

Abstract
Heteromeric channel assembly is a potential source of physiological variability. The potential significance of Kir2 subunit heterotetramerization has been controversial, but recent findings suggest that heteromultimerization of Kir2.1-3 may be significant. This study was designed to investigate whether the recently described Kir2.4 subunit can form heterotetramers with the important subunit Kir2.1, and if so, to investigate whether the resulting heterotetrameric channels are functional. Co-expression of either dominant negative Kir2.1 or Kir2.4 subunits in Xenopus oocytes with either wild-type Kir2.1 or 2.4 strongly decreased resulting current amplitude. To examine physical association between Kir2.1 and Kir2.4, Cos-7 cells were co-transfected with a His6-tagged Kir2.1 subunit (Kir2.1-His6) and a FLAG-tagged Kir2.4 subunit (Kir2.4-FLAG). After pulldown with a His6-binding resin, Kir2.4-FLAG could be detected in the eluted cell lysate by Western blotting, indicating co-assembly of Kir2.1-His6 and Kir2.4-FLAG. Expression of a tandem construct containing covalently linked Kir2.1 and 2.4 subunits led to robust current expression. Kir2.1-Kir2.4 tandem subunit expression, as well as co-injection of Kir2.1 and Kir2.4 cRNA into Xenopus oocytes, produced currents with barium sensitivity greater than that of Kir2.1 or Kir2.4 subunit expression alone. These results show that Kir2.4 subunits can co-assemble with Kir2.1 subunits, and that co-assembled channels are functional, with properties different from those of Kir2.4 or Kir2.1 alone. Since Kir2.1 and Kir2.4 mRNAs have been shown to co-localize in the CNS, Kir2.1 and Kir2.4 heteromultimers might play a role in the heterogeneity of native inward rectifier currents.
Inward rectifier potassium channels play a key role in setting the membrane potential and regulating excitability in various tissues including the central nervous system and the heart (Nichols & Lopatin, 1997). Despite their obvious importance, little is known about the molecular basis of native inward rectifier currents. Subunits of the Kir2 family are thought to underlie the inward rectifier current (IK1) in the heart and play an important role in the central nervous system. Kir2.1 was first cloned from a macrophage cell line in 1993 (Kubo et al. 1993). Over the past few years, the properties of currents carried by heterologous expression of Kir2.1, 2.2 and 2.3 subunits cloned from various tissues including the heart (Ishii et al. 1994; Raab-Graham et al. 1994; Ashen et al. 1995; Wible et al. 1995; Wood et al. 1995) and the brain (Koyama et al. 1994; Makhina et al. 1994; Morishige et al. 1994; Perier et al. 1994; Tang & Yang, 1994; Tang et al. 1995) have been studied in detail. Recently, a fourth subunit of the Kir2 group, with somewhat different properties from the other Kir2 subunits, was cloned from rat brain (Topert et al. 1998) and human retina (Hughes et al. 2000) and designated Kir2.4. A human genomic clone corresponding to Kir2.4 was assigned to chromosome 19q13 and designated KCNJ14 (Topert et al. 2000).
Biochemical and electrophysiological experiments on cardiac myocytes support the theory of the diversity of inward rectifier K+ channels contributing to cardiac IK1 (Josephson & Brown, 1986; Wible et al. 1995; Wang et al. 1998). During myocardial development, different IK1 channels with distinct unitary conductances and properties are present (Josephson & Sperelakis, 1990; Chen et al. 1991; Wahler, 1992). Kir2.1 transcripts are about 10 times more abundant than those of Kir2.2 or 2.3 in human atrium and ventricle, with similar concentrations in each, despite a much larger IK1 current density in the ventricle (Wang et al. 1998). In the central nervous system, co-localization of various Kir2 subunits has been noted (Fink et al. 1996; Horio et al. 1996; Karschin et al. 1996). The ability of different subunits to form heteromultimers could partly explain the great diversity observed among native inward rectifier channels in various cells and tissues. Heteromultimerization among inward rectifier subunits of the Kir3 family has been shown to occur and to be functionally important in the heart and the central nervous system (Lesage et al. 1994; Krapivinsky et al. 1995; Lesage et al. 1995). The results of studies on Kir2 heteromultimerization are conflicting. Fink et al. (1996) studied co-assembly between Kir2.1 and Kir2.3 with the use of a dominant negative chimera. The results of co-injection of chimeric constructs with either Kir2.1 or Kir2.3 into Xenopus oocytes suggested that co-assembly occurs if the N-terminus is preserved (Fink et al. 1996), similar to findings with voltage gated K+ channel (Kv) subunits (Lee et al. 1994; Green & Millar, 1995). On the other hand, Tinker et al. (1996) found that the C-terminus and a part of the M2 segment are essential determinants of co-assembly among Kir2 channels, and their results were not consistent with important heteromultimerization between Kir2.1 and Kir2.3 (Tinker et al. 1996). However, strong evidence has recently been provided that suggests that co-assembly among Kir2.1-3 subunits might contribute to inward rectifier diversity in the guinea-pig heart (Preisig-Müller et al. 2002).
Co-localization between the important subunit Kir2.1 and the recently cloned Kir2.4 occurs in various tissues (Kubo et al. 1993; Takahashi et al. 1994; Topert et al. 1998; Derst et al. 2001). The goals of our study were (1) to determine whether Kir2.4 can co-associate with Kir2.1, (2) to assess whether channels formed by co-assembled Kir2.1 and 2.4 subunits are functional and (3) to compare Ba2+-blocking properties of currents carried by channels composed of co-assembled Kir2.1-2.4 subunits with those of homomeric Kir2.1 and 2.4 channels.
METHODS
Construction of dominant negative (dn) Kir2.4 and Kir2.1 constructs
A PCR-based approach was used to engineer Kir2.4 subunits with the GYG motif important for ion selectivity and permeability (Heginbotham et al. 1994; Slesinger et al. 1996) replaced by three alanine residues (AAA). A 5′ and a 3′ fragment were generated and combined by overlap extension. Primers used to synthesize the 5′ fragment were:
ACAGAATTCAGCATGGGCTTGGCCAGGGCCCTGCGCC
(sense),
and:
GACGCTGCGCACAGCAGCGGCTATGGACGTCTG
(antisense).
Primers used to generate the 3′ fragment were:
CAGACGTCCATAGCCGCTGCTGTGCGCAGCGTC (sense),
and:
ACACTCGAGTCATGGAGGCAGGGTCAGTGCCAG
(antisense).
Both fragments were combined by overlap extension PCR using the 5′ sense and the 3′ antisense primer. The resulting construct was subcloned into the Xenopus oocyte expression vector psGEM. The sequence and the presence of the mutation were verified by sequencing. Dn-Kir2.1 was a generous gift from Dr Andrew Tinker, Centre for Clinical Pharmacology, Rayne Institute, London.
Co-precipitation of Kir2.1 and Kir2.4
To evaluate potential physical association between Kir2.4 and Kir2.1 a His-pulldown approach was used. A six-histidine tag was engineered to the N-terminus of Kir2.1 as previously described (Tinker et al. 1996), allowing high affinity specific purification by a His6-binding resin. Kir2.1-His6 was generously provided by Dr A. Tinker, London. Kir2.4 was tagged with an eight amino acid sequence (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) known as ‘FLAG sequence’ that can be recognized by a commercially available monoclonal antibody. Cos-7 cells (American Type Culture Collection (ATCC), Manassas, VA, USA) were then co-transfected with Kir2.1-His6 and Kir2.4-FLAG. Kir2.1-His6-containing complexes were purified under non-denaturing conditions from the detergent-solubilized cell homogenate, washed and eluted from the His6-binding resin. Proteins were then subjected to Western blotting. If protein-protein interactions occurred between Kir2.1 and Kir2.4 the two proteins should copurify (Hoffmann & Roeder, 1991), allowing Kir2.4-FLAG detection via the FLAG epitope.
Construction of Kir2.4-FLAG
Kir2.4 in pcDNA3 (Invitrogen) was a friendly gift from Dr Andreas Karschin, Göttingen, Germany. Using the PCR technique, an EcoRI restriction enzyme site and a sequence coding for the FLAG protein (GACTATAAAGACGACGACGACAAA) were introduced by the sense primer to the 5′ end of Kir2.4. The antisense primer introduced an Eco47III restriction enzyme site which is present only once in Kir2.4 and not present in pcDNA3 (Invitrogen). The Kir2.4-pcDNA3 construct as well as the Kir2.4-FLAG PCR construct was digested with EcoRI and Eco47III (Boehringer Mannheim) and fragments were cleaned on a 1 % agarose gel. Kir2.4-FLAG was then ligated into pcDNA3 with T4 DNA polymerase (Promega). After transformation of competent JM109 E. coli cells (Promega), culture in ampicillin-containing Luria-Bertani (LB) medium, and plasmid isolation, the presence of the construct was verified by restriction enzyme digestion with EcoRI and Eco47III (Boehringer Mannheim). Cos-7 cells were transfected with Kir2.4-FLAG and after subsequent cell lysis the presence of Kir2.4 was shown by Western blot. The sense primer for construction of Kir2.4-FLAG was:
TTAGAATTC*ACGATG†GACTATAAAGACGACGACGACAAA‡
ATGGGCTTGGCCAGG,
with the ends of the EcoRI, Kozac and FLAG sequences designated by *, † and ‡ respectively, followed by the Kir2.4 sequence. The antisense primer consisted of:
ACAGGCGCT*GCGGTCTCGCAGAGCCAC,
with the Eco 47III sequence identified by *, followed by the Kir2.4-specific sequence.
Cos-7 cell maintenance and transfection
Cos-7 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10 % heat-inactivated fetal bovine serum (FBS) and 100 units ml−1 sodium penicillin-G-100 μg ml−1 streptomycin sulfate. Cells (2 × 105) were plated into 35 mm culture dishes with DMEM for 24 h (37 °C, 5 % CO2) to reach ≈70 % confluence. Sample cDNA (1 μg of Kir2.4-FLAG or Kir2.1-His or 1 μg of each) was mixed with 5.5 μl lipofectamine, brought to a final volume of 200 μl with DMEM and added to an 800 μl FBS- and antibiotic-free DMEM cell suspension. After 6 h incubation at 37 °C in 5 % CO2, 1 ml of 20 % FBS was added to achieve a final concentration of 10 % FBS.
His-pulldown
Forty-eight to seventy-two hours after transfection, Cos-7 cells were incubated in RIPA buffer ((octylphenoxy)polyethoxyethanol (Igepal) 1 % v/v, sodium deoxycholate 0.5 %, SDS 0.1 % and β-mercaptoethanol 10 mm) with protease inhibitors (0.1 mg ml−1 benzamidine, 1 μg ml−1 leupeptin, 0.1 mm phenylmethylsulfonyl fluoride (PMSF) and 2 μg ml−1 aprotinin; Sigma Chemical Co., St Louis, MO, USA) for 20 min on ice to solubilize membrane proteins. The lysates were then passed through a HiTrap Chelating Column (Amersham Biosciences Corp., USA) that binds His-tagged proteins. The washing and elution buffers contained 20 and 40 mm imidazole, respectively, to eliminate non-specific binding. After elution, the collected fractions were concentrated by centrifugation in Ultrafree-MC filter units (Millipore Ltd).
Western blotting
The solubilized membrane proteins were fractionated on 8 % SDS-polyacrylamide gels. The proteins were electrophoretically transferred to Immobilon-P polyvinylidene fluoride membranes (Millipore Ltd) in 25 mm Tris-base, 192 mm glycine and 5 % methanol at 0.07 A for 16 h. The membranes were blocked using 5 % non-fat dry milk (Bio-Rad Laboratories, USA), in Tris-buffered saline (TBS: Tris-HCl 50 mm, NaCl 500 mm, pH 7.5) containing 0.05 % Tween-20 (TTBS) for 2 h at room temperature. Membranes were then incubated for 1 h in primary antibody solutions in 1 % non-fat dry milk in TTBS. The primary antibody against the histidine tag (anti-His; Amersham Biosciences Corp., USA) was used at a dilution of 1:3000, the anti-FLAG M2 antibody (Sigma Chemical Co.) was used at a dilution of 1:1000 and the anti-Kir2.1 antibody (Alomone Labs, Jerusalem, Israel) was used at a dilution of 1:1000. After incubation, the membranes were washed in TTBS and reblocked in 1 % non-fat dry milk in TTBS for 10 min. They were then incubated with horseradish peroxidase-conjugated anti-mouse IgG (1:3000) in 5 % non-fat dry milk in TTBS for 30 min and washed in TTBS three more times. Antibody detection was performed with Western Blot Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences Inc., USA).
Construction of Kir2.1-Kir2.4 tandem
A BamHI and a SacII restriction enzyme site were introduced at the N-terminus and after 10 amino acids of the 5′-untranslated region of Kir2.4 by PCR. A XhoI restriction enzyme site was introduced at the C-terminus. A stop codon in the 5′-untranslated region was mutated to glycine by site-directed mutagenesis. The PCR product and pcDNA3.1+ (Invitrogen Corp., USA) were then digested with BamHI and XhoI and the PCR product was subcloned into pcDNA3.1+. After transformation of competent JM109 cells (Promega US, USA) with the construct and amplification, the presence of the insert was verified by digestion with BamHI and XhoI. A BamHI restriction enzyme site was engineered to the N-terminus of Kir2.1 by PCR. A BamHI and a SacII restriction enzyme site were introduced to the N-terminus. The stop codon of Kir2.1 was mutated by site-directed mutagenesis to GGA (glycine;). The PCR product and the previously obtained Kir2.4-pcDNA3.1+ construct were digested with BamHI. The Kir2.1 sequence was then introduced upstream of the Kir2.4 sequence into pcDNA3.1+. The correct orientation of Kir2.1 was verified by restriction enzyme digestion and subsequent electrophoresis on a 1 % agarose gel. The presence of Kir2.1 and Kir2.4 was verified by complete sequence confirmation. A 10-amino acid sequence of the 5′ untranslated region (UTR) of Kir2.4 (GQIGKGSPHL) was preserved as a linker between Kir2.1 and Kir2.4, based on previous studies showing a 10-amino acid linker to be sufficient for functional integration of linked subunits in the membrane (Lee et al. 1994). A second stop codon in this linker region was mutated to glycine.
The primers for tandem construction were:
GGATCCAGCATGGGCAGTGTGMGAAC (Kir2.1, sense),
GGATCCCCGCGGTCCTATCTCCGAYTCTCGCCG
(Kir2.1, antisense),
GGATCCCCGCGGGGACAAATCGGGAAGGGGTCTCC
(Kir2.4, sense),
and:
CTCGAGTCATGGAGGCAGGGTCAGTGC
(Kir2.4, antisense).
For heterologous expression in Xenopus oocytes the Kir2.1-2.4 tandem was subcloned into the polyadenylation transcription vector pSGEM. The BamHI site between Kir2.1 and Kir2.4 was removed by restriction enzyme digestion with SacII. After purification on a 1 % agarose gel with the QIAquick Gel Extraction Kit (Qiagen Inc., Mississauga, Ontario, Canada), the construct was religated with T4 DNA polymerase (Promega US). After transformation of competent JM109 cells (Promega US) with the construct and amplification, the tandem was harvested by digestion with BamHI and XhoI, isolated on a 1 % agarose gel and further subcloned into pSGEM.
Kir2.1/Kir2.4 co-injection
We then investigated the properties of channels formed by spontaneous Kir2.1 and Kir2.4 co-assembly. To ensure comparable expression of Kir2.1 and 2.4, a sequence based on the tandem construct but containing a stop codon in place of glycine at the distal end of Kir2.1 was used.
The primers were:
GGATCCAGCATGGGCAGTGTGMGAAC (Kir2.1 sense),
CCGCGGTCCTATCTCCGAYTCTCGCCG (Kir2.1 antisense),
GGATCCCCGCGGGGACAAATCTAGAAGGGGTCTCC
(Kir2.4 sense),
and:
CTCGAGTCATGGAGGCAGGGTCAGTGC
(Kir2.4 antisense).
Electrophysiology
The procedures followed for surgery and maintenance of frogs were approved by the Animal Research Ethics Committee of the Montreal Heart Institute. All frogs were humanely killed after a series of experiments. Female Xenopus laevis were anaesthetized in 0.13 % w/v tricaine (Sigma Chemical Co.) for 30 min at 4 °C. Segments of the ovarian lobe were removed through a small abdominal incision. The follicular layer was removed by digestion with 2 U ml−1 collagenase type V (Sigma Chemical Co.) in OR2 solution (mm: NaCl, 82.5; KCl, 2; MgCl2, 1; Hepes, 5, pH 7.6, containing 50 mg l−1 gentamicin solution). Oocytes were stored at 17 °C in SOS solution (mm: NaCl, 100; KCl, 2; CaCl2, 1.8; MgCl2, 1; Hepes, 5, pH 7.6, containing 50 mg l−1 gentamicin solution). For heterologous expression, 6–12 ng of cRNA was injected into stage IV-V Xenopus oocytes, followed by two-electrode voltage clamp 1 to 3 days later. When co-injected with dominant negative constructs, equal amounts of dominant negative and wild-type cRNA were injected. Currents were elicited at room temperature by 750 ms voltage steps from a holding potential of −60 mV with a GeneClamp-500 amplifier and pClamp 6.0 software (Axon Instruments Inc., USA). The external solution contained (mm): KCl, 5; NaCl, 100; MgC12, 2; Hepes, 10; CaC12, 0.3; the pH was adjusted to 7.4 with NaOH. Niflumic acid (10 μM) was added to block Ca2+-dependent Cl− current. Ba2+-containing solutions were superfused until steady state block occurred (generally ≈10 min) before repeating full voltage-clamp protocols. Glass microelectrodes (3 m KCl-filled) had resistances of 1.3–1.8 MΩ.
Data analysis
Data analysis was conducted using Axon Clampfit 6.0, GraphPad Prism 3, SPSS 10 and Microsoft Excel 2000. Data are presented as means ± s.e.m. Statistical comparisons were made with Student's t test. Analysis of variance (ANOVA) was used for multiple comparison. A two-tailed probability of P < 0.05 was taken to indicate statistical significance.
RESULTS
Co-expression of Kir2.1 or 2.4 channels with dominant negative subunits
Figure 1 compares representative currents (IKir2.x) recorded from Xenopus oocytes injected with Kir2.1 or Kir2.4 cRNA (left panels) with those of oocytes co-injected with dn-Kir2.1/Kir2.1 or dn-Kir2.1/Kir2.4 cRNA (middle panels). Corresponding mean current-voltage relations are shown in the right panels. Injection of either subunit alone produced robust inward rectifier currents (left panels). The middle panels show the results of co-injection of dn-Kir2.1 with Kir2.1 or 2.4, into the same sets of oocytes studied on the same days as the corresponding homomeric wild-type constructs. Mean data are shown at the right. As expected, co-expression of Kir2.1 with dn-Kir2.1 (A) strongly suppressed Kir2.1 currents (P < 0.001). Co-injection of Kir2.4 with dn-Kir2.1 (B) substantially reduced IKir2.4 (P < 0.001) in a fashion quite similar to the co-expression of Kir2.1 with dn-Kir2.1 shown in A.
Figure 1. Currents recorded from Xenopus oocytes injected with Kir2.1 or 2.4 cRNA alone (left panels) or together with dn-Kir2.1 (middle panels) and corresponding mean current-voltage relationships (right panels).
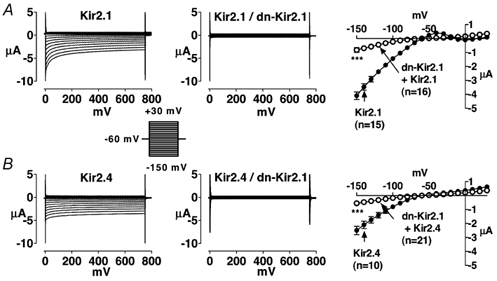
A, results for Kir2.1 and B, results for Kir2.4. Mean ± s.e.m. current-voltage relations for 15 (Kir2.1), 16 (Kir2.1/dn-Kir2.1), 10 (Kir2.4), and 21 (Kir2.4/dn-Kir2.1) oocytes are illustrated on the right. Filled symbols represent currents recorded from oocytes injected with Kir2.x cRNA alone, whereas corresponding open symbols represent currents recorded from oocytes co-injected with equal amounts of Kir2.x cRNA and dn-Kir2.1 cRNA. *** P < 0.001 for significance of difference between current density in co-injected oocytes vs. Kir2.x.
Figure 2 shows results of complementary experiments in which dn-Kir2.4 was co-injected with either Kir2.1 or Kir2.4 subunits. Once again, results obtained following co-injection (middle panels) are compared with results obtained upon injection of the corresponding homomeric wild-type constructs (left panels), with injection and study occurring on the same days and into the same batches of oocytes. Mean data are shown on the right. Co-expression of dn-Kir2.4 with Kir2.1 and Kir2.4 strongly suppressed Kir2.x currents (A, Kir2.1/dn-Kir2.1 P < 0.001; B, Kir2.4/dn-Kir2.4 P < 0.001). The results in Fig. 1 and Fig. 2 suggest co-assembly of Kir2.1 and Kir2.4 subunits.
Figure 2. Currents recorded from Xenopus oocytes injected with Kir2.1or 2.4 cRNA alone (left panels) or together with dn-Kir2.4 (middle panels) and corresponding mean current-voltage relationships (right panels).
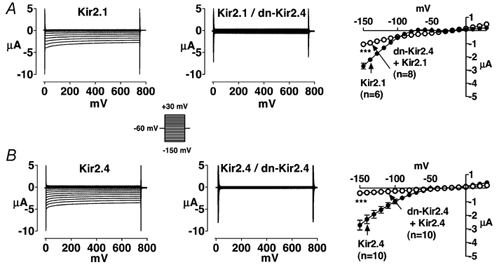
A, results for Kir2.1 and B, results for Kir2.4. Means ± s.e.m. current-voltage relations for 6 (Kir2.1), 8 (Kir2.1/dn-Kir2.4), 10 (Kir2.4), and 10 (Kir2.4/dn-Kir2.4) oocytes are illustrated on the right. Filled symbols represent currents recorded from oocytes injected with Kir2.x cRNA alone, whereas corresponding open symbols represent currents recorded from oocytes co-injected with equal amounts of Kir2.x cRNA and dn-Kir2.4 cRNA. *** P < 0.001 for significance of difference between current density in co-injected oocytes vs. Kir2.x.
Kir2.1-His6 / Kir2.4-FLAG co-precipitation
Figure 3 shows Western blots of lysates obtained from Cos-7 cells transfected with Kir2.1-His6 alone, Kir2.4-FLAG alone or both. A cell lysate of Kir2.4-FLAG-transfected cells probed with anti-FLAG antibody (lane 1) revealed a single band of molecular mass 55 kDa. Lanes 2–7 were obtained with His6-pulldown. Lanes 2 and 3 were obtained from cells transfected with Kir2.1-His6 and probed with antibodies directed against Kir2.1 (anti-Kir2.1, lane 2) or the His6-epitope (lane 3). Both antibodies detected a specific band with a molecular mass of 67 kDa, corresponding to Kir2.1. Tinker et al. (1996) have published a molecular mass of 50 kDa for Kir2.1 expressed in HEK cells. The noted difference in molecular mass might be due to differences in the post-translational modification of Kir2.1 in HEK versus Cos-7 cells. Incubation and probing with the anti-FLAG antibody of the cell lysate of co-transfected cells after His-pulldown (lane 4) detected a band with a molecular mass of 55 kDa corresponding to Kir2.4, indicating protein-protein interactions between His-tagged Kir2.1 (purified by His-pulldown) and FLAG-tagged Kir2.4. In addition to detecting FLAG-tagged Kir2.4 in the Kir2.1-His6/Kir2.4-FLAG co-transfected cell His-pulldown experiment (lane 4), the FLAG antibody also detected a band with a molecular mass of Kir2.1 (67 kDa). We suspected that this was due to cross-reactivity of the anti-FLAG antibody with the Kir2.1-His6 product. Lane 5 shows an experiment to test this notion. The band shown was obtained from cells transfected with Kir2.1-His6 alone, subjected to His-pulldown. The anti-FLAG antibody detected a single band with a molecular mass of ≈67 kDa, corresponding to Kir2.1 suggesting that the band with a molecular mass of 67 kDa in lane 4 was indeed Kir2.1 detected by the anti-FLAG antibody.
Figure 3. Western blots of cell lysates from Cos-7 cells transfected with Kir2.1-His6, Kir2.4-FLAG or both.
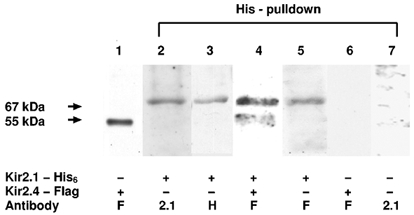
The constructs transfected and antibody probes used are indicated at the bottom. + indicates construct transfected, – indicates construct not present. Primary antibodies are designated by F for anti-FLAG, 2.1 for anti-Kir2.1 and H for anti-His. Arrows on the left indicate molecular masses of specific bands detected by specific primary antibodies. Molecular masses are given in kilodaltons (kDa). Lane 1 is a Western blot of Cos-7 cell lysate transfected with Kir2.4-FLAG and purified on a FLAG-Affinity gel (Sigma Chemical Co.). Cell lysates in lanes 2 to 7 were subjected to His6 -pulldown prior to incubation with the primary antibody. Lanes 2 and 3 were obtained with a lysate of cells transfected with Kir2.1-His6 only and lane 4 was obtained with a lysate of cells co-transfected with Kir2.4-FLAG and Kir2.1-His6. Lane 5 was obtained from cells transfected with Kir2.1-His6 and incubated with anti-FLAG after His-pulldown. Lane 6 is a Western blot of lysate of Cos-7 cells transfected with Kir2.4-FLAG only incubated with anti-FLAG. Lane 7 is a Western blot with crude (non-transfected) Cos-7 cell lysate incubated with anti-Kir2.1.
Lane 6 was obtained with His-pulldown of lysate from cells transfected with Kir2.4-FLAG alone, followed by probing with FLAG antibody. The lack of a signal indicates that association with His-tagged Kir2.1 is essential for FLAG-tagged Kir2.4 to be detected in the sample obtained by His-pulldown. Furthermore, the results shown in lane 6 exclude non-specific binding of the FLAG sequence or Kir2.4 to the His-binding resin. Lane 7 shows a Western blot of crude (non-transfected) Cos-7 cell lysate incubated with anti-Kir2.1 antibody. The band with a molecular mass of ≈67 kDa shown in lane 1 was not detected, indicating that it does not represent non-specific binding of anti-Kir2.1 antibody. Western blotting was also performed with anti-Kir2.1 preincubated with the antigenic peptide supplied by the manufacturer. After pre-incubation with antigen, anti-Kir2.1 no longer identified the band at 67 kDa (data not shown). Similar results were obtained in four such experiments.
Properties of co-expressed Kir2.1 and Kir2.4 and of a covalently linked Kir2.1-2.4 tandem construct
The experiments shown in Figs 1-3 strongly suggest co-assembly of Kir2.1 and 2.4, but provide no information about the ability of heteromeric channels to carry current. We therefore addressed the issue of whether co-assembled channels can conduct current by studying the results of expression of a covalently linked Kir2.1-Kir2.4 tandem construct. Figure 4 shows currents carried by Kir2.1 alone (Fig. 4A), Kir2.4 alone (Fig. 4B), Kir2.1-Kir2.4 tandem constructs (Fig. 4C) and Kir2.1/2.4 co-expression resulting from co-injection (Fig. 4D) under control conditions (left panels) and then in the presence of 10 and 100 μM and 1 mm Ba2+. Recordings for each construct were obtained under all four conditions in each oocyte studied. From the data shown in Fig. 4C it is clear that channels consisting of co-assembled, covalently-linked Kir2.1 and Kir2.4 carry substantial currents. Currents carried by Kir2.1-Kir2.4 tandem constructs (Fig. 4C), as well as by co-injected Kir2.1/2.4 (Fig. 4D) are more sensitive to Ba2+ than currents carried by Kir2.1 (Fig. 4A) or Kir2.4 (Fig. 4B) alone. Currents carried by Kir2.1-Kir2.4 tandem constructs appear smaller than those carried by Kir2.1 or Kir2.4 alone, or by those resulting from co-injection of Kir2.1 and Kir2.4. Corresponding mean current-voltage relationships at the onset of the pulse (left panels) and under steady state conditions (right panels) are shown in Fig. 5. These data confirm the fact that currents carried by the tandem construct are smaller than those carried by homomeric Kir2.1 or 2.4 injection, or by co-injected (but not covalently linked) Kir2.1 and 2.4. They also suggest that co-injected and tandem constructs are more sensitive to Ba2+ than homomeric channels. For example, 10 μM Ba2+ produced near-maximal inhibition of tandem and co-injected constructs, shown in the bottom panels of Fig. 5C and D, whereas it clearly did not for homomeric Kir2.1 and 2.4 shown in the corresponding panels of Fig. 5A and B.
Figure 4. Original current recordings obtained from oocytes injected with Kir2.1 (A), Kir2.4 (B), the Kir2.1-Kir2.4 tandem (C) and co-injected Kir2.1 and 2.4 (D).
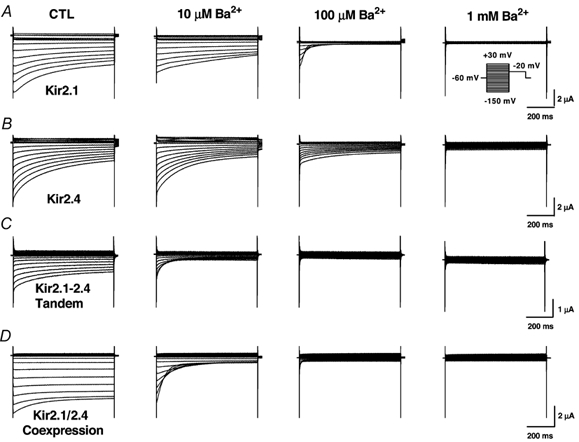
Results are shown for oocytes under control conditions (left panels) and in the presence of 10 μM (left middle panels), 100 μM (right middle panels) and 1 mm (right panels) Ba2+. Currents were elicited by voltage steps from a holding potential of −60 mv to step potentials between −150 mV and + 30 mV in 10 mV increments as shown by the voltage protocol in the inset.
Figure 5. Mean ± s.e.m. current-voltage relations just after the onset of the voltage step (top panels) and under steady state conditions (bottom panels).
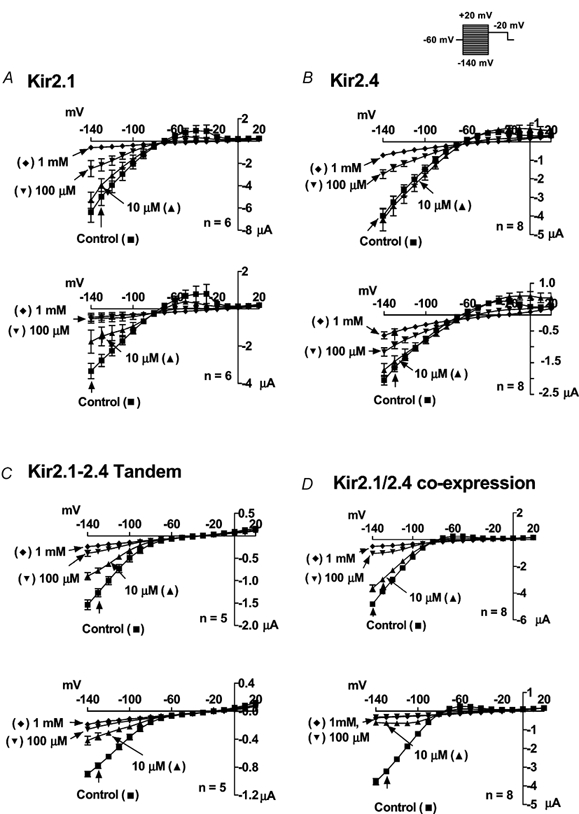
n = 6, 8, 5 and 8 oocytes for Kir2.1 (A), Kir2.4 (B), tandem (C) and co-injected constructs (D), respectively.
We have previously shown that the concentration and voltage dependence of Ba2+ blockade is a signature property of various Kir2.x clones and native IK1 channels (Schram et al. 1999). We therefore determined the voltage-dependent Ba2+-blocking properties of Kir2.1 and Kir2.4 and compared them with Ba2+-block of the tandem construct and co-expressed Kir2.1/2.4. Figure 6 (left panels) shows currents recorded upon stepping to −120 mV in oocytes expressing Kir2.1 (A), Kir2.4 (B), the tandem construct (C), or co-expressed Kir2.1 and Kir2.4 (D). Results are shown under control conditions (○) and after exposure to 0.01 (⋄), 0.1 (▪), 1 (•) and 10 (★) mm Ba2+. Note that 10 μM Ba2+ produced about 30 % inhibition of end-pulse Kir2.1 current in the example shown in Fig. 6A, whereas the same Ba2+ concentration (10 μM) produced much weaker inhibition of the Kir2.4 current shown in 6B. Current resulting from the tandem construct (C) and co-expressed Kir2.1/2.4 (D) was inhibited strongly by 10 μM Ba2+. Corresponding mean Ba2+ concentration- response relations at −120 mV based on all cells studied are shown for each construct in the middle panels. The curves in the middle panels are best-fit relations of the form:
where B is the Ba2+-block at a concentration C. The Ba2+ IC50 at −120 mV for Kir2.1 (15.9 μM, Fig. 6A, middle panel) is about one-quarter of the value for Kir2.4 (72.3 μM, Fig. 6B, middle). On the other hand, the values for the Kir2.1-2.4 tandem (Fig. 6C) and co-expressed Kir2.1 and 2.4 (Fig. 6D) are of the same order, in the range of 4-5 μM, a value one-third that of Kir2.1 and less than one-tenth that of Kir2.4. Significant differences were found between groups (analysis of variance, P < 0.0001) and the different test potentials (analysis of variance, P < 0.0001). There was also a significant group × voltage interaction (analysis of variance, P = 0.0003), indicating significant voltage dependence of intergroup differences. The right panels of Fig. 6 show mean IC50 values for each construct as a function of test potential in comparison to Kir2.1, represented by the circles and dashed line. Significant differences between Kir2.4, Kir2.1-2.4 tandem and Kir2.1/2.4 co-expression, on one hand, and Kir2.1, on the other, are denoted by asterisks. Kir2.4 is clearly less sensitive to Ba2+ than Kir2.1 over the entire voltage range. The tandem and co-expressed channels are significantly more sensitive to Ba2+ than either Kir2.1 or 2.4 alone. Ba2+ block of Kir2.4 (Fig. 6B) is not clearly voltage dependent and shows limited time dependence, in contrast to block of Kir2.1, which is clearly time and voltage dependent, suggesting that the principal Ba2+-blocking site in Kir2.4 may be much more shallow than that in Kir2.1. The tandem (Fig. 6C) and co-expressed constructs (Fig. 6D) show a direct voltage-dependent Ba2+ sensitivity, like that of Kir2.1 and different from that of Kir2.4. This observation may provide supplementary evidence for Kir2.1-2.4 heteromultimer formation.
Figure 6. Ba2+ block of currents resulting from the expression of Kir2.1 (A), Kir2.4 (B), Kir2.1-2.4 tandem (C), co-expressed Kir2.1 and 2.4 (D).
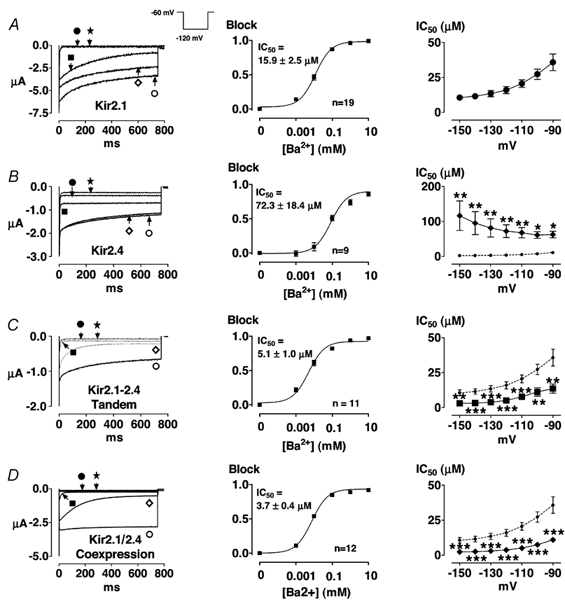
Left panels show original recordings from one oocyte under control conditions (○) and in the presence of Ba2+ at the following mm concentrations: 0.01 (⋄), 0.1 (▪), 1 (•) and 10 (★). The middle panels show corresponding mean ± s.e.m. concentration-response curves based on end-pulse block at each concentration upon hyperpolarization to −120 mV (n = 19, 9, 11 and 12 oocytes for Kir2.1, Kir2.4, tandem and co-injected constructs, respectively). The right panels show mean IC50 values obtained from the type of analysis illustrated in the middle panels, at each test potential studied (n = 19, 9, 11 and 12 oocytes for Kir2.1, Kir2.4, tandem and co-injected constructs, respectively). To facilitate direct comparison of data obtained in oocytes following injection of Kir2.4 and engineered constructs with data for Kir2.1, data for Kir2.1 are reproduced in the right panels, as indicated by the dashed lines. * P < 0.05, ** P < 0.01, *** P < 0.001vs. Kir2.1 at the same voltage.
DISCUSSION
In the present study, we showed that Kir2.4 associates with Kir2.1 subunits, that heteromeric Kir2.1-2.4 channels conduct robust inward rectifying currents and that they have Ba2+-blocking properties different from those of homomeric channels resulting from expression of either individual subunit alone.
Heteromultimer formation in inward rectifier channels
The formation of heterotetrameric K+ channels has been well described for the Shaker-related voltage gated K+ (Kv) channels. Heteromultimerization usually occurs only within a subfamily, but not between members of different Kv subfamilies (Li et al. 1992; Shen et al. 1993). It is believed that the main regions responsible for co-assembly of Kv family subunits are located in the N-terminus and the S1 segment (Li et al. 1992; Shen et al. 1993). Deletion of the N-terminus has the potential to remove the specificity of subfamily co-assembly (Lee et al. 1994).
Co-assembly in the inward rectifier group is less well understood. Heteromultimerization of inward rectifier K+ channels has been shown to occur and to be functionally important in the Kir3 group. The molecular basis of the G-protein-gated inward rectifier potassium and acetylcholine-dependent current (IK,ACh) is a heterotetramer of the inward rectifier channels Kir3.1 and Kir3.4 (Krapivinsky et al. 1995). Functional heteromultimers consisting of Kir3.1 and Kir3.2 exist in the central nervous system (Lesage et al. 1994; Lesage et al. 1995). Tucker et al. (1996) showed that Kir4.1 subunits form heteromeric channels when co-expressed with members of the Kir3 subfamily, but that the resulting channels are not functional. The inhibition of Kir4.1 current is likely to be due to degradation of the co-assembled channels rather than to the formation of stable non-conducting heteromeric channels (Tucker et al. 1996). Glowatzki et al. (1995) showed that Kir1.1 and 4.1 subunits can co-assemble, and that co-assembled channels have distinct functional properties. Oocytes co-injected with cRNA for Kir4.2 and Kir5.1 displayed currents with properties distinct from those expressing Kir4.2 alone. Co-injected oocytes displayed larger currents than Kir4.2, with novel kinetic properties and an increased sensitivity to Ba2+ block at negative potentials, suggesting that Kir4.2 forms functional heteromultimeric channels with Kir5.1, as has been shown for Kir4.1 (Pearson et al. 1999).
Fakler et al. (1996) reported the formation of functionally distinct heterotetramers between Kir2.1 and Kir4.1. When co-expressed in Xenopus oocytes, Kir5.1 and Kir2.1 are efficiently targeted to the cell surface but form electrically silent channels in a stoichiometry-dependent manner. In vivo there is overlapping expression of Kir5.1 and Kir2.1 mRNA in the brain and the kidney. Kir5.1 thus appears to function as a negative regulator of Kir2.1 channel activity in native cells (Derst et al. 2001). Pessia et al. (1996) showed that co-expression of Kir4.1 subunits with Kir5.1 subunits results in functionally distinct channels (Pessia et al. 1996). In contrast to Kir4.1 subunits, which form homomeric channels with a single-channel conductance of 12 pS, and Kir5.1, which does not form conducting channels upon heterologous expression, Kir4.1/5.1 co-expression produces channels with a single-channel conductance of 43 pS. The properties of Kir4.1-5.1 heteromeric channels depend on the order of subunits, and Kir5.1 co-injection did not modify the properties of currents carried by Kir1.1, 2.2, 2.3, 3.1, 3.2 or 3.4 (Pessia et al. 1996). Heteromeric Kir4.1-5.1 channels may function as a proton sensor in the kidney (Tanemoto et al. 2000) and play a role in CO2 chemoreception in central nervous system neurons (Yang et al. 2000). Co-injection of Kv1.1 and Kir2.1 subunits produces currents compatible with the sum of homomeric channels formed by each alone, and injection of a Kir2.1-Kv1.1 tandem construct fails to produce measurable currents despite detection by Western blot of protein with a molecular weight corresponding to the predicted size of the tandem (Tytgat et al. 1996). These results suggest that Kir2.1 and Kv1.1 subunits do not co-assemble to produce functional channels.
Conflicting data have been reported for heteromultimerization within the Kir2 family. Tinker et al. (1996) addressed the regions responsible for homo- and heteromultimer formation among Kir2.1, Kir2.2 and Kir2.3 subunits. Their results indicate that the formation of homomultimers is by far the preferred reaction when Kir2.1 and Kir2.2 or Kir2.3 are co-expressed. The principal regions responsible for the co-assembly of homomeric channels and for the lack of co-assembly between Kir2.1 and Kir2.2, 2.3 and 6.1 appear to be the M2 segment and the proximal C-terminus (Tinker et al. 1996). Fink et al. (1996) studied the structural determinants that define the specificity of co-assembly of Kir2 channels and preclude the formation of heteromers between Kir2.3 and Kir3.2 subunits. They found that chimeric constructs containing the N-terminal of Kir2.3 and the M1-M2-proximal C-terminal moiety of Kir3.2 have a dominant negative effect upon co-injection with Kir2.3, irrespective of whether the C-terminal of the chimeric protein corresponds to Kir2.3 or Kir3.2. Fink et al. (1996) also noted suppression of Kir2.1 currents when Kir2.1 was co-injected along with chimeras containing the Kir2.3 N-terminal and the Kir3.2 M1-M2-proximal C-terminal, irrespective of whether the distal C-terminal was derived from Kir2.3 or 3.2. These findings, along with the co-localization of Kir2.1 and 2.3 in similar brain regions, led the authors to conclude that the N-terminus of Kir2.3 determines co-assembly and that Kir2.1 and 2.3 probably form heterotetramers in vivo (Fink et al. 1996). Very recently Preisig-Müller et al. (2002) showed that Kir2.1, Kir2.2 and Kir2.3 can form heterotetramers with distinct properties and suggested that diversity of inward rectifier channel co-assembly might underlie different phenotypes of Andersen's syndrome. Using a yeast two-hybrid system they found that co-assembly occurs between the C- and N-termini of Kir2 subunits.
Our findings were similar to those of Preisig-Müller et al. (2002), in that we found that Kir2.1 can co-assemble with Kir2.4 to form functional heterotetramers and that channels consisting of different subunits may have specific properties different from those of homomeric channels composed of either subunit alone. Furthermore, we found that co-assembly of Kir2.1 and Kir2.4, whether via co-injection or expression of concatemers, led to currents with Ba2+ IC50 values lower than those of Kir2.1 and Kir2.4 alone. Preisig-Müller et al. (2002) found that co-assembled Kir2.1-3 channels had Ba2+ sensitivities of the same order as those of the more sensitive homomeric constituent subunit.
Novelty and potential significance
Heteromultimerization is a potentially important contributor to the functional diversity of K+ channels (Coetzee et al. 1999). The native IK,ACh current, an ion-conducting pathway of central importance to parasympathetic nervous system effector pathways, is composed of heteromultimeric Kir3.1 and Kir3.4 subunits (Krapivinsky et al. 1995). There is evidence that the important transmembrane gradient in the slow delayed rectifier current IKs, a key determinant of the cardiac ventricular transmural gradient in action potential duration, (Liu & Antzelevitch, 1995) is caused by differential transmural distribution of a splice variant of KvLQT1 with dominant negative function (Pereon et al. 2000). Heteromultimer formation may contribute to the properties of cardiac transient outward currents in tissues that express both Kv1.4 and Kv1.5 proteins (Po et al. 1993). Differential heteromultimerization among Kir2.1-3 subunits might underlie the different phenotypes of Andersen's syndrome (Preisig-Müller et al. 2002).
In the present study, we demonstrate with both biochemical and electrophysiological methods that Kir2.4 subunits co-assemble with Kir2.1 subunits. Kir2.1 and 2.4 were co-purified by His6-pulldown when only Kir2.1 was covalently bound to His6, dominant negative Kir2.1 suppressed Kir2.4 currents upon co-expression and a tandem Kir2.1-2.4 construct carried Ba2+-sensitive currents with voltage-dependent Ba2+ sensitivity similar to that of co-expressed Kir2.1 and 2.4. The results of our Kir2.1/2.4 co-expression and Kir2.1-2.4 chimeric channel studies suggest that heteromeric Kir2.1-2.4 channels have properties that are different from those of homomeric channels formed by either subunit alone. In particular, the Ba2+ sensitivity of heteromeric channels is clearly greater than that of homomeric Kir2.1 channels and much greater (by an order of magnitude) than that of Kir2.4 channels. This is similar to the findings of Pearson et al. (1999), who showed that co-expression of Kir4.2 and Kir5.1 results in currents with higher Ba2+ sensitivity than when Kir4.2 is expressed alone. Currents resulting from Kir2.1 and Kir2.4 co-expression were of the same magnitude as Kir2.1 or Kir2.4 currents whereas currents carried by Kir2.1-2.4 chimeras were reduced by approximately 50 %, possibly due to structural constraints limiting functional insertion.
Overlapping expression of Kir2.1-Kir2.3 mRNA in the rat brain has been reported (Morishige et al. 1993; Fink et al. 1996; Horio et al. 1996; Karschin et al. 1996). Northern blot analysis and RT-PCR have shown that Kir2.1 and Kir2.4 mRNA is expressed in the brain, heart, kidney and skeletal muscle (Kubo et al. 1993; Takahashi et al. 1994; Topert et al. 1998; Derst et al. 2001). In situ hybridization studies of rat brain tissue have shown both Kir2.1 and Kir2.4 to be expressed in the pons, the facial nucleus and strongly in the midbrain (Karschin et al. 1996; Topert et al. 1998). The fact that Kir2.1 and Kir2.4 channels are expressed in the same regions, as well as their ability to co-assemble, raises the possibility that Kir2.1-2.4 heterotetramers may contribute to inward rectifier function in the CNS. A recent study by Liu et al. (2001) found Kir2.4 to be present in guinea-pig heart, but immunocytochemical studies showed Kir2.4 to be present only in neuronal elements. More experiments are warranted to determine whether Kir2.4 is present in myocardial tissues of other species.
Potential limitations
Changes in subunit arrangement can affect channel properties, as has been shown for Kir4.1 and Kir5.1 (Pessia et al. 1996). The formation of heteromeric channels with different Kir2.1/Kir2.4 subunit arrangements and stoichiometry might therefore lead to the expression of channels with functionally different properties. Further studies are required to assess the effects of different subunit arrangements and stoichiometries on the properties of currents carried by systems co-expressing Kir2.1 and 2.4 subunits. There are discrepant observations regarding the structural determinants of Kir2.x subunit co-assembly (Fink et al. 1996; Tinker et al. 1996; Preisig-Müller et al. 2002). Further work is necessary to establish the structural basis for the ability of Kir2.4 subunits to co-assemble with Kir2.1, as well as to establish whether Kir2.4 can also co-assemble with other inward rectifier channels. The structural basis for the differing Ba2+ sensitivities of Kir2 subfamily channels, as well as the mechanisms that lead to enhanced Ba2+ sensitivity of Kir2.1-2.4 heteromultimers, are subjects of considerable potential interest that our study raises but cannot address. Single-channel experiments to evaluate the detailed biophysical properties of the channels formed by the tandem construct in comparison with homomeric Kir2.1 and 2.4 would be quite interesting, but are beyond the scope of the present work. Finally, more detailed studies of the quantity and localization of Kir2.x protein expression would be of interest in order to assess the potential physiological importance of Kir2.4-Kir2.1 heteromultimerization.
Acknowledgments
The authors wish to thank Drs Terence E. Hebert and Bruce G. Allen, Montreal Heart Institute for helpful discussions on construction of Kir2.4-FLAG and His-pulldown, and Chantal St-Cyr, Evelyn Landry and Xiao Fan Yang for their excellent and reliable technical assistance. They thank Dr Lily Y. Jan, San Francisco for Kir2.1, Dr Barbara A. Wible, Cleveland for Kir2.2, Dr Carol A. Vandenberg, Santa Barbara for Kir2.3, Dr Andreas Karschin, Göttingen, Germany for the gift of Kir2.4 and Dr Andrew Tinker, London, England for the generous gift of dn-Kir2.1 and Kir2.1-His6. Dr Schram wishes to thank the Ernst and Berta Grimmke Foundation, Düsseldorf, Germany, the Canadian Institutes of Health Research (CIHR) and Aventis Pharma for fellowship support, as well as CIHR and the Quebec Heart and Stroke Foundation for operating support awarded to Dr Nattel.
REFERENCES
- Ashen MD, O'Rourke B, Kluge KA, Johns DC, Tomaselli GF. Inward rectifier K+ channel from human heart and brain: cloning and stable expression in a human cell line. American Journal of Physiology. 1995;268:H506–511. doi: 10.1152/ajpheart.1995.268.1.H506. [DOI] [PubMed] [Google Scholar]
- Chen F, Wetzel GT, Friedman WF, Klitzner TS. Single-channel recording of inwardly rectifying potassium currents in developing myocardium. Journal of Molecular and Cellular Cardiology. 1991;23:259–267. doi: 10.1016/0022-2828(91)90062-q. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz DM, Rudy B. Molecular diversity of K+ channels. Annals of the New York Academy of Sciences. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Derst C, Karschin C, Wischmeyer E, Hirsch JR, Preisig-Muller R, Rajan S, Engel H, Grzeschik K, Daut J, Karschin A. Genetic and functional linkage of Kir5. 1 and Kir2.1 channel subunits. FEBS Letters. 2001;491:305–311. doi: 10.1016/s0014-5793(01)02202-5. [DOI] [PubMed] [Google Scholar]
- Fakler B, Bond CT, Adelman JP, Ruppersberg JP. Heterooligomeric assembly of inward-rectifier K+ channels from subunits of different subfamilies: Kir2. 1 (IRK1) and Kir4.1 (BIR10) Pflügers Archiv. 1996;433:77–83. doi: 10.1007/s004240050251. [DOI] [PubMed] [Google Scholar]
- Fink M, Duprat F, Heurteaux C, Lesage F, Romey G, Barhanin J, Lazdunski M. f chimeras provide evidence for homo and heteromultimeric assembly of inward rectifier K+ channel proteins via their N-terminal end. FEBS Letters. 1996;378:64–68. doi: 10.1016/0014-5793(95)01388-1. [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Fakler G, Brandle U, Rexhausen U, Zenner HP, Ruppersberg JP, Fakler B. Subunit-dependent assembly of inward-rectifier K+ channels. Proceedings of the Royal Society. 1995;261:251–611. doi: 10.1098/rspb.1995.0145. [DOI] [PubMed] [Google Scholar]
- Green WN, Millar NS. Ion-channel assembly. Trends in Neurosciences. 1995;18:280–287. [PubMed] [Google Scholar]
- Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophysical Journal. 1994;66:1061–1077. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Roeder RG. Purification of His-tagged proteins in non-denaturing conditions suggests a convenient method for protein interaction studies. Nucleic Acids Research. 1991;19:6337–6338. doi: 10.1093/nar/19.22.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horio Y, Morishige K, Takahashi N, Kurachi Y. Differential distribution of classical inwardly rectifying potassium channel mRNAs in the brain: comparison of IRK2 with IRK1 and IRK3. FEBS Letters. 1996;379:239–243. doi: 10.1016/0014-5793(95)01519-1. [DOI] [PubMed] [Google Scholar]
- Hughes BA, Kumar G, Yuan Y, Swaminathan A, Yan D, Sharma A, Plumley L, Yang-Feng TL, Swaroop A. Cloning and functional expression of human retinal Kir2. 4, a pH- sensitive inwardly rectifying K+ channel. American Journal of Physiology – Cell Physiology. 2000;279:C771–784. doi: 10.1152/ajpcell.2000.279.3.C771. [DOI] [PubMed] [Google Scholar]
- Ishii K, Yamagishi T, Taira N. Cloning and functional expression of a cardiac inward rectifier K+ channel. FEBS Letters. 1994;338:107–111. doi: 10.1016/0014-5793(94)80126-6. [DOI] [PubMed] [Google Scholar]
- Josephson IR, Brown AM. Inwardly rectifying single-channel and whole cell K+ currents in rat ventricular myocytes. Journal of Membrane Biology. 1986;94:19–35. doi: 10.1007/BF01901010. [DOI] [PubMed] [Google Scholar]
- Josephson IR, Sperelakis N. Developmental increases in the inwardly-rectifying K+ current of embryonic chick ventricular myocytes. Biochimica et Biophysica Acta. 1990;1052:123–127. doi: 10.1016/0167-4889(90)90066-m. [DOI] [PubMed] [Google Scholar]
- Karschin C, Dissmann E, Stuhmer W, Karschin A. IRK(1–3) and GIRK(1–4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. Journal of Neuroscience. 1996;16:3559–3570. doi: 10.1523/JNEUROSCI.16-11-03559.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama H, Morishige KI, Takahashi N, Zanelli JS, Fass DN, Kurachi Y. Molecular cloning, functional expression and localization of a novel inward rectifier potassium channel in the rat brain. FEBS Letters. 1994;341:303–307. doi: 10.1016/0014-5793(94)80478-8. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–411. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Lee TE, Philipson LH, Kuznetsov A, Nelson DJ. Structural determinant for assembly of mammalian K+ channels. Biophysical Journal. 1994;66:667–673. doi: 10.1016/s0006-3495(94)80840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Duprat F, Fink M, Guillemare E, Coppola T, Lazdunski M, Hugnot JP. Cloning provides evidence for a family of inward rectifier and G-protein coupled K+ channels in the brain. FEBS Letters. 1994;353:37–42. doi: 10.1016/0014-5793(94)01007-2. [DOI] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Heurteaux C, Fosset M, Romey G, Barhanin J, Lazdunski M. Molecular properties of neuronal G-protein-activated inwardly rectifying K+ channels. Journal of Biological Chemistry. 1995;270:28660–28667. doi: 10.1074/jbc.270.48.28660. [DOI] [PubMed] [Google Scholar]
- Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1300. doi: 10.1126/science.1519059. [DOI] [PubMed] [Google Scholar]
- Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell Circulation Research. 1995;76:351–365. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- Liu GX, Derst C, Schlichthorl G, Heinen S, Seebohm G, Brüggemann A, Kummer W, Veh RW, Daut J, Preisig-Muller R. Comparison of cloned Kir2 channels with native inward rectifier K+ channels from guinea-pig cardiomyocytes. Journal of Physiology. 2001;532:115–126. doi: 10.1111/j.1469-7793.2001.0115g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhina EN, Kelly AJ, Lopatin AN, Mercer RW, Nichols CG. Cloning and expression of a novel human brain inward rectifier potassium channel. The Journal of Biological Chemistry. 1994;269:20468–20744. [PubMed] [Google Scholar]
- Morishige K, Takahashi N, Findlay I, Koyama H, Zanelli JS, Peterson C, Jenkins NA, Copeland NG, Mori N, Kurachi Y. Molecular cloning, functional expression and localization of an inward rectifier potassium channel in the mouse brain. FEBS Letters. 1993;336:375–380. doi: 10.1016/0014-5793(93)80840-q. [DOI] [PubMed] [Google Scholar]
- Morishige K, Takahashi N, Jahangir A, Yamada M, Koyama M, Zanelli JS, Kurachi Y. Molecular cloning and functional expression of a novel brain-specific inward rectifier potassium channel. FEBS Letters. 1994;346:251–256. doi: 10.1016/0014-5793(94)00483-8. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annual Review of Physiology. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Pearson WL, Dourado M, Schreiber M, Salkoff L, Nichols CG. Expression of a functional Kir4 family inward rectifier K+ channel from a gene cloned from mouse liver. Journal of Physiology. 1999;514:639–653. doi: 10.1111/j.1469-7793.1999.639ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereon Y, Demolombe S, Baro I, Drouin E, Charpentier F, Escande D. Differential expression of KvLQT1 isoforms across the human ventricular wall. American Journal of Physiology – Heart and Circulatory Physiology. 2000;278:H1908–1915. doi: 10.1152/ajpheart.2000.278.6.H1908. [DOI] [PubMed] [Google Scholar]
- Perier F, Radeke CM, Vandenberg CA. Primary structure and characterization of a small-conductance inwardly rectifying potassium channel from human hippocampus. Proceedings of the National Academy of Sciences of the USA. 1994;91:6240–6244. doi: 10.1073/pnas.91.13.6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO Journal. 1996;15:2980–2987. [PMC free article] [PubMed] [Google Scholar]
- Po S, Roberds S, Snyders DJ, Tamkun MM, Bennett PB. Heteromultimeric assembly of human potassium channels. Molecular basis of a transient outward current? Circulation Research. 1993;72:1326–1336. doi: 10.1161/01.res.72.6.1326. [DOI] [PubMed] [Google Scholar]
- Preisig-Müller R, Schlichthörl G, Goerge T, Heinen S, Bruggemann A, Rajan S, Derst C, Veh RW, Daut J. Heteromerization of Kir2. x potassium channels contributes to the phenotype of Andersen's syndrome. Proceedings of the National Academy of Sciences of the USA. 2002;99:7774–7779. doi: 10.1073/pnas.102609499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab-Graham KF, Radeke CM, Vandenberg CA. Molecular cloning and expression of a human heart inward rectifier potassium channel. NeuroReport. 1994;5:2501–2555. doi: 10.1097/00001756-199412000-00024. [DOI] [PubMed] [Google Scholar]
- Schram G, Wang Z, Nattel S. Molecular composition of human cardiac IK1 evaluated by barium blocking profile. Circulation. 1999;100 I633. [Google Scholar]
- Shen NV, Chen X, Boyer MM, Pfaffinger PJ. Deletion analysis of K+ channel assembly. Neuron. 1993;11:67–76. doi: 10.1016/0896-6273(93)90271-r. [DOI] [PubMed] [Google Scholar]
- Slesinger PA, Patil N, Liao YJ, Jan YN, Jan LY, Cox DR. Functional effects of the mouse weaver mutation on G protein-gated inwardly rectifying K+ channels. Neuron. 1996;16:321–311. doi: 10.1016/s0896-6273(00)80050-1. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Morishige K, Jahangir A, Yamada M, Findlay I, Koyama H, Kurachi Y. Molecular cloning and functional expression of cDNA encoding a second class of inward rectifier potassium channels in the mouse brain. Journal of Biological Chemistry. 1994;269:23274–23299. [PubMed] [Google Scholar]
- Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5. 1 with Kir4.1. Journal of Physiology. 2000;525:587–592. doi: 10.1111/j.1469-7793.2000.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Qin CL, Yang XC. Cloning, localization, and functional expression of a human brain inward rectifier potassium channel (hIRK1) Receptors and Channels. 1995;3:175–833. [PubMed] [Google Scholar]
- Tang W, Yang XC. Cloning a novel human brain inward rectifier potassium channel and its functional expression in Xenopus oocytes. FEBS Letters. 1994;348:239–433. doi: 10.1016/0014-5793(94)00612-1. [DOI] [PubMed] [Google Scholar]
- Tinker A, Jan YN, Jan LY. Regions responsible for the assembly of inwardly rectifying potassium channels. Cell. 1996;87:857–868. doi: 10.1016/s0092-8674(00)81993-5. [DOI] [PubMed] [Google Scholar]
- Topert C, Doring F, Derst C, Daut J, Grzeschik KH, Karschin A. Cloning, structure and assignment to chromosome 19q13 of the human Kir2. 4 inwardly rectifying potassium channel gene (KCNJ14) Mammalian Genome. 2000;11:247–249. doi: 10.1007/s003350010047. [DOI] [PubMed] [Google Scholar]
- Topert C, Doring F, Wischmeyer E, Karschin C, Brockhaus J, Ballanyi K, Derst C, Karschin A. Kir2. 4: a novel K+ inward rectifier channel associated with motoneurons of cranial nerve nuclei Journal of Neuroscience. 1998;18:4096–1055. doi: 10.1523/JNEUROSCI.18-11-04096.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Bond CT, Herson P, Pessia M, Adelman JP. Inhibitory interactions between two inward rectifier K+ channel subunits mediated by the transmembrane domains. Journal of Biological Chemistry. 1996;271:5866–5870. doi: 10.1074/jbc.271.10.5866. [DOI] [PubMed] [Google Scholar]
- Tytgat J, Buyse G, Eggermont J, Droogmans G, Nilius B, Daenens P. Do voltage-gated Kv1.1 and inward rectifier Kir2.1 potassium channels form heteromultimers? FEBS Letters. 1996;390:280–244. doi: 10.1016/0014-5793(96)00674-6. [DOI] [PubMed] [Google Scholar]
- Wahler GM. Developmental increases in the inwardly rectifying potassium current of rat ventricular myocytes. American Journal of Physiology. 1992;262:C1266–1272. doi: 10.1152/ajpcell.1992.262.5.C1266. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yue L, White M, Pelletier G, Nattel S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation. 1998;98:2422–2428. doi: 10.1161/01.cir.98.22.2422. [DOI] [PubMed] [Google Scholar]
- Wible BA, De Biasi M, Majumder K, Taglialatela M, Brown AM. Cloning and functional expression of an inwardly rectifying K+ channel from human atrium. Circulation Research. 1995;76:343–500. doi: 10.1161/01.res.76.3.343. [DOI] [PubMed] [Google Scholar]
- Wood LS, Tsai TD, Lee KS, Vogeli G. Cloning and functional expression of a human gene, hIRK1, encoding the heart inward rectifier K+-channel. Gene. 1995;163:313–317. doi: 10.1016/0378-1119(95)00244-z. [DOI] [PubMed] [Google Scholar]
- Yang Z, Xu H, Cui N, Qu Z, Chanchevalap S, Shen W, Jiang C. Biophysical and molecular mechanisms underlying the modulation of heteromeric Kir4.1-Kir5.1 channels by CO2 and pH. Journal of General Physiology. 2000;116:33–45. doi: 10.1085/jgp.116.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]