biophysical Journal. 1998;75:2302–2312. doi: 10.1016/S0006-3495(98)77674-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circulation Research. 2001;88:84–87. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- Zahradnikova A, Palade P. Procaine effects on single sarcoplasmic reticulum Ca2+ release channels. Biophysical Journal. 1993;64:991–1003. doi: 10.1016/S0006-3495(93)81465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhuGe R, Fogarty KE, Tuft RA, LifshitZ LM, Sayar K, Walsh JV., Jr Dynamics of signaling between Ca2+ sparks and Ca2+-activated K+ channels studied with a novel image-based method for direct intracellular measurement of ryanodine receptor Ca2+ current. Journal of General Physiology. 2000;116:845–864. doi: 10.1085/jgp.116.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhuGe R, Tuft RA, Fogarty KE, Bellve K, Fay FS, Walsh JV. The influence of sarcoplasmic reticulum Ca2+ concentration on Ca2+ sparks and spontaneous transient outward currents in single smooth muscle cells. Journal of General Physiology. 1999;113:215–228. doi: 10.1085/jgp.113.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]