

Abstract
Adenosine, prostaglandins (PG) and nitric oxide (NO) have all been implicated in hypoxia-evoked vasodilatation. We investigated whether their actions are interdependent. In anaesthetised rats, the PG synthesis inhibitors diclofenac or indomethacin reduced muscle vasodilatation evoked by systemic hypoxia or adenosine, but not that evoked by iloprost, a stable analogue of prostacyclin (PGI2), or by an NO donor. After diclofenac, the A1 receptor agonist CCPA evoked no vasodilatation: we previously showed that A1, but not A2A, receptors mediate the hypoxia-induced muscle vasodilatation. Further, in freshly excised rat aorta, adenosine evoked a release of NO, detected with an NO-sensitive electrode, that was abolished by NO synthesis inhibition, or endothelium removal, and reduced by ≈50 % by the A1 antagonist DPCPX, the remainder being attenuated by the A2A antagonist ZM241385. Diclofenac reduced adenosine-evoked NO release by ≈50 % under control conditions, abolished that evoked in the presence of ZM241385, but did not affect that evoked in the presence of DPCPX. Adenosine-evoked NO release was also abolished by the adenyl cyclase inhibitor 2′,5′-dideoxyadenosine, while dose-dependent NO release was evoked by iloprost. Finally, stimulation of A1, but not A2A, receptors caused a release of PGI2 from rat aorta, assessed by radioimmunoassay of its stable metabolite, 6-keto PGF1α, that was abolished by diclofenac. These results suggest that during systemic hypoxia, adenosine acts on endothelial A1 receptors to increase PG synthesis, thereby generating cAMP, which increases the synthesis and release of NO and causes muscle vasodilatation. This pathway may be important in other situations involving these autocoids.
It is generally accepted that adenosine plays a major role in vasodilatation evoked by hypoxia in several different tissues including skeletal muscle, heart and brain (Berne et al. 1983). Indeed, when the actions of adenosine are prevented in these tissues, hypoxia-induced dilatation is greatly reduced or even abolished (Berne et al. 1983; Bryan & Marshall, 1999a; Coney & Marshall, 1998; Nakhostine & Montagne, 1994). However, there is also substantial evidence that hypoxia-induced dilatation is mediated by newly synthesised prostaglandins (PGs): when PG synthesis is blocked hypoxia-induced dilatation is reduced or abolished (Busse et al. 1984; Fredricks et al. 1994b; Messina et al. 1992). This raises the possibility that adenosine- and PG-induced dilatation are synergistic, or that the dilatation induced by one is somehow dependent on the other.
The main aim of the present study was to investigate the possible interrelationships between adenosine and PGs more fully, by investigating the vasodilatation evoked in skeletal muscle of the rat by systemic hypoxia. This response we have previously attributed in part to adenosine acting on A1 but not A2A receptors, even though exogenous adenosine produces muscle vasodilatation by stimulating both A1 and A2A receptors (Bryan & Marshall, 1999a). Experiments described herein produced the novel finding that a large part of the hypoxia-induced muscle vasodilatation that can be attributed to adenosine is dependent on PG synthesis. Moreover, in agreement with this, muscle vasodilatation evoked by exogenous adenosine was greatly reduced by inhibition of PG synthesis. Since there is evidence, including our own, that adenosine causes vasodilatation, at least in part, by releasing NO from the endothelium (e.g. Merkel et al. 1992; Vials & Burnstock, 1993; Skinner & Marshall, 1996; Danialou et al. 1997; Bryan & Marshall, 1999b), we investigated the interrelationships between adenosine and PG synthesis on freshly excised arterial vessels in which we directly measured the release of NO. The results showed directly that adenosine does indeed release NO and PG from vascular endothelium. They also showed for the first time that the release of NO caused by the action of adenosine on A1 receptors is dependent on PG synthesis.
METHODS
All experiments were performed in accordance with the UK Animals (Scientific Procedures) Act 1986.
In vivo studies
Experiments were performed on 41 male Wistar rats (body weight 227.5 ± 3.9 g, mean ±s.e.m.) anaesthetised with Saffan (7-12 mg kg−1 h−1i.v., Plough Animal Health, UK) using techniques that have been described before (Bryan & Marshall, 1999a; Edmunds & Marshall, 2001a). Briefly, the animals spontaneously breathed 21 % O2 in N2 delivered by a gas rotameter system via a tracheal cannula, except when the response to systemic hypoxia was tested: they then breathed 8 % O2 in N2 for 5 min. Arterial samples (150 μl) taken from a brachial artery were analysed for arterial partial pressure of O2 (Pa,O2), CO2 (Pa,CO2) and pH; each sample was replaced with an equal volume of saline. Arterial blood pressure (ABP) was recorded from the right femoral artery. Pharmacological antagonists (see below) were given via the right femoral vein in a volume of ≈1 ml kg−1, while agonists were infused close-arterially into the left hind-limb via a cannula in the ventral tail artery. Femoral blood flow (FBF) was recorded from the left femoral artery using a 0.5 V Transonic flow probe and meter (T106, Transonic Systems Inc., Ithaca, NY, USA). ABP and FBF were sampled by a MacLab/8S at a frequency of 100 Hz and collected by a PowerMac 4400/200 computer by Chart (AD Instruments Ltd, Hastings, UK). Mean arterial pressure and heart rate (HR) were derived from the ABP signal and femoral vascular conductance (FVC) was computed on-line by division of FBF and ABP.
Protocols
Effects of diclofenac. In 18 rats, after a stabilisation period of 45 min, responses were evoked by 5 min periods of: (i) breathing 8 % O2, and/or close-arterial infusion into the left hind-limb of: (ii) adenosine (1 mg kg−1 min−1); (iii) SNP (sodium nitroprusside; 0.02 mg kg−1 min−1); and (iv) the stable PGI2 analogue iloprost (1 mg kg−1 min−1). The doses of adenosine, SNP and iloprost were each chosen so that they evoked increases in FVC of similar magnitude to that evoked by breathing 8 % O2. Each rat received no more than three of the four stimuli in random order at intervals of at least 5 min so that cardiovascular variables had time to stabilise.
The cyclooxygenase inhibitor diclofenac was then given at 1 mg kg−1i.v. and after 10–15 min the stimuli were repeated in random order. Assuming the drug was freely distributed in body water this dose was ≈3 times that used in perfused heart preparations in vitro (Nakhostine & Lamontagne, 1994), to completely block the release of PGs evoked by bradykinin, and to attenuate the associated coronary dilator responses. In our own preliminary studies, diclofenac given at >1 mg kg−1 had no greater effect. Arterial blood gases were analysed during air breathing (normoxia) before and after diclofenac and in the fifth minute of each period of 8 % O2 or agonist infusion.
Since the adenosine component of the hypoxia-evoked muscle vasodilatation is mediated via A1 receptors (Bryan & Marshall, 1999a), at the end of three of these experiments the A1 receptor agonist 2-chloro-N6-cyclopentyladenosine (CCPA) was infused at 0.35 μg kg−1 min−1i.a. In our earlier study (Bryan & Marshall, 1999a) this dose of CCPA evoked a long-lasting increase in FVC and a pronounced bradycardia that was completely reversed by a selective A1 receptor antagonist.
Effects of diclofenac followed by 8-sulphophenyltheophylline (8-SPT)
In five rats, responses evoked by 8 % O2 for 5 min were tested as described above before and after diclofenac (1 mg kg−1i.v.) and then after subsequent administration of the adenosine receptor antagonist 8-SPT (10 mg kg−1i.v.). This dose of 8-SPT virtually abolishes an adenosine-evoked increase in FVC comparable in magnitude with that evoked by hypoxia (Skinner & Marshall, 1996).
Effects of indomethacin
In six rats responses evoked by 8 % O2 for 5 min were tested before and 5 min after a different cyclooxygenase inhibitor: indomethacin at 5 mg kg−1i.v. A further dose of 5 mg kg−1i.v. was given and the response to 8 % O2 was re-tested.
Effects of 8-SPT followed by indomethacin
In 12 rats, responses evoked by breathing 8 % O2 were tested before and after 8-SPT (10 mg kg−1i.v.) and then after subsequent administration of indomethacin (5 mg kg−1i.v.).
At the end of these experiments, all animals were killed with an overdose of anaesthetic.
In vitro studies: NO recordings
The output of NO from the rat thoracic aorta was recorded continuously with a NO-sensitive electrode (ISO-NOP, WPI, FL, USA) with a 2 mm diameter tip, connected to a meter (ISO-NO Mark II, WPI), essentially as described by Guo et al. (1996) who demonstrated that this system is selective for NO. Lengths of thoracic aorta (10.6 ± 0.21 mm) were removed from 60 male Wistar rats (287.6 ± 3.6 g) immediately after they had been killed by cervical dislocation under anaesthesia achieved with 3.5 % halothane in O2. Each length of aorta was placed in Krebs solution containing (mm): 118 NaCl, 4.7 KCl, 1.5 CaCl2, 25 NaHCO3, 1.2 KH2PO4, 1.1 MgSO4, 10 Hepes and 5.6 glucose. It was opened longitudinally, care being taken to preserve the endothelium, and pinned, endothelial surface upwards, to a Petri dish covered with dental impression material (President, Coltene, NJ, USA). The dish was filled with 10 ml Krebs solution and placed on a magnetic stirrer. A magnetic flea placed in the Krebs solution at the edge of the dish facilitated mixing of drugs added to the dish by a micropipette, as shown in control experiments in which dye was added.
The experimental apparatus was then placed in a Faraday cage to reduce electrical noise. The electrode tip was placed as close as possible to the endothelial surface with the aid of a micromanipulator. The redox current produced at the electrode and recorded by the meter was passed across a resistor and recorded as a voltage change with a data acquisition system (MacLab/2e, AD Instruments Ltd, UK; sample rate: 10 Hz) connected to a computer (Power Macintosh 6100/60). The NO electrode was calibrated on each experimental day by chemical generation of NO according to the equation:
In our experiments, the daily calibration of the electrode (Fig. 1A and B) produced sensitivities that typically ranged from 0.12 to 0.8 mV nm−1 or 1.19 to 1.78 pA nm−1 over the 15 day life-time of a single electrode membrane. Guo et al. (1996) showed that this electrode system allowed reproducible NO measurements with successive calibrations; our calibrations showed similar reproducibility on and between successive experimental days.
Figure 1. Calibration of the NO electrode.
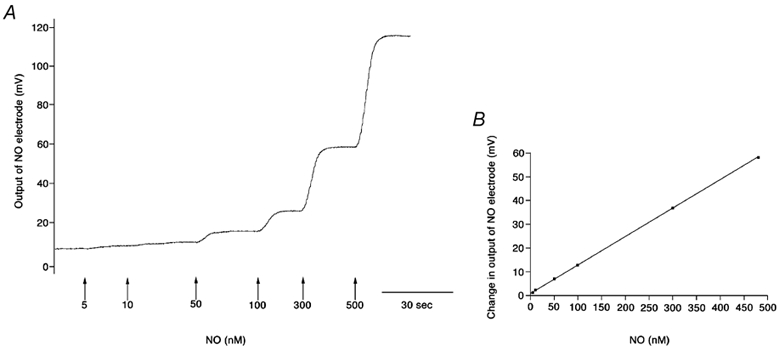
A, original trace showing recording of NO electrode calibration. Arrows represent concentration of NO generated on the addition of NaNO2 to 0.1 m KI and 0.1 m H2SO4 solution. B, linear regression analysis of relationship between NO generated and change in voltage output of electrode: y= 0.11x+ 0.14, R2= 0.99, electrode sensitivity 0.11 mV nm−1 or 1.10 pA nm−1.
Protocols
Group 1
The NO release evoked by graded concentrations of adenosine, 10 μM to 5 mm, was recorded by adding appropriate volumes of a stock solution of adenosine. At least 5 min was allowed between additions so that the response to adenosine was completed and the electrode output stabilised again (see Fig. 4A). Responses were measured as the maximum change in NO output from the electrode at each concentration of adenosine. This protocol produced essentially a non-cumulative dose-response curve.
Figure 4. Responses evoked by adenosine in the absence (A) and presence (B) of EHNA and NBTI.
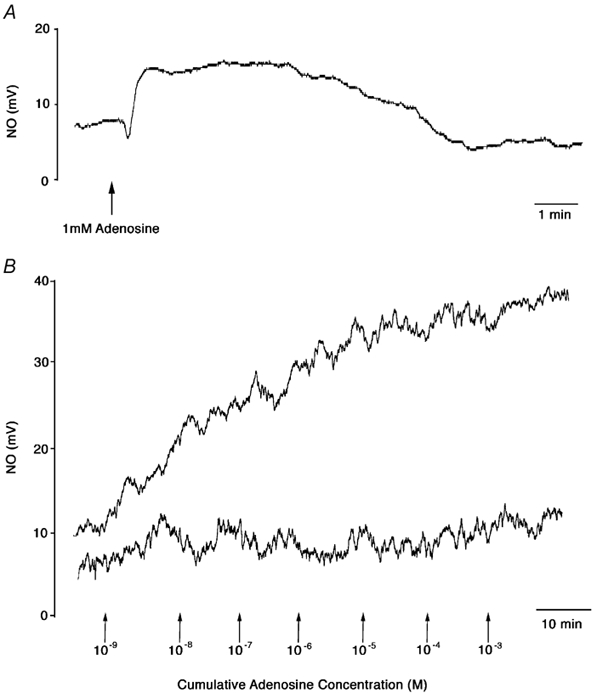
A, original trace showing the NO release evoked by 1 mm adenosine. B, original trace showing NO release evoked by cumulative concentrations of adenosine (indicated by arrows) before (upper trace) and 30 min after l-NAME (100 μM, lower trace) in the presence of EHNA (adenosine deaminase inhibitor, 10 μM) and NBTI (adenosine uptake inhibitor, 10 μM).
Group 2
In order to test the integrity of the endothelium the response to 1 μM acetylcholine (ACh) was recorded: in pre-constricted mesenteric artery rings this concentration of ACh caused an increase in the output of NO and a relaxation that were 50 % of their respective maxima (Simonsen et al. 1999). Then, 10 μM erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA), an adenosine deaminase inhibitor, and 10 μM S-(p-nitrobenzyl)-6-thio-inosine (NBTI), an adenosine uptake inhibitor, were added to the Krebs solution and 20 min later the NO responses evoked by cumulative concentrations of adenosine, 1 nm to 1 mm, were recorded. Such concentrations of EHNA and NBTI have been found to decrease the concentration of adenosine required to release a given amount of NO from endothelial cells in culture (Li et al. 1998) and to cause a leftward shift in the dose-response relationship of adenosine acting to relax coronary artery rings in vitro (Rubin et al. 2000). In these experiments at least 10 min was allowed between additions of adenosine so that the NO response could fully develop (see Fig. 4B).
This whole protocol was repeated after 30 min incubation of the vessel with 100 μM l-NAME. This concentration of l-NAME abolished the adenosine-evoked increase in cGMP in human umbilical vein endothelial cells (HUVEC; Sobrevia et al. 1997).
Responses were measured as the mean increase in NO release over the 10 min period between each addition of agonist, all values being measured from the original baseline. This protocol gave a cumulative dose-response curve.
Group 3
The response to 1 mm adenosine was tested as in Group 1, before and 5 min after adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) was given at 100 nm, a concentration shown to be highly selective for A1 receptors but without effect on A2 receptors (Daly et al. 1985). Then the A2A receptor antagonist 4-(2-[7-amino-2-(2-furyl)[1,2,4]-triazolo[2,3-a][1,3,5]triazin-5-yl-amino]ethyl) phenol (ZM241385, 100 nm) was added and the response to 1 mm adenosine was re-tested. ZM241385 is highly selective against A2A receptors (Poucher et al. 1995) and at this concentration it abolished the A2A receptor-mediated effect of adenosine on cGMP in human umbilical vein endothelial cells (Sobrevia et al. 1997).
Group 4
The response to 1 mm adenosine was tested as in Group 1, before and 5 min after diclofenac at 1 μM, the dose that blocked release of PGs and reduced vasodilatation evoked by hypoxic perfusion in rabbit heart (see above, Nakhostine & Lamontagne, 1994).
Groups 5 and 6
In Group 5, the response to 1 mm adenosine was tested before and after DPCPX (100 nm) and then after subsequent addition of diclofenac (1 μM). In Group 6 ZM241385 (100 nm) was used instead of DPCPX.
Group 7
The NO release evoked by iloprost was tested, doses being added cumulatively to give a final concentration of 0.1-500 nm.
Groups 8 and 9
In Group 8, the response to 1 mm adenosine was tested before and after DPCPX (100 nm) and then after the subsequent addition of 2′,5′-dideoxyadenosine (DDA) at 50 μM. This concentration selectively blocks agonist-stimulated adenyl cyclase (AC) activity in vascular smooth muscle (Sabouni et al. 1991). In Group 9, ZM241385 (100 nm) was used instead of DPCPX.
Control experiments
In all the above protocols, each drug was added carefully to the edge of the bath to avoid the effect of shear stress on NO release. To test whether this was successful, Krebs solution was added to the bath in the same way during the protocol for Group 1: no measurable release of NO was detected. To test whether agonists or antagonists directly affected the output of the NO electrode, each drug used in the experiments described above was added to the bath at least twice in the absence of arterial tissue: no change in the output of the electrode was detected. In five separate experiments, the endothelium was gently removed from the luminal surface of the aorta by mechanical rubbing: application of 1 mm adenosine, as described above, evoked no measurable change in the output of the NO electrode.
Prostaglandin assays
In a further group of 25 rats (279.6 ± 9.9 g) the release of PGI2 from the aorta was assessed by radioimmunoassay (RIA) of its stable breakdown product 6-keto prostaglandin F1α (6-keto PGF1α). Briefly, the rats were killed by anaesthetic overdose and the aorta was excised from the rats as described above, opened and divided into four equal lengths (5-7 mm), care being taken to maintain the integrity of the endothelium. They were then placed in Krebs solution at room temperature and equilibrated for 2 h.
Group A
Vessels from seven rats were then randomly assigned to one of four sets: (a) control; (b) + adenosine; (c) + diclofenac; and (d) adenosine + diclofenac. They were preincubated for 30 min, vessels (a) and (b) in tubes containing 1 ml Krebs solution and vessels (c) and (d) in tubes containing 1 μM diclofenac in 1 ml Krebs solution. At 30 min, vessels (b) and (d) were stimulated by the addition of 1 mm adenosine and vessels (a) and (c) received an equivalent volume of the vehicle for adenosine. After a further 30 min the vessels were removed and placed in an oven to allow determination of their dry weight, while the supernatant was immediately frozen and kept at −20 °C for the later determination of 6-keto PGF1α, as described by Salmon (1978). Briefly, RIA measurement of 6-keto PGF1α was based on the comparison of the experimental samples to a standard curve constructed over a range of 0–250 pg (0.1 ml)−1 of authentic 6-keto PGF1α. Four replicates were carried out for each standard concentration and each experimental sample: the production of PGI2 was expressed as picograms of 6-keto PGF1α per milligram dry weight of tissue.
Groups B and C
Vessels from 18 rats were randomly assigned to one of four sets (a-d, as in Group A) and assayed for the release of PGI2 in the presence of DPCPX (100 nm, Group B, n= 9) or ZM241385 (100 nm, Group C, n= 9) in order to elucidate the adenosine receptor subtype involved.
Drugs
For in vivo studies, all drugs except CCPA were dissolved in 0.9 % NaCl. The vehicle for CCPA, 3 % DMSO in 0.9 % NaCl, had no effect on cardiovascular variables (Bryan & Marshall, 1999a). For in vitro studies, adenosine, l-NAME, l-arginine, diclofenac and iloprost were dissolved in Krebs solution. The vehicle for DPCPX and DDA was 10 % DMSO and 0.1 m NaOH diluted 50:50 in Krebs solution; NBTI and EHNA were dissolved in 10 % DMSO; stock solutions were then diluted in Krebs solution to give final concentrations of DMSO in the bath of 0.0002, 0.02, 0.05 and 0.05 %, respectively. Li et al. (1998) showed that 0.045 % DMSO had no effect on the basal, or agonist-stimulated release of NO from cultured endothelial cells. The vehicle for ZM241385 was 3 % polytheylene glycol (PEG) and 0.1 m NaOH diluted 50:50 in Krebs solution. Adenosine, SNP, l-NAME, l-arginine, diclofenac, DMSO, indomethacin, EHNA, NBTI, anti-6-keto PGF1α and 6-keto PGF1α were obtained from Sigma (Poole, UK); 8-SPT, DPCPX, CCPA and ZM241385 from Research Biochemicals Inc. (Natick, MA, USA); iloprost from Schering Health Care (Burgess Hill, UK); PEG from BDH Chemicals (Poole, UK); DDA from Calbiochem (Nottingham, UK); and [3H]-6-keto PGF1α from Amersham Pharmacia Biotech (Amersham, UK).
Statistical analyses
All results are expressed as means ±s.e.m. For the cardiovascular variables, changes were computed using Chart software as the integral of the variable during the 5 min stimulus minus the integral over the 5 min before the stimulus. To assess the effect of diclofenac or 8-SPT on the muscle vasodilator responses evoked by hypoxia, and by the infusion of each agonist except CCPA, changes from baseline before and after the antagonist were compared by using Student's paired t test. For the CCPA infusion, the value recorded at the fifth minute of the CCPA infusion was compared with the baseline before the infusion by paired t test and this change was compared by using an unpaired t test with that recorded in our previous study (Bryan & Marshall, 1999a), in which no diclofenac was given. For the in vitro studies, all responses were analysed by ANOVA followed by Fisher's test when appropriate. For all analyses P < 0.05 was considered significant. In all cases, n is the number of animals.
RESULTS
In vivo studies
Effects of diclofenac
As expected, breathing 8 % O2 for 5 min induced a substantial fall in Pa,O2 and Pa,CO2 (Table 1), a fall in ABP, an increase in FVC indicating muscle vasodilatation but no change in FBF (see Bryan & Marshall, 1999a, Fig. 2 and Fig. 3). Similar falls in ABP and increases in FVC were evoked by the chosen infusion rates of adenosine, SNP and iloprost (Fig. 3). Adenosine caused a small increase in Pa,O2 and pH and decrease in Pa,CO2, reflecting an increase in ventilation (Bryan & Marshall, 1999a), whereas SNP and iloprost had no effect on blood gases (data not shown).
Table 1.
Arterial blood gas and pH values (mean ±s.e.m.) recorded during air breathing (control) and in the fifth minute of breathing 8% O2 (hypoxia) before and after diclofenac
| Before diclofenac | After diclofenac | |||||
|---|---|---|---|---|---|---|
| Pa,O2(mmHg) | Pa,CO2(mmHg) | pH | Pa,O2(mmHg) | Pa,CO2 | pH | |
| Control | 88.1 ± 2.4 | 35.9 ± 1.5 | 7.40 ± 0.01 | 90.1 ± 3.5 | 32.4 ± 1.8 | 7.37 ± 0.03 |
| Hypoxia | 31.3 ± 1.3*** | 26.7 ± 1.3** | 7.45 ± 0.03 | 33.2 ± 2.2*** | 24.4 ± 1.6*** | 7.44 ± 0.04 |
P < 0.01
P < 0.001, Control vs. Hypoxia.
Figure 2. Differential effects of diclofenac on muscle vasodilator responses evoked by different stimuli.
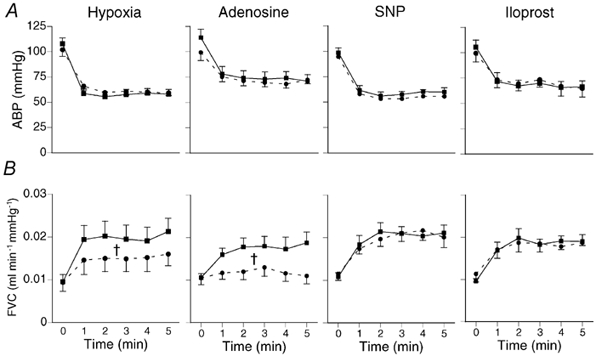
Each panel, from left to right, shows mean (±s.e.m.) ABP (A) and FVC (B) at time 0 and after five 1 min intervals of breathing 8 % O2 (hypoxia, left-hand panel) or infusion of agonist as indicated above panels, before (▪) and after (•) diclofenac. Diclofenac (1 mg kg−1i.v.) reduced muscle dilator response evoked by hypoxia (n= 8) and adenosine (n= 6), but not that evoked by SNP (n= 7) or iloprost (n= 5). †P < 0.05 vs. control, Student's paired t test on integral of change in variable.
Figure 3. Effects of diclofenac followed by adenosine receptor blockade on cardiovascular responses evoked by systemic hypoxia.
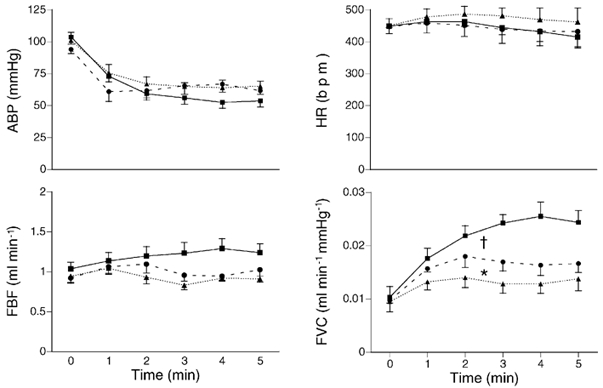
Each graph shows mean values (±s.e.m.) recorded at time 0 and at five 1 min intervals of breathing 8 % O2 before (▪) and after diclofenac (•; 1 mg kg−1i.v.), and after subsequent administration of the adenosine receptor antagonist, 8-SPT (▴; 10 mg kg−1i.v., n= 5). Variables are indicated by ordinates. The muscle vasodilatation (increase in FVC) evoked by hypoxia was reduced by diclofenac and further reduced by 8-SPT, but there was no effect on the other variables. †P < 0.05 vs. control, *P < 0.05 vs. after diclofenac, Student's paired t test on integral of change in variable.
Diclofenac had no effect on the baselines of the cardiovascular variables, or on the blood gas values recorded during air breathing, during 8 % O2 (Table 1) or agonist infusions (data not shown). However, diclofenac substantially reduced the increase in FVC evoked by hypoxia and by adenosine (Fig. 2). By contrast diclofenac had no effect on the increases in FVC evoked by SNP or iloprost (Fig. 2).
In experiments in which the A1 receptor agonist CCPA was given after diclofenac, it had no significant effect on FVC (FVC was 0.015 ± 0.002 and 0.016 ± 0.003 ml min−1 mmHg−1 before and at the fifth minute of CCPA infusion, respectively). This contrasts with the 35 % increase in FVC evoked in the absence of diclofenac (see Bryan & Marshall, 1999a). It may be noted that in two further animals prepared like those of Group 1, but not given diclofenac, CCPA prepared from the same drug batch as that used for Group 1 evoked a comparable increase in FVC to that described previously (Bryan & Marshall, 1999a, data not shown).
Effects of diclofenac followed by 8-SPT
As in Group 1, diclofenac substantially reduced the increase in FVC evoked by 8 % O2. Subsequent administration of the adenosine receptor antagonist 8-SPT caused a further reduction in the hypoxia-evoked increase in FVC (Fig. 3).
Effects of indomethacin
Indomethacin (5 mg kg−1) had a similar effect to diclofenac on the responses evoked by 8 % O2: the increase in FVC was substantially reduced (by 32.84 ± 27.16 % of the control response, P < 0.05, by Student's paired t test), but there was no effect on the other variables (data not shown). Indomethacin at 10 mg kg−1 had no greater effect (the increase in FVC was reduced by 27.16 ± 4.83 %, P < 0.05 vs. control response).
Effects of 8-SPT followed by indomethacin
As expected (Skinner & Marshall, 1996; Bryan & Marshall, 1999a), 8-SPT reduced the increase in FVC evoked by 8 % O2 (by 22.68 ± 7.21 %, P < 0.05 vs. control response). Indomethacin given after 8-SPT had no further effect on the increase in FVC evoked by 8 % O2; it was reduced by 14.75 ± 6.55 % (P < 0.05) of the original control response.
In vitro studies on NO output
Group 1
Adenosine evoked dose-dependent increases in NO release, a maximal response being reached at 1 mm adenosine (Fig. 5A). The peak of each response was reached within 10 s and was usually preceded by a small decrease in NO output (Fig. 4A). The NO output remained at its peak level for 1–3 min and then gradually returned towards baseline over the following 3–6 min.
Figure 5. Adenosine evokes dose-dependent release of NO from endothelial surface of thoracic aorta.
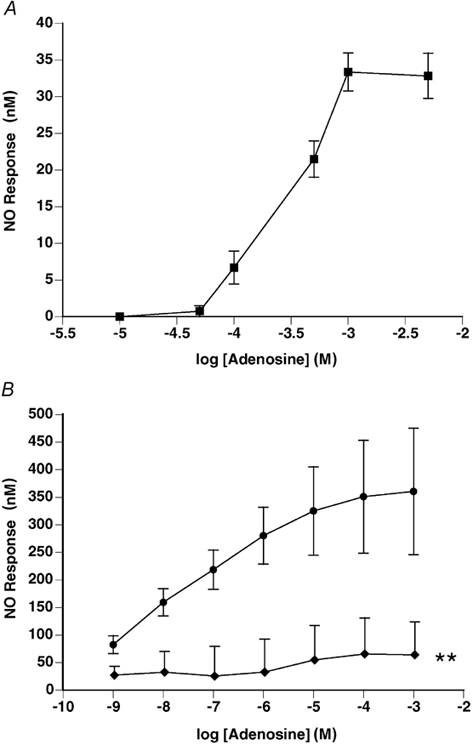
A, non-cumulative dose-response curve to adenosine: 1 mm adenosine evoked maximum NO release (n= 8). B, cumulative dose-response curve to adenosine in the absence (•) and presence of l-NAME (100 μM, ♦), both curves being obtained in the presence of EHNA (adenosine deaminase inhibitor, 10 μM) and NBTI (adenosine uptake inhibitor, 10 μM). Adenosine (10-100 μM) evoked a peak release of NO, which was significantly attenuated by l-NAME. In both A and B responses are shown as mean change in NO output ±s.e.m.; **P < 0.01, ANOVA followed by Fisher's test.
Group 2
Acetylcholine (1 μM) evoked a gradual increase in the output of NO from the endothelium, to a mean value of 100.1 ± 22.5 nm. In the presence of the enzyme inhibitors EHNA and NBTI, adenosine also caused a gradual increase in the release of NO similar to that seen with ACh, which reached a maximum between 8 and 10 min after the addition of each agonist concentration (Fig. 4B). Cumulative concentrations of adenosine caused a dose-dependent increase in the release of NO from the endothelium reaching a peak at 10–100 μM adenosine (Fig. 5B). The NO synthase (NOS) inhibitor l-NAME (100 μM) significantly decreased the NO response curve to adenosine (Fig. 5B).
Group 3
The A1 receptor antagonist DPCPX reduced the response to adenosine (1 mm). The remaining response was reduced by subsequent addition of the A2A receptor antagonist ZM241385 (Fig. 6A).
Figure 6. Nitric oxide release evoked by adenosine from rat aorta is mediated partly by A1 and partly by A2A receptors.
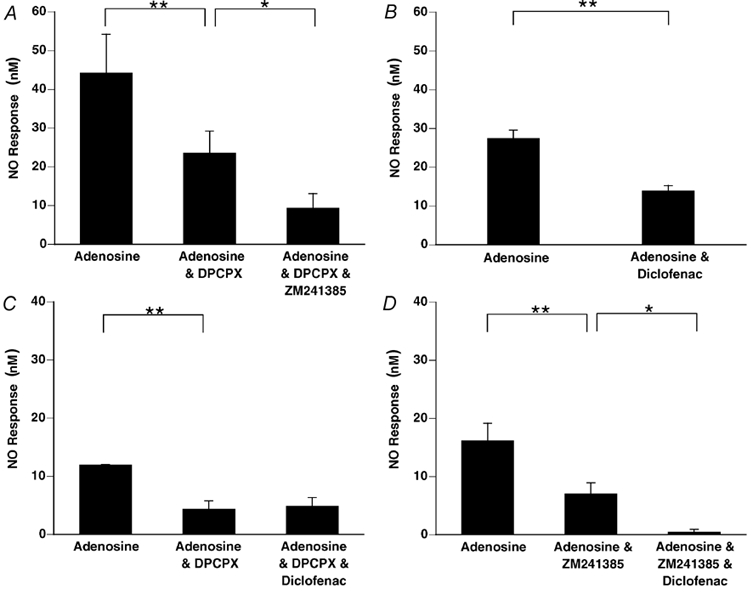
Diclofenac reduces NO release evoked from rat aorta by adenosine when A1 receptors are functionally active, but not when they are blocked. A, the response evoked by 1 mm adenosine was reduced by the A1 receptor antagonist DPCPX (100 nm) and further reduced after subsequent addition of the A2A receptor antagonist ZM241385 (100 nm, n= 5). B, the control response to adenosine (1 mm) was reduced by ≈50 % by diclofenac (1 μM, n= 8). C, diclofenac (1 μM) had no effect on the response to adenosine (1 mm) evoked in the presence of DPCPX (100 nm, n= 5), but (D) attenuated that evoked in the presence of ZM241385 (100 nm, n= 6). Columns show mean change in NO output (±s.e.m.); **P < 0.01, *P < 0.05, ANOVA followed by Fisher's test.
Groups 4, 5 and 6
In Group 4, diclofenac substantially reduced the response evoked by 1 mm adenosine (Fig. 6B). In Group 5, the response to adenosine that remained after DPCPX was not affected by subsequent addition of diclofenac (Fig. 6C). In Group 6, the response to adenosine that remained after ZM241385 was greatly reduced by subsequent addition of diclofenac (Fig. 6D).
Group 7
Iloprost evoked a dose-dependent increase in the output of NO (Fig. 7). In contrast to the effect of adenosine in Group 1, but in a similar fashion to ACh and adenosine added in the presence of EHNA and NBTI in Group 2, cumulative additions of iloprost evoked increases in NO output that reached a peak at ≈0.1 μM.
Figure 7. Iloprost causes dose-dependent release of NO from rat aorta.
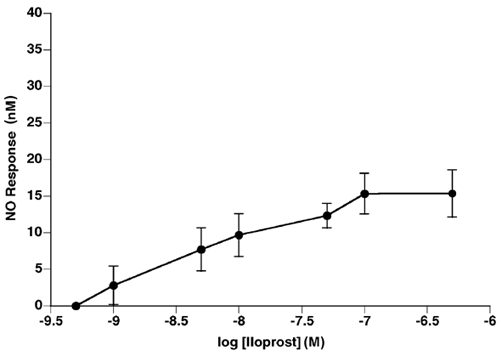
Data are shown as mean change in NO output (±s.e.m.). n= 5.
Groups 8 and 9
In Group 8, the response to adenosine that remained after DPCPX was significantly reduced by DDA (Fig. 8A). Similarly, in Group 9, the response to adenosine that remained after ZM241385 was attenuated by DDA (Fig. 8B).
Figure 8. The activation of adenyl cyclase is required in order that adenosine acting at A1 and A2A receptors can evoke NO release.
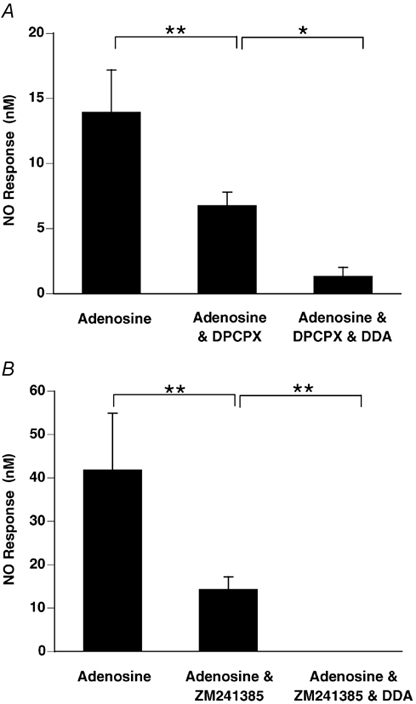
NO release evoked by adenosine (1 mm) in the presence of the A1 receptor antagonist DPCPX (A) and the A2A receptor antagonist ZM241385 (B) was significantly attenuated by adenyl cyclase inhibition with DDA (50 μM, n= 6). Columns show mean increase in NO output (±s.e.m.); **P < 0.01, *P < 0.05, ANOVA followed by Fisher's test.
Prostaglandin assays
Group A
Adenosine evoked an increase in the release of PGI2 from the vessel sections as measured by RIA of 6-keto PGF1α (Fig. 9). The cyclooxygenase inhibitor diclofenac reduced the basal release of PGI2 from the vessel and also abolished the increase in PGI2 release evoked by adenosine (Fig. 9).
Figure 9. Adenosine increases the generation of 6-keto PGF1α by rat aorta by stimulating A1 receptors but not A2A receptors.
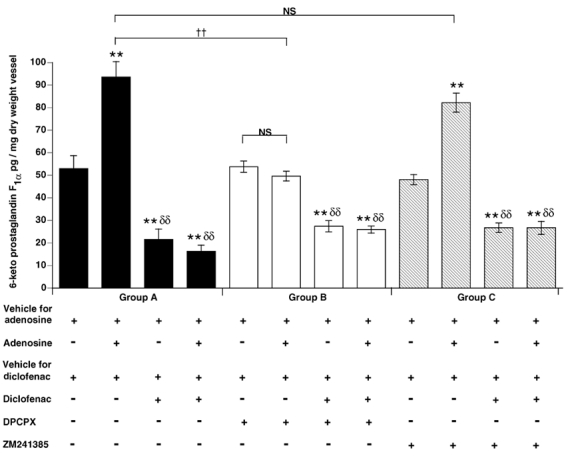
Columns show 6-keto PGF1α (mean ±s.e.m.) assayed in supernatant under control conditions (Group A, filled columns), in the presence of DPCPX (Group B, open columns), and in the presence of ZM241385 (Group C, hatched columns). Symbols below the chart show the presence (+) or absence (-) of vehicle for adenosine, adenosine, vehicle for diclofenac, diclofenac, DPCPX and ZM241385, in the assay tube. Within groups: ** significantly different from basal 6-keto PGF1α generation (first column); δδ significantly different from adenosine-evoked 6-keto PGF1α generation (second column). P < 0.01 in both cases. NS indicates no significant difference and †† indicates significant difference (P < 0.01) between groups, as indicated by brackets.
Groups B and C
In Group B, in the presence of DPCPX, the release of PGI2 evoked by adenosine was attenuated, whereas basal release remained unchanged (Fig. 9). In Group C, in the presence of ZM241385, adenosine evoked a release of PGI2 that was not significantly different from that evoked in Group A in the absence of antagonists (Fig. 9). In Group B, in the presence of DPCPX, diclofenac reduced the basal release of PGI2 and in Group C, in the presence of ZM241385, diclofenac abolished the adenosine-evoked increase in PGI2 release (Fig. 9).
DISCUSSION
The in vivo experiments of the present study showed that a large part of the vasodilatation evoked in skeletal muscle by systemic hypoxia is dependent on PG synthesis, in that it is sensitive to cyclooxygenase inhibition. They also showed that the muscle vasodilatation evoked by adenosine, which makes its contribution to the vasodilatation of systemic hypoxia by acting on A1 receptors (Bryan & Marshall, 1999a), is dependent on PG synthesis. These findings are supported and elucidated by the results of our in vitro experiments. We showed that adenosine can stimulate the release of NO from the endothelium of arterial vessels by acting on either A1 or A2A receptors, and that the A1, but not the A2A receptor-mediated response is dependent on PG synthesis. We further demonstrated that NO release can be evoked by a stable analogue of PGI2 and that the A1 receptor-mediated release of NO is dependent on AC activity which generates cAMP, the second messenger for PGs. Finally, we showed that adenosine can increase the release of PGI2 by stimulating A1 receptors, but not A2A receptors, and that the A1-stimulated PGI2 release is sensitive to cyclooxygenase inhibition.
Involvement of PGs in muscle vasodilatation
The finding that the muscle vasodilatation of systemic hypoxia is dependent on PG synthesis is novel. However, it is compatible with evidence that dilatation evoked by hypoxia in vitro, in arteries and arterioles of skeletal muscle (Busse et al. 1984; Messina et al. 1992; Fredricks et al. 1994a), diaphragm muscle (Ward, 1999), brain (Fredricks et al. 1994b) and coronary circulation (Nakhostine & Lamontagne, 1994) is dependent on endothelial synthesis of PGs. It is also consistent with evidence that hypoxia increased the synthesis and release of PGs, particularly PGI2, from cultured endothelial cells (Michiels et al. 1993). Indeed, since in systemic hypoxia the endothelial cells must experience the fall in PO2 of the blood, and since in isolated lengths of arterial vessel PGs (mainly PGI2) were released by a fall in intraluminal PO2, but not by a fall in extraluminal PO2 (Busse et al. 1984), it is probable that in vivo the endothelial cells both ‘sense’ the fall in PO2 and respond by releasing PGI2. It is generally accepted that PGI2 is the major PG released from endothelial cells and, as shown in the present study, iloprost, the stable analogue of PGI2, evoked substantial muscle vasodilatation.
Importantly, a comparable chain of events has been proposed for the dilators adenosine and NO: both are released in an O2-dependent manner and induce vasodilatation, thereby improving O2 delivery from the endothelium of skeletal muscle, coronary circulation and isolated, perfused arteries (Deussen et al. 1986; Pohl & Busse, 1989; Bryan & Marshall, 1999a; Edmunds & Marshall, 2001a, b).
Prostaglandin synthesis triggered by an increase in shear rate (Koller et al. 1994) may have contributed to the hypoxia-evoked muscle vasodilatation of the present study. However, at most, this probably made a relatively small contribution in individual arterioles. Firstly, systemic hypoxia had no significant effect on gross FBF: the increase in FVC balanced the fall in ABP such that FBF did not change significantly. Maintenance of blood flow associated with vasodilatation would have been unlikely to produce a significant increase in blood velocity and shear stress. Secondly, diclofenac had no effect on the comparable increases in FVC evoked by SNP or iloprost, even though they are likely to have had comparable effects on shear rates through the muscle.
It may be noted that in the present study, diclofenac caused a larger reduction in the muscle vasodilatation evoked by systemic hypoxia than indomethacin. This may be related to their different modes of action and to their relative efficacies as antagonists of PG synthesis. For example, diclofenac produces less shunting of arachidonic acid from the cyclooxygenase pathway towards the lipoxygenase pathway than indomethacin (Ku et al. 1986). However, it is more likely the discrepancy reflected a real difference between groups of animals in the dependence of the hypoxia-evoked muscle vasodilatation upon PG synthesis. In the animals in which diclofenac was given, it reduced the hypoxia-evoked dilatation by ≈50 %, which is similar to the reduction we achieved with adenosine receptor antagonists in previous studies (Skinner & Marshall, 1996; Bryan & Marshall, 1999a). By contrast, the experiments in which indomethacin or 8-SPT were given were performed several months after the diclofenac experiments, and in them indomethacin caused only a ≈30 % reduction in the hypoxia-evoked dilatation, which was similar to the 23 % reduction caused by 8-SPT. In other words, the antagonism caused by diclofenac or indomethacin was closely related to the contribution made by adenosine to the hypoxia-evoked muscle vasodilatation.
Interaction between adenosine and PGs
This apparent relationship between the magnitude of the contributions of adenosine and PGs to hypoxia-evoked dilatation is obviously relevant to the major new findings of the present study. Although inhibition of PG synthesis, or blockade of adenosine receptors, each caused a large reduction in the hypoxia-evoked dilatation when the antagonists were given separately, indomethacin had no effect when given in the presence of 8-SPT, while 8-SPT had only a small effect when given in the presence of diclofenac. These results clearly indicated that the roles of PGs and adenosine in evoking vasodilatation are interdependent. This idea is fully supported by our findings that diclofenac greatly reduced muscle vasodilatation evoked by exogenous adenosine and prevented dilatation from being evoked by the selective A1 receptor agonist, CCPA (cf. Bryan & Marshall, 1999a).
In previous in vitro studies on rabbit and guinea-pig heart, the contributions made by adenosine and prostaglandins to hypoxia-induced coronary dilatation were simply additive (Park et al. 1992; Nakhostine & Lamontagne, 1994). However, in these studies hypoxia was achieved by reducing the PO2 of the Krebs solution perfusing the coronary circulation from > 500 to ≈100 mmHg. The O2 content of the fluid perfusing the heart was therefore considerably less, both in ‘normoxia’ and hypoxia, than in the present study in which the skeletal muscle was perfused with whole blood containing haemoglobin. Moreover, since the heart was beating, its O2 consumption must have been much greater than that of resting skeletal muscle. Thus, it is likely that in the heart, hypoxic perfusion not only caused hypoxia of the endothelium, but also of the cardiac myocytes, and that they too released adenosine which acted directly on the vascular smooth muscle (see Deussen et al. 1986): this would have reduced the proportional effect of adenosine released from and acting on the endothelium.
Interdependence of PG- and adenosine-evoked dilatation such as we observed might be explained simply by the mutual facilitatory interaction between cAMP- and cGMP-mediated dilator responses within vascular smooth muscle (de Wit et al. 1994). Prostaglandins act on vascular smooth muscle to induce vasodilatation by increasing intracellular cAMP. On the other hand, hypoxia- and adenosine-evoked vasodilatation has been attributed, at least partly, to increased endothelial synthesis of NO (Pohl & Busse, 1989; Bryan & Marshall, 1999b; Edmunds & Marshall, 2001a, b and the present study) and the second messenger for NO is cGMP. However, interaction between cAMP and cGMP is unlikely to be the sole explanation, for diclofenac had no effect on baseline FVC, or on the increase in FVC evoked by the NO donor, SNP. This suggests that the cAMP generated by PG synthesis was not required to facilitate the dilator influence of tonically released NO, or of increased levels of NO and cGMP. Nevertheless, we have seen evidence of possible synergism between cGMP and cAMP under the conditions of our experiments, in that blockade of NO synthesis substantially reduced baseline FVC and the increases in FVC evoked by adenosine. However, increases in FVC to adenosine returned when a tonic dilator influence of NO was restored by infusion of the NO donor, SNP (Edmunds & Marshall, 2001a). Thus, at least when NO synthesis is blocked, adenosine can increase FVC by mechanisms that are facilitated by a basal level of cGMP. This is consistent with the present findings and is given further consideration below.
A further possible explanation for our results might be that NO generated by the action of adenosine increased PG synthesis, as described in endothelial cells in vitro (Davidge et al. 1995). This is very unlikely, for diclofenac had no effect on SNP-evoked muscle vasodilatation. The results of our in vitro experiments also argue against this possibility (see below).
Adenosine, PGs and NO synthesis
Thus, having considered the alternatives, the most likely explanation for the present in vivo results is that adenosine released by hypoxia and exogenous adenosine both acted on endothelial cells to generate PGs, which then stimulated NO synthesis. This is the pathway that was identified by our in vitro experiments. We directly showed that adenosine can cause dose-dependent synthesis and release of NO from the endothelium of freshly excised rat aorta, by using an NO-selective electrode: we have recently obtained similar results in rat iliac artery (C. J. Ray & J. M. Marshall, unpublished observations). Our findings confirm much indirect evidence from the effects of NO synthesis inhibitors on vasodilator responses evoked by adenosine, both in vivo and in vitro (Vials & Burnstock, 1993; Danialou et al. 1997; Bryan & Marshall, 1999b) and the report that adenosine increased cGMP and nitrate/nitrite levels in endothelial cells in vitro (Li et al. 1995; Sobrevia et al. 1997).
It may be noted that the adenosine concentrations required to increase NO output from the aorta in our experiments were 10−4 to 10−3m. This deserves comment. Although high, these concentrations can be compared with the adenosine concentrations of 10−7 to 10−3m required to evoke dose-dependent dilatation of skeletal muscle arterioles (Mian & Marshall, 1991) and diaphragm muscle arterioles (Danialou et al. 1997) in vivo, and the concentrations of 10−6 to 10−4m required to evoke graded relaxation of porcine coronary artery rings (Rubin et al. 2000). In the present study, inhibition of adenosine transport and adenosine deaminase with the selective antagonists NBTI and EHNA greatly prolonged the NO response to single doses of adenosine and increased the sensitivity range to 10−9 to 10−4m. This finding is consistent with, but extends the observation of, Li et al. (1998), that the NO output evoked from cultured human and porcine endothelial cells by a standard concentration of 10−4m adenosine was accentuated and prolonged from 2 min to 7 min by EHNA and an adenosine kinase inhibitor. Moreover, it is also consistent with the findings of Rubin et al. (2000) who showed that the dose-response curves for the relaxing effect of adenosine on coronary artery rings were considerably leftward shifted by either NBTI or EHNA. However, even though the activity of the adenosine transporter is at least two orders of magnitude greater in endothelial cells than vascular smooth muscle, Rubin et al. (2000) attributed the effect of NBTI on coronary artery responsiveness predominantly to blockade of adenosine transport into the vascular smooth muscle because endothelial cells represented a relative small proportion of the total tissue volume. Thus, the present study is the first to demonstrate the very large impact adenosine transport and deaminase activity can have on NO responses evoked in intact endothelium by exogenous adenosine.
The very fact that the activities of the adenosine transport and degradation processes are so high, not just for endothelial and vascular smooth muscle cells, but also for skeletal muscle cells (Cheng et al. 2000), means that it is impossible to use the adenosine concentrations that have been measured in plasma and interstitial space during systemic hypoxia or muscle contraction (10−7 to10−6m; Mo & Ballard, 2001), or the concentrations of exogenous adenosine required to evoke vascular responses (up to 10−3m, see above) to estimate the concentrations that might be reached in the micro-environment of the adenosine receptors under conditions when the release of adenosine is increased. This problem is compounded for the A1 receptors because adenosine deaminase and A1 receptors co-localise on the sarcolemma and the enzyme not only degrades adenosine but facilitates the binding of adenosine to the A1 receptor (Saura et al. 1998). Thus, considering all of this information, and given the strong evidence that adenosine is released from endothelial cells of skeletal muscle during systemic hypoxia and that it produces vasodilatation, at least in part by acting on adenosine receptors on the endothelium (Marshall, 2000; Mo & Ballard, 2001), we argue that our approach of using a standard adenosine concentration of 10−3m to investigate the mechanisms of action of adenosine on the endothelium is reasonable.
Our finding that the adenosine-evoked NO release was attenuated by either an A1- or an A2A-selective antagonist demonstrates that both A1 and A2A receptors can be coupled to NO synthesis. This accords with deductions from in vivo and in vitro studies on arterial vessels from several different tissues, including rat skeletal and diaphragm muscle, rabbit and porcine coronary circulation and rat brain (Merkel et al. 1992; Vials & Burnstock, 1993; Danialou et al. 1997; Coney & Marshall, 1998; Bryan & Marshall, 1999b). By contrast, Li et al. (1998) showed that A2A receptor stimulation increased, but A1 receptor stimulation decreased, NO output from arterial endothelial cells. These results cannot be directly compared with ours as the cultured cells they used were in passage 3-5, or 13-18. Our finding that diclofenac blocked the NO release evoked by adenosine in the presence of an A2A receptor antagonist, but not that evoked in the presence of an A1 receptor antagonist, is novel and clearly suggests A1 receptors, but not A2A receptors, are coupled to PG synthesis.
In several published studies, ATP and ADP, but not adenosine, were shown to increase PG synthesis in endothelial cells in culture (Needham et al. 1987; de Nucci et al. 1988). However, it was also shown in these studies that endothelial cells under culture may ‘lose’ their functional responses to agonists of several of the receptor types they express in vivo (Needham et al. 1987; de Nucci et al. 1988). The present results raise the possibility that adenosine A1 receptors are members of this vulnerable group. This may be explained by the fact that A1 receptors readily desensitise and internalise (Saura et al. 1998). Our in vitro assays indicate that adenosine can indeed increase the synthesis and release of PGI2 from rat aorta by stimulating A1 but not A2A receptors.
There is already evidence of the cellular mechanisms that would explain A1 receptor stimulation of PG synthesis. In vitro studies on Chinese hamster ovary cells and a vascular smooth muscle cell line have shown that A1 receptor stimulation can activate phospolipase A2 (PLA2) and liberate arachidonic acid, the substrate for PG synthesis, and can greatly augment phosphoinositide hydrolysis and arachidonic acid release caused by other agonists (Akbar et al. 1994; Schachter et al. 1995). Moreover, in endothelial cells in vitro, both cAMP and iloprost can stimulate NO production in a Ca2+-independent, as well as in a Ca2+-dependent manner, by activating cAMP-dependent protein kinase, which phosphorylates NOS (Butt et al. 2000). This last finding accords with our observations that iloprost caused a dose-dependent release of NO from the endothelium of the aorta and that A1-evoked NO release was blocked by inhibition of AC with DDA. It is not surprising that the NO release evoked by adenosine in the presence of DPCPX was also inhibited by DDA, for A2A receptor stimulation has generally been shown to produce its effects by stimulating AC.
A composite view of muscle vasodilatation in systemic hypoxia
Thus, our overall proposals for skeletal muscle are that during systemic hypoxia, adenosine is released from the endothelial cells and acts on endothelial A1 receptors (see Bryan & Marshall, 1999a, b) in an autocrine fashion, to increase the synthesis of PGs, mainly PGI2. The PGs then act via cAMP to stimulate NOS, generate NO and cause vasodilatation by increasing cGMP in the smooth muscle. Since a small component of hypoxia-evoked vasodilatation remained after diclofenac and was reduced by 8-SPT, endogenous adenosine can apparently evoke muscle vasodilatation independently of PG synthesis, presumably by (i) acting on endothelial A1 receptors opening K+ channels, causing hyperpolarisation, Ca2+ influx (Luckoff & Busse, 1990) and directly increasing NO synthesis, or (ii) acting on smooth muscle A1 receptors to cause vasodilatation by opening ATP-sensitive K+ (KATP) channels (Dart & Standen, 1993; Bryan & Marshall, 1999b). On the other hand, since indomethacin had no effect on hypoxia-evoked dilatation when given after 8-SPT, any PG synthesis stimulated by hypoxia independently of adenosine apparently plays little role. This remaining dilatation may be due to adrenaline acting on β2-adrenoceptors (Mian et al. 1990).
In our other studies, the hypoxia-evoked muscle vasodilatation that was restored after NOS inhibition by infusion of SNP (see above, Edmunds & Marshall, 2001a), was reduced by ≈50 % by DPCPX (Edmunds & Marshall, 2000b) and by ≈30 % by indomethacin (N. J. Edmunds & J. M. Marshall, unpublished observations). These results are consistent with adenosine acting on smooth muscle A1 receptors to open KATP channels (Dart & Standen, 1993) and with adenosine acting on endothelial A1 receptors to stimulate PG release which then causes dilatation by increasing smooth muscle cAMP or, more directly, by opening KATP channels (Lombard et al. 1999). It is known that dilatation induced by KATP channel opening is facilitated by a basal level of cGMP (Kubo et al. 1994), as would be achieved by infusion of SNP, while the dependence of cAMP-mediated dilatation on a basal level of cGMP is consistent with the known synergism between cGMP and cAMP (de Wit et al. 1994).
As indicated above, adenosine, PGs and NO have been implicated in hypoxia-evoked vasodilatation in several different tissues and organs. The very fact that our in vitro experiments were performed on the thoracic aorta and yet showed cellular mechanisms that explain the vascular responses observed in skeletal muscle in vivo, raises the strong possibility that our proposals have a wider significance. The vasodilatation evoked by systemic hypoxia in, for example, the coronary circulation, may be similarly dependent on interaction between adenosine A1 receptor stimulation, PG and NO synthesis in endothelial cells. Moreover, the ability of adenosine to stimulate PG synthesis as well as NOS, may be important in other situations in which these mediators have been implicated, e.g. in inhibiting platelet aggregation and activation, in inhibiting neutrophil adherence and extravasation and in protecting against ischaemia-reperfusion injury.
REFERENCES
- Akbar M, Okajima F, Tomura H, Shimegi S, Kondo Y. A single species of A1 adenosine receptor expressed in Chinese hamster ovary cells not only inhibits cAMP accumulation but also stimulates phospholipase-C and arachidonate release. Molecular Pharmacology. 1994;45:1036–1042. [PubMed] [Google Scholar]
- Berne RM, Knabb RM, Ely SW, Rubio R. Adenosine in the local regulation of blood flow : a brief overview. Federation Proceedings. 1983;42:3136–3142. [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia : a role for A1 receptors. Journal of Physiology. 1999a;514:151–162. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. Journal of Physiology. 1999b;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Forsterman U, Matsuda H, Pohl U. The role of prostaglandins in the endothelium-mediated vasodilatory response to hypoxia. Pflügers Archiv. 1984;401:77–83. doi: 10.1007/BF00581536. [DOI] [PubMed] [Google Scholar]
- Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, Meyer HE, Lohmann SM, Schmidt HH. Endothelial nitric oxide synthase (Type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. Journal of Biological Chemistry. 2000;275:5179–5187. doi: 10.1074/jbc.275.7.5179. [DOI] [PubMed] [Google Scholar]
- Cheng B, Essackjee HC, Ballard HJ. Evidence for control of adenosine metabolism in rat oxidative skeletal muscle by changes in pH. Journal of Physiology. 2000;522:467–477. doi: 10.1111/j.1469-7793.2000.t01-1-00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coney AM, Marshall JM. Role of adenosine and its receptors in the vasodilatation induced in the cerebral cortex of the rat by systemic hypoxia. Journal of Physiology. 1998;509:507–518. doi: 10.1111/j.1469-7793.1998.507bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly JW, Padgett W, Shamin MT, Butts-Lamb P, Waters J. 1,3-Dialkyl-8-(p-sulphophenyl) xanthines : potent water soluble antagonists for A1 and A2 adenosine receptors. Journal of Medicinal Chemistry. 1985;28:487–492. doi: 10.1021/jm00382a018. [DOI] [PubMed] [Google Scholar]
- Danialou G, Vicaut E, Sambe A, Aubier M, Boczkowski J. Predominant role of A1 adenosine receptors in mediating adenosine induced vasodilatation of rat diaphragmatic arterioles : involvement of nitric oxide and the ATP-dependent K+ channels. British Journal of Pharmacology. 1997;121:1355–1363. doi: 10.1038/sj.bjp.0701247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart C, Standen NB. Adenosine-activated potassium current in smooth muscle isolated from the pig coronary artery. Journal of Physiology. 1993;471:767–786. doi: 10.1113/jphysiol.1993.sp019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidge ST, Baker PN, McLaughlin MK, Roberts JM. Nitric oxide produced by endothelial cells increases production of eicosanoids through activation of prostaglandin H synthase. Circulation Research. 1995;77:274–283. doi: 10.1161/01.res.77.2.274. [DOI] [PubMed] [Google Scholar]
- de Nucci G, Gryglewski RJ, Warner TD, Vane JR. Receptor mediated release of endothelium-derived relaxing factor and prostacyclin from bovine aortic endothelial cells is coupled. Proceedings of the National Academy of Sciences of the USA. 1988;85:2334–2338. doi: 10.1073/pnas.85.7.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deussen A, Moser G, Schrader J. Contribution of coronary endothelial cells to cardiac adenosine production. Pflügers Archiv. 1986;406:608–614. doi: 10.1007/BF00584028. [DOI] [PubMed] [Google Scholar]
- De Wit C, von Bismarck P, Pohl U. Synergistic action of vasodilators that increase cGMP and cAMP in hamster cremaster microcirculation. Cardiovascular Research. 1994;28:1513–1518. doi: 10.1093/cvr/28.10.1513. [DOI] [PubMed] [Google Scholar]
- Edmunds NJ, Marshall JM. Vasodilatation, oxygen delivery and oxygen consumption in rat hindlimb during systemic hypoxia: roles of nitric oxide. Journal of Physiology. 2001a;532:251–259. doi: 10.1111/j.1469-7793.2001.0251g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds NJ, Marshall JM. Oxygen delivery and oxygen consumption in rat hindlimb during systemic hypoxia: role of adenosine. Journal of Physiology. 2001b;536:927–935. doi: 10.1111/j.1469-7793.2001.00927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredricks KT, Liu Y, Lombard JH. Response of extra parenchymal resistance arteries of rat skeletal muscle to reduced PO2. American Journal of Physiology. 1994a;267:H706–715. doi: 10.1152/ajpheart.1994.267.2.H706. [DOI] [PubMed] [Google Scholar]
- Fredricks KT, Liu Y, Rusch NJ, Lombard JH. Role of endothelium and arterial K+ channels in mediating hypoxic dilation of middle cerebral arteries. American Journal of Physiology. 1994b;267:H580–586. doi: 10.1152/ajpheart.1994.267.2.H580. [DOI] [PubMed] [Google Scholar]
- Guo JP, Murohara T, Buerke M, Scalia R, Lefer AM. Direct measurement of nitric oxide release from vascular endothelial cells. Journal of Applied Physiology. 1996;81:774–779. doi: 10.1152/jappl.1996.81.2.774. [DOI] [PubMed] [Google Scholar]
- Koller A, Sun D, Huang A, Kaley G. Co-release of nitric oxide and prostaglandins mediates flow-dependent dilation of rat gracilis muscle arterioles. American Journal of Physiology. 1994;267:H326–332. doi: 10.1152/ajpheart.1994.267.1.H326. [DOI] [PubMed] [Google Scholar]
- Ku EC, Lee W, Kothari HV, Scholer DW. Effect of diclofenac sodium on the arachidonic acid cascade. American Journal of Medicine. 1986;80:18–23. doi: 10.1016/0002-9343(86)90074-4. [DOI] [PubMed] [Google Scholar]
- Kubo M, Nakaya Y, Matsuoka S, Saito K, Kuroda Y. Atrial natriuretic factor and isosorbide dinitrate modulate the gating of ATP-sensitive K+ channels in cultured vascular smooth muscle cells. Circulation Research. 1994;74:471–476. doi: 10.1161/01.res.74.3.471. [DOI] [PubMed] [Google Scholar]
- Li JM, Fenton RA, Wheeler Brownell H, Powell CC, Peyton BD, Cutler BS, Dobson JG. Adenosine A2A receptors increase endothelial cell nitric oxide. Journal of Surgical Research. 1998;80:357–364. doi: 10.1006/jsre.1998.5439. [DOI] [PubMed] [Google Scholar]
- Li JM, Fenton RA, Cutler BS, Dobson JG. Adenosine enhances nitric oxide production by vascular endothelial cells. American Journal of Physiology. 1995;269:C519–523. doi: 10.1152/ajpcell.1995.269.2.C519. [DOI] [PubMed] [Google Scholar]
- Lombard JH, Liu YP, Fredricks KT, Bizub DM, Roman RJ, Rusch NJ. Electrical and mechanical responses of rat middle cerebral arteries to reduced PO2and prostacyclin. American Journal of Physiology. 1999;276:H509–516. doi: 10.1152/ajpheart.1999.276.2.H509. [DOI] [PubMed] [Google Scholar]
- Luckoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium derived relaxing factor is controlled by the membrane potential. Pflügers Archiv. 1990;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Adenosine and muscle vasodilatation in acute systemic hypoxia. Acta Physiologica Scandinavica. 2000;168:561–573. doi: 10.1046/j.1365-201x.2000.00709.x. [DOI] [PubMed] [Google Scholar]
- Merkel LA, Lappe RW, Rivera LM, Cox BF, Perrone MH. Demonstration of vasorelaxant activity with an A1-selective adenosine agonist in porcine coronary artery involvement of potassium channels. Journal of Pharmacology and Experimental Therapeutics. 1992;260:437–443. [PubMed] [Google Scholar]
- Messina EJ, Sun D, Koller A, Wolin MS, Kaley G. Role of endothelium-derived prostaglandins in hypoxia-elicited arteriolar dilation in rat skeletal muscle. Circulation Research. 1992;71:790–796. doi: 10.1161/01.res.71.4.790. [DOI] [PubMed] [Google Scholar]
- Mian R, Marshall JM. The role of adenosine in dilator responses induced in arterioles and venules of rat skeletal muscle in systemic hypoxia. Journal of Physiology. 1991;443:499–511. doi: 10.1113/jphysiol.1991.sp018847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian R, Marshall JM, Kumar P. Interactions between K+ and β2-adrenoreceptors in determining muscle vasodilatation induced in the rat by systemic hypoxia. Experimental Physiology. 1990;75:407–410. doi: 10.1113/expphysiol.1990.sp003416. [DOI] [PubMed] [Google Scholar]
- Michiels C, Arnould T, Knott I, Dieu M, Remacle J. Stimulation of prostaglandin synthesis by human endothelial cells exposed to hypoxia. American Journal of Physiology. 1993;264:C866–874. doi: 10.1152/ajpcell.1993.264.4.C866. [DOI] [PubMed] [Google Scholar]
- Mo FM, Ballard HJ. The effect of systemic hypoxia on interstitial and blood adenosine, AMP, ADP and ATP in skeletal muscle. Journal of Physiology. 2001;536:593–603. doi: 10.1111/j.1469-7793.2001.0593c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhostine N, Lamontagne D. Contribution of prostaglandins in hypoxia-induced vasodilatation in isolated rabbit hearts. Relation to adenosine and KATP channels. Pflügers Archiv. 1994;428:526–532. doi: 10.1007/BF00374574. [DOI] [PubMed] [Google Scholar]
- Needham L, Cusack NJ, Pearson JD, Gordon JL. Characteristics of the P2 purinoceptor that mediates prostacyclin production by pig aortic endothelial cells. European Journal of Pharmacology. 1987;134:199–209. doi: 10.1016/0014-2999(87)90166-x. [DOI] [PubMed] [Google Scholar]
- Park KH, Rubin LE, Gross SS, Levi R. Nitric oxide is a mediator of hypoxic coronary vasodilatation : relation to adenosine and cyclooxygenase-derived metabolites. Circulation Research. 1992;71:992–1001. doi: 10.1161/01.res.71.4.992. [DOI] [PubMed] [Google Scholar]
- Pohl U, Busse R. Hypoxia stimulates release of endothelium-derived relaxant factor. American Journal of Physiology. 1989;256:H1595–1600. doi: 10.1152/ajpheart.1989.256.6.H1595. [DOI] [PubMed] [Google Scholar]
- Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PWR, Jones G, Collis MG. The in vitro pharmacology of ZM241385, a potent, non-xanthine, A2A selective adenosine receptor antagonist. British Journal of Pharmacology. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin LJ, Johnson LR, Dodam JR, Dhalla AK, Magliola L, Laughlin MH, Jones AW. Selective transport of adenosine into porcine coronary smooth muscle. American Journal of Physiology - Heart and Circulatory Physiology. 2000;279:H1397–1410. doi: 10.1152/ajpheart.2000.279.3.H1397. [DOI] [PubMed] [Google Scholar]
- Sabouni MH, Cushing DJ, Makujina SR, Mustafa SJ. Inhibition of adenylate cyclase attenuates adenosine receptor-mediated relaxation in coronary artery. Journal of Pharmacology and Experimental Therapeutics. 1991;259:508–512. [PubMed] [Google Scholar]
- Salmon JA. A radioimmunoassay for 6 keto-prostaglandin F1α. Prostaglandins. 1978;15:383–397. doi: 10.1016/0090-6980(78)90122-3. [DOI] [PubMed] [Google Scholar]
- Saura CA, Mallol J, Canela EI, Lluis C, Franco R. Adenosine deaminase and A1 adenosine receptors internalise together following agonist-induced receptor desensitization. Journal of Biological Chemistry. 1998;273:17610–17617. doi: 10.1074/jbc.273.28.17610. [DOI] [PubMed] [Google Scholar]
- Schachter JB, Yasuda RP, Wolfe BB. Adenosine receptor activation potentiates phosphoinositide hydrolysis and arachidonic acid release in DDT1-MF2 cells: putative interrelations. Cell Signalling. 1995;7:659–668. doi: 10.1016/0898-6568(95)00037-p. [DOI] [PubMed] [Google Scholar]
- Simonsen U, Wadsworth RM, Buus NH, Mulvaney MJ. In vitro simultaneous measurements of relaxation and nitric oxide concentration in rat superior mesenteric artery. Journal of Physiology. 1999;516:271–282. doi: 10.1111/j.1469-7793.1999.271aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MR, Marshall JM. Studies on the roles of ATP, adenosine and nitric oxide in mediating muscle vasodilatation evoked in the rat by acute systemic hypoxia. Journal of Physiology. 1996;495:553–560. doi: 10.1113/jphysiol.1996.sp021615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevia L, Yudilevich DL, Mann GE. Activation of A2-purinoceptors by adenosine stimulates l-arginine transport (system y+) and nitric oxide synthesis in human fetal endothelial cells. Journal of Physiology. 1997;499:135–140. doi: 10.1113/jphysiol.1997.sp021916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vials A, Burnstock G. A2-purinoceptor-mediated relaxation in the guinea-pig coronary vasculature : a role for nitric oxide. British Journal of Pharmacology. 1993;109:424–429. doi: 10.1111/j.1476-5381.1993.tb13586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME. Dilation of rat diaphragmatic arterioles by flow and hypoxia: roles of nitric oxide and prostaglandins. Journal of Applied Physiology. 1999;86:1644–1650. doi: 10.1152/jappl.1999.86.5.1644. [DOI] [PubMed] [Google Scholar]