

Abstract
The ROMK subtypes of inward-rectifier K+ channels mediate potassium secretion and regulate NaCl reabsorption in the kidney. Loss-of-function mutations in this pH-sensitive K+ channel cause Bartter's disease, a familial salt wasting nephropathy. One disease-causing mutation truncates the extreme COOH-terminus and induces a closed gating conformation. Here we identify a region within the deleted domain that plays an important role in pH-dependent gating. The domain contains a structural element that functionally interacts with the pH sensor in the cytoplasmic NH2-terminus to set a physiological range of pH sensitivity. Removal of the domain shifts the pKa towards alkaline pH values, causing channel inactivation under physiological conditions. Suppressor mutations within the pH sensor rescued channel gating and trans addition of the cognate peptide restored pH sensitivity. A specific interdomain interaction was revealed in an in vitro protein-protein binding assay between the NH2- and COOH-terminal cytoplasmic domains expressed as bacterial fusion proteins. These results provide new insights into the molecular mechanisms underlying Kir channel regulation and channel gating defects that are associated with Bartter's disease.
A definitive link between the Kir1.1 (ROMK or KCNJ1) gene product (Ho et al. 1993), the renal secretory K+ channel, and kidney function (Simon et al. 1996b;Karaloyi & International Collaborative Study Group for Bartter-like Syndromes, 1997) has been established by a familial salt wasting nephropathy called Bartter's syndrome (Bartter et al. 1962). Reflecting genetic defects in the renal concentrating mechanism, the disorder is characterized by a constellation of fluid and electrolyte abnormalities, including polyuria, hypokalaemia, metabolic alkalosis and a tendency to develop hypotension despite elevated aldosterone levels (Guay-Woodford, 1998). In fact, loss-of-function mutations in each of the major components of the NaCl reabsorbtive machinery in the thick ascending limb of Henle's loop (TAL) have been linked with this genetically heterogenous disorder (Simon et al. 1996a, b, 1997; Karaloyi & International Collaborative Study Group for Bartter-like Syndromes, 1997). Consistent with an essential role of the secretory K+ channel in the TAL (Giebisch, 1998), loss-of-function mutations in the KCN1 (Kir1.1) gene co-segregate with Bartter's syndrome.
The discovery of disease-causing mutations in Kir1.1 not only provides valuable insights into the role of this channel in health and disease, but also lends important clues about structural determinants of function. Like other inward rectifiers, Kir1.1 channels are formed by the tetrameric arrangement of identical subunits. The core domain of each subunit consists of two transmembrane domains and a ‘P’ loop that contribute to the permeation pathway and the potassium selectivity filter (Nichols & Lopatin, 1997). The cytoplasmic NH2- and COOH-termini, flanking the core domain, contain determinants of channel regulation (Xu et al. 1996; Choe et al. 1997; MacGregor et al. 1998; Huang et al. 1998), conduction (Lopatin et al. 1994; Wible et al. 1994; Yang et al. 1995) and oligomerization (Tinker et al. 1996; Koster et al. 1998). Mutations within each of these domains have pointed to residues that regulate channel permeation and gating (Derst et al. 1997, 1998; Karaloyi & International Collaborative Study Group for Bartter-like Syndromes, 1997; Schulte et al. 1999). Interestingly, many of the mis-sense mutations appear to cluster around residues that are known to be required for regulation by intracellular pH and PKA (Schulte et al. 1999).
Like the native channel (Wang et al. 1990), Kir1.1 channels are gated by intracellular pH, closing with cytoplasmic acidification (Tsai et al. 1995, Choe et al. 1997; McNicholas et al. 1998). Recent studies have begun to illuminate the pH-sensing mechanism, whereby two arginine residues in the cytoplasmic NH2- and COOH-termini, R41 and R311, (Fakler et al. 1996; Schulte et al. 1999) functionally interact with the pH sensor, K80, in the cytoplasmic NH2-terminus (Fakler et al. 1996; Schulte et al. 1999) to adjust the titration of the residue into a physiological range of pH sensitivity. Underscoring the critical role of pH-dependent gating in physiological regulation of channel activity, recent studies of Leipziger et al. (2001) indicate that PKA activation of Kir1.1, involving phosphorylation of residues near the R41-K80-R311 triad (Xu et al. 1996), is facilitated by an acid shift in the pKa. Interestingly, Bartter's disease-causing mutations in Kir1.1 that cluster around the triad, particularly C49Y, I51T, D74Y and L220F, shift the pKa into the alkaline range and cause channel closure at physiological pHi (Schulte et al. 1999).
We previously reported that a Bartter's disease-causing mutation in Kir1.1a (T332 frameshift) that removes a more distal structure – the extreme COOH-terminal 60 amino acids – disrupts channel function by locking the channel in a long-lived closed state (Flagg et al. 1999). Here we show that the extreme COOH-terminus acts as an important determinate of Kir1.1 channel gating by intracellular pH. The COOH-terminus contains a structure that appears to be required for an interdomain interaction with the cytoplasmic NH2-terminus, which in turn determines the physiological range of pH sensitivity. Deletion of the domain shifts the open probability-pHi relation in an alkaline direction, causing channel closure at normal pHi. These results provide new insights into the structural determinants of pHi gating in the Kir1.1 channel.
METHODS
cDNAs and mutagenesis
Mutagenesis was performed by overlap extension PCR (Ho et al. 1989). Deletion mutants (Kir1.1a 351X, Kir1.1a 361X and Kir1.1a 388X) were constructed by introducing three stop codons in frame at the appropriate location, as described previously (Flagg et al. 1999). All constructs were cloned between 5′ and 3′ untranslated regions of the Xenopus β-globin gene in the modified pSD64 vector to increase expression efficiency (Krieg & Melton, 1984). This vector also contains a poly-adenylate sequence in the 3′UTR (dA23dC30). Appropriate cDNA sequences were verified by dye termination sequencing (Applied Biosystem, Foster City, CA, USA).
cRNA synthesis
Complementary RNA was transcribed in vitro in the presence of capping analogue G(5′)ppp(5′)G using PstI or SmaI linearized cDNA templates. SP6 RNA polymerase was used in all reactions (mMESSAGE mMACHINE; Ambion, Austin, TX, USA). Following DNaseI treatment, cRNA was purified by phenol- chloroform extraction and ammonium acetate/isopropanol precipitation. Yield and concentration were measured spectrophotometrically and confirmed by agarose gel electrophoresis.
Oocyte isolation and injection
Female Xenopus laevis frogs were obtained from NASCO (Fort Atkinson, WI, USA). Standard protocols were followed for the isolation and care of Xenopus laevis oocytes. Briefly, frogs were anaesthetized by immersion in 0.15 % 3-aminobenzoate and a partial oophorectomy was performed through an abdominal incision. Oocyte aggregates were manually dissected from the ovarian lobes and then incubated in a calcium-free ORII medium (mm): 82.5 NaCl, 2 KCl, 1 MgCl2, 5 N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (Hepes)) containing collagenase (Sigma type IA, 2 mg ml−1) for ∼2 h at room temperature to remove the follicular layer. After oocytes were washed extensively with collagenase-free ORII, they were placed in a modified L15 medium (50 % Leibovitz's medium, 10 mm Hepes, pH 7.4) and stored at 19 °C. Twelve to 24 h after isolation, healthy looking Dumont stage V-VI oocytes were pneumatically injected (PV820 picopump, World Precision Instruments) with 50 nl of water containing 0-50 ng of cRNA and then stored in L15 medium at 19 °C. Channel activity was assessed 1-5 days post-injection. After the final oocyte collection, frogs were humanely killed under anaesthesia. Treatment of frogs followed procedures approved by the Institutional Animal care and use Committee.
Peptide synthesis and injection
A peptide corresponding to amino acids 332-362 of Kir1.1a (TKEGKYRVDFHNFGKTVEVETPHCAMCLYNE) was synthesized by the Biopolymer Laboratory at the University of Maryland School of Medicine. To increase peptide stability, both ends were capped with an amino group. The lyophilized peptide was resuspended in deionized water and the stock solution (1 mg ml−1) stored at −20 °C.
The effects of both acute (injected < 2 h prior to experiment) and chronic (injected 12-24 h prior to experiment) application of the synthetic peptide were examined. In all cases, 50 ng of the peptide was injected. This quantity represents a several order of magnitude molar excess of the peptide compared with the channel based on estimates of channel number from total macroscopic current. Since both acute and chronic peptide application produced the same effect, the data were combined for analysis.
Recording solutions
The present study is focused on the pH-dependent gating of wild type and mutant forms of Kir1.1a. For the reliable control of intracellular pH in Xenopus oocytes, extracellular pH was changed in the presence of a permeant weak acid, butyric acid, as described by Leipziger et al. (2001). After incubation in the control 45 mm K+ solution (mm): 45 KCl, 45 N-methyl-d-glucamine (NMDG)-Cl, 1 CaCl2, 1 MgCl2 and 5 Hepes, pH 7.4), oocytes were exposed to a modified 45 mm K+ solution containing 3 mm sodium butyrate. Decreasing the pH (7.4, 6.9, 6.4 and 5.4) of the bathing solution (containing butyrate) caused a concomitant decrease in the intracellular pH as assessed with pH-sensitive microelectrodes. Where required, barium acetate was added at a concentration of 1 mm.
pH microelectrodes and measurement of intracellular pH
Ion-selective microelectrodes were used to monitor intracellular pH (pHi) of oocytes, as previously described (Sciortino & Romero, 1999; Romero et al. 1997, 2001). Intracellular ion activity is measured as the difference between the pH electrode and a KCl voltage electrode impaled into the oocyte; and membrane potential is the potential difference between the KCl microelectrode and an extracellular calomel electrode (Romero et al. 1997). Briefly, pH microelectrodes were fabricated using filamented borosilicate glass pulled to 0.5 μm tips, silanized at 210 °C with bis-(dimethylamino)-dimethylsilane (Fluka Chemical, Ronkonkoma, NY, USA) and shanks coated with Sylgard (Dow Corning, Midland, MI, USA). Micropipettes were cooled under vacuum, and the tips filled with a H+ ionophore I-cocktail B ion-selective resin (Fluka Chemical, Ronkonkoma, NY) and back-filled with (mm): 40 KH2PO4, 23 NaOH, 15 NaCl, pH 7.0. The pH electrodes were calibrated using pH 6.0 and 8.0 (traceable National Bureau Standards, Fisher Scientific, Pittsburgh, PA, USA) followed by point calibration. These pH microelectrodes had slopes of at least −56 mV (decade change)−1.
Electrophysiology
Whole cell oocyte currents were monitored using a two-microelectrode voltage clamp equipped with a bath-clamp circuit (OC-725B; Warner, New Haven, CT, USA), as described before (Welling, 1997). Voltage-sensing and current-injecting electrodes had resistances of 0.5-1.5 MΩ when back-filled with 3 m KCl. After a stable impalement was attained, such that both electrodes measured the same membrane potential, pulse protocols were conducted. Stimulation and data acquisition were performed with a Macintosh Centris 650 computer using an Instrutech ITC16 analog-to-digital, digital-to-analog converter and Pulse software (HEKA, Southboro, MA, USA). Data were filtered at 1 kHz and digitized on-line at 2 kHz to the hard disk using Pulse and IGOR (Wave Metrics, Inc., Lake Oswego, OR, USA) for later analysis.
For macroscopic titration experiments, oocytes were sequentially perfused with a series of gradually decreasing pH solutions. Following stable impalement and assessment of baseline current at Vm = −100 mV (500 ms pulse from Vhold = −20 mV) in the control solution (no butyrate), butyrate-containing solutions at different pH were perfused in succession. Both inward (Vm = −100 mV) and outward (Vm = +40 mV) currents were monitored every 5 s throughout each experiment (Vhold = −20 mV). After perfusion of each solution, the current was allowed to reach a new steady state (≈5 min after solution exchange) and then measured.
Single-channel properties were assessed 1-5 days post-injection with the patch-clamp technique (Hamill et al. 1981). In these studies, the vitelline membrane was removed from oocytes following hyperosmotic shrinking (Methfessel et al. 1986). Patch-clamp electrodes, pulled from Corning-Kovar 7052 borsilicate glass, had resistances of 1-5 MΩ. Single-channel currents were measured with an Axopatch 200A patch-clamp amplifier, digitized at a sampling rate of 47 kHz using a VR-10B digital data recorder (Instrutech, Great Neck, NY, 11021) and stored on videotape. Data were acquired and analysed using the Acquire and TAC family of programs (Bruxton Corporation, Seattle, WA USA). Data were replayed, filtered with an eight-pole Bessel filter (Frequency Devices 900) at a cut-off frequency of 1 kHz, and sampled at least five times the filtering frequency. A 50 % threshold criterion was used to detect events. Channel open probability, Po, was calculated from 1-3 min continuous records using the equation:
where tn is the fraction open time spent at each of the observed levels and N is the number of levels. In all experiments with mutant channels, data analysis was limited to patches that contained one channel. All analysis of the wild-type channel was limited to patches that contained fewer than or equal to five channels, as determined by counting the maximum number of current levels during the course of the experiment and/or by determining the final current amplitude following patch excision and channel rundown at the end of an experiment. For Po diary plots, continuous records were broken into sequential 5-10 s segments, and single-channel open probability was assessed as above. Open and closed dwell-time histograms (logarithmic time scale, 10 bins decade−1) were constructed from 15-60 s records containing one channel and fitted to exponential distributions using the Maximum Likelihood Method (Sigworth & Sine, 1987). The single-channel current magnitude was estimated by fitting Gaussian distributions to the current amplitude histograms or by measuring the amplitudes directly from analogue current traces. Inward slope conductance was assessed from such current measurements at −120 to −40 mV.
Data analysis
To determine pH-sensitivity, relative current values, I/Imax, were plotted as a function of pH for each titration experiment and fitted with the modified Hill equation:
where Ka is the half-maximal inhibitory proton concentration and nH is the Hill coefficient. pH sensitivity is reported as the apparent pKa, where pKa = −log(Ka). All fits were performed using the IGOR analysis program (Wave Metrics, Inc.) using a non-linear, least squares, iterative algorithm (Levenberg-Marquardt). For the assessment of statistical differences in pH sensitivity, the two numerical determinants of the Hill equation, Ka and n, were compared for each of the channels tested. Statistical evaluation of all data was performed with the GB-Stat v5.0.6 for Macintosh statistics package (Dynamic Microsystems, Inc., Silver Spring, MD, USA). Student's t test was used for two-group comparisons. ANOVA followed by a Bonferroni test were used for larger group comparisons. The P values are listed in the text and figure legends. All data are presented as means ± s.e.m.
Fusion protein constructs of Kir1.1a
To test for a direct interaction between the cytoplasmic domains of Kir1.1a, recombinant Kir1.1a NH2- and COOH-terminal domains were produced in bacteria as fusion proteins of either glutathione S-transferase (GST) or the maltose binding protein (MBP). A PCR product encoding the cytoplasmic NH2-terminus of Kir1.1 (amino acids 1-60 of Kir1.1b) was cloned in frame with GST in the fusion expression vector, pGEX-5x-3 (Amersham Pharmacia Biotech, Piscutaway, NJ, USA). The GST-Kir1.1 N protein or a GST fusion of the Shaker B NH2-terminus (Li et al. 1992) was produced in E. coli (BL21-RIL codon plus strain, Stratagene, La Jolla, CA, USA), and purified under non-denaturing conditions using glutathione-sepharose 4B affinity chromatography, as recommended by the manufacturer. A PCR product corresponding to the entire cytoplasmic COOH-terminal domain of Kir1.1a, encoding amino acids 183-391, was subcloned in frame with MBP in the MBP-fusion expression vector, pMal-C2 (New England Biolabs, Beverly, MA, USA). The MBP-Kir1.1a C protein was produced in E. coli (Top 10 cells, Invitrogen, Carlsbad, CA, USA) and then purified under non-denaturing conditions using amylose resin affinity chromatography, as recommended by the manufacturer.
Overlay assay
Interaction between the two cytoplasmic domains of Kir1.1 was determined in vitro by performing an overlay assay following the methods of Wyszynski & Sheng (1999), as have been employed to elucidate the oligomerization domains in Shaker potassium channels (Li et al. 1992) and interdomain interactions in Kir6.2 (Tucker & Ashcroft, 1999; Jones et al. 2001) and Kir2.3 channels (Qu et al. 2000). Briefly, GST fusion proteins (≈0.2 nmol) were resolved by SDS-PAGE (10 % SDS acrylamide) and electrophoretically transferred onto a nitrocellulose membrane (Hybond-ECL, Amersham Pharmacia Biotech). Proteins immobilized on the filter were renatured in gradually decreasing concentrations of guanidine HCl (from 6 m to zero), and then overlaid with MBP-Kir1.1a-C (0.3 μM) and extensively washed, as described by Wyszynski & Sheng (1999). Blots were probed with Anti-MBP at 1:10 000 (NEB Biolabs) followed by donkey anti-rabbit IgG conjugated to horseradish peroxidase at 1:10 000 (Amersham). An enhanced chemiluminescence system (Amersham) was used to detect bound MBP-Kir1.1a-C according to the manufacturer's instructions.
RESULTS
We recently reported that truncation of the carboxyl-terminal 60 amino acids of Kir1.1 (Kir1.1 331X) causes a complete loss of channel function (Flagg et al. 1999), as would be predicted by the link to Bartter's syndrome. 331X channels reside in an aberrant closed gating conformation (Flagg et al. 1999). In order to define the structural basis for the defect, we extended our earlier analysis of mutant channels bearing shorter COOH-terminal truncations, spanning amino acid 331 to the end in roughly 10-residue increments (Fig. 1).
Figure 1. Channel properties of Kir1.1 COOH-terminal truncation mutants.
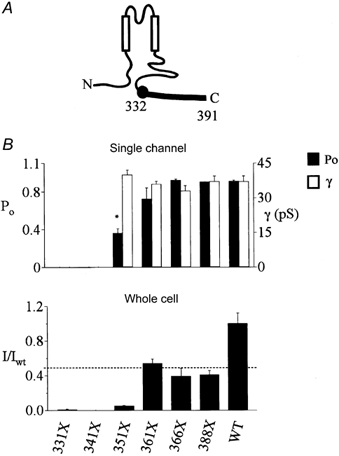
A, cartoon of Kir1.1 highlighting the cytoplasmic COOH-terminal domain of interest. A series of progressively shorter deletion mutants, from the Bartter's truncation (331X) to the PDZ binding site (388X), were studied. Amino acid 391 is the last amino acid in Kir1.1, B). The upper panel summarizes the single-channel open probability (Po) and single-channel conductance (γ) of the mutants. The lower panel summarizes the whole-cell potassium currents of the mutants relative to wild-type at −100 mV (* P < 0.01).
As characterized in Xenopus oocytes at the single-channel level by patch-clamp analysis, extension of Kir1.1 331X by 20 amino (351X) was sufficient to rescue minimal channel function, as we reported previously (Flagg et al. 1999), and addition of 10 more residues increased open channel probability to a level that is indistinguishable from the wild-type channel. These data define amino acids 332-361 as a structural element that is essential for wild-type channel gating. At the whole cell level, as measured with the two-microelectrode voltage clamp, minimal activity was detected in the 351X channel and macroscopic currents increased further with 361X, qualitatively consistent with what was observed at the single-channel level. However, there was a quantitative discordance between the open probability and the whole cell activity. None of the truncation mutants ever acquired the same level of whole cell activity as the wild-type channel. In fact, removing as little as the last three amino acids (388X), forming a type 1 PDZ binding motif (Fanning & Anderson, 1996), reduced whole cell currents to about one-half those of the wild-type channel. As the single-channel conductance and open probability of 361X, 366X and 388X are identical to the wild-type channel, the decrease in macroscopic activity reflects a significant reduction in the number of active channels at the plasma membrane.
Collectively, these studies reveal two distinct structural determinants of channel function. The distal domain, requiring at least the last three amino acids, is required for maintaining the number of active channels at the plasma membrane, possibly reflecting the presence of an endoplasmic reticulum (ER) export signal (Ma et al. 2001) and PDZ-based trafficking and retention signals (Fanning & Anderson, 1996). In contrast, the proximal domain (332-361) appears to be an important determinant of channel gating behaviour. In the remainder of the study, we focus on elucidating the molecular mechanism for altered gating by studying the basis for the defect in the minimally active 351X channel.
The altered gating behaviour of the Kir1.1a 351X channel that we previously reported was confirmed in single-channel, cell-attached patch-clamp recordings (Fig. 2). In contrast to the sustained high open probability kinetics of the wild-type channel, the Kir1.1a 351X mutant exhibited bursts of channel activity interrupted by sojourns in a closed gating mode. In some cases, the 351X channel permanently entered a closed state. As a consequence, the time-averaged (3 min) open probability of the channel was reduced (0.55 ± 0.14, n = 6) compared with the wild-type channel (0.90 ± 0.01, n = 6).
Figure 2. COOH-terminal truncation induces an aberrant gating mode.
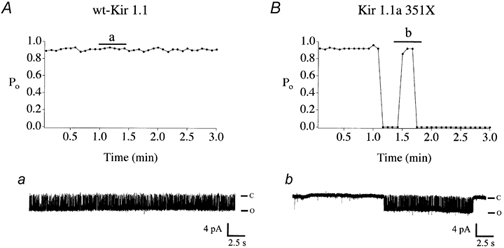
Diary plots of single-channel open probability, Po of wild-type Kir1.1a (A) vs. the truncated mutant, Kir1.1a 351X (B). Shown are plots of Po measured in 5 s intervals over the duration of a 3 min recording. Representative channel records corresponding to the indicated time are shown in a and b. Single-channel recordings were obtained in the cell-attached mode (Vm = −80 mV) from oocytes injected with either wild-type or mutant channel cRNA.
Kir1.1a 351X is more sensitive to intracellular pH than the wild-type channel
The altered gating phenotype of the Kir1.1a 351X channel is reminiscent of the wild-type channel undergoing pH-dependent channel closure. To determine whether the COOH-terminal domain might be playing a role in pH-dependent gating, we compared the pHi sensitivity of the 351X channel to the wild-type Kir1.1a.
Channel activity was determined in intact cells to safeguard against any possibility of excision-induced channel rundown phenomena (McNicholas et al. 1994; Huang et al. 1998). Because Kir1.1 is insensitive to extracellular pH (Tsai et al. 1995), pHi-dependent changes in channel activity can be accurately determined in intact oocytes by changing extracellular pH in the presence of a permeant weak acid like butyrate (Leipziger et al. 2001). Intracellular pH measurements were performed to construct a titration curve. A representative experiment is shown in Fig 3A. There were no significant differences between pHi measured in oocytes expressing either the mutant or the wild-type channel. Consequently, average pHi data were pooled to construct a pHi-pHo curve for all subsequent titration experiments (Fig. 3B).
Figure 3. Intracellular pH in oocytes as a function of extracellular pH and butyrate.
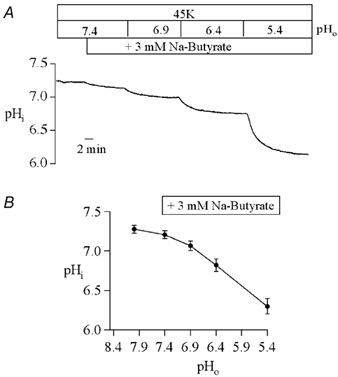
A, typical titration in a Xenopus oocyte injected with wild-type Kir1.1a cRNA. Identical results were obtained for oocytes injected with Kir1.1a 351X cRNA. Butytrate was added and the pH of the 45 mm K+ bathing solution was changed as indicated. Intracellular pH was measured with intracellular pH-sensitive microelectrodes every 2 s for the duration of the record. B, the mean steady-state pHi in each of the bathing solutions is summarized.
Macroscopic currents were measured at the different internal pH values using two-microelectrode voltage clamp. As shown in Fig. 4, Kir1.1a 351X was markedly more sensitive to intracellular acidification than the wild-type channel. The 351X mutation shifted the apparent pKa from 6.63 ± 0.04 (n = 16) to 7.00 ± 0.04 (wild-type, n = 15) without significantly changing the highly co-operative process (h = 5.15 ± 0.73, 351X, and 6.90 ± 2.25, wild-type).
Figure 4. Kir1.1a 351X channels are more sensitive to gating by cytoplasmic pH than wild-type channels.
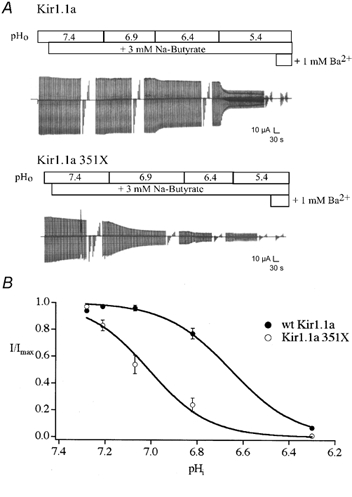
A, titration of macroscopic channel activity in oocytes injected with either Kir1.1a (250 pg) or Kir1.1a 351X (2.5 ng) cRNA. Representative experiments are shown. The modified 45 mm K+ solutions containing butyrate, at different pH values, were perfused as indicated, and current was monitored throughout the course of the solution change (Vhold = −20 mV, ≈EK, stepped to −100 and +40 mV every 5 s). Continuous current record was occasionally interrupted for current-voltage assessment (−100 to −40 mV in 20 mV steps). B, steady-state macroscopic currents (I/Imax, Vm = −100 mV) were plotted as a function of pHi for each titration experiment (n = 15-16) and fitted with the modified Hill equation: I/Imax = 1 / 1 + ([H+]/KanH), where Ka is the proton concentration at half-maximal inhibition and nH is the Hill coefficient. Kir1.1a 351X channels are significantly more sensitive to pHi (pKa = 7.00 ± 0.04) than wild-type Kir1.1a (pKa = 6.63 ± 0.04).
The result was confirmed at the single-channel level. In these studies, open probability was monitored in cell-attached patches at resting intracellular pH (≈7.2) in control solution and then at an intracellular pH of 6.68 in the 3 mm sodium butyrate, pH 6.4 solution. Typical results are shown in Fig. 5. As predicted by the macroscopic titration curves, the Kir1.1a 351X open probability was significantly reduced in the pH 6.4 solution (ΔPo = −0.41 ± 0.14, n = 3), while the open probability of the wild-type channel remained nearly unchanged (ΔPo = −0.14 ± 0.12, n = 3).
Figure 5. Increased pH sensitivity of Kir1.1a 351X at the single-channel level.
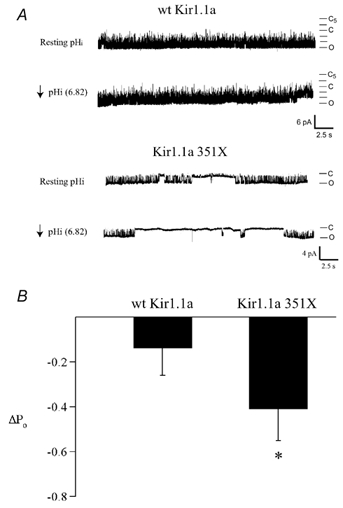
A, representative cell-attached patch recordings at resting pHi (7.28) and upon intracellular acidification (pHi = 6.82; Vm = −80 mV) from oocytes expressing either wild-type or mutant channels. B, summarized data showing the change in open probability upon intracellular acidification for wild-type and mutant channel (351X; * P < 0.01).
These data demonstrate that the extreme COOH-terminus plays an important role in determining pH-dependent gating. Removal of the domain shifts the apparent pKa in an alkaline direction, as would be predicted if the COOH-terminus normally participates in shielding the pH sensor from proton attack. In this case, the altered gating kinetics of the Kir1.1a 351X channel reflects the increased probability of K80 protonation and thus inactivation at physiological pHi. The hypothesis predicts that rendering the Kir1.1a 351X channel insensitive to pHi should rescue normal channel function.
A suppressor mutation within the pH sensor (K80M) restores channel function
Replacement of K80 with the corresponding non-titratable methionine residue of Kir2.1, a pH-insensitive inward rectifying K+ channel, completely abolished pH-dependent gating of Kir1.1 (Fakler et al. 1996). The reverse mutation, M84K in Kir2.1, introduced dependence on pHi similar to the wild-type Kir1.1a channel (Fakler et al. 1996). Accordingly, we introduced a K80M mutation in Kir1.1a 351X channels (Kir1.1a 351X/K80M) to suppress the pH sensor and remove the pH-dependent gating process.
We first compared the macroscopic current density in Xenopus oocytes injected with either Kir1.1a 351X, Kir1.1a 351X/K80M or wild-type Kir1.1a cRNA. As predicted by our hypothesis, substitution of methionine for lysine 80 removed channel pH sensitivity (data not shown) and rescued Kir1.1a 351X function (Fig. 6). The Ba2+-sensitive inward current measured in oocytes expressing Kir1.1a 351X/K80M was significantly increased (I = −13.09 ± 1.93 μA, n = 9) compared with Kir1.1a 351X (I = −1.09 ± 0.16 μA, n = 10) at Vm = −100 mV. In fact, the macroscopic current density of the Kir1.1a 351X/ K80M increased to about one-half the level of the wild-type channel, as would be predicted if the suppressor mutation, K80M, specifically rescued a gating defect involving the proximal domain. The increase in channel activity was specific for Kir1.1a 351X; replacement of lysine 80 with methionine had no effect on wild-type current density because the wild-type channel is normally active at physiological pH. The observation was confirmed at the single-channel level in long duration cell-attached patch recordings (3 min). In contrast to the lower average Po of Kir1.1a 351X (0.55 ± 0.10, n = 6), Kir1.1a 351X/K80M exhibit an average Po (0.89 ± 0.01, n = 6) which is identical to the wild-type channel (0.90 ± 0.01, n = 6). Moreover, the mean open time (16.32 ± 1.09 vs. 15.98 ± 1.03 ms), mean closed time (1.05 ± 0.08 vs. 1.28 ± 0.06 ms) and single-channel conductance (37.5 ± 2 vs. 36.9 ± 5 pS) did not differ between the K80M/351X and the wild-type channel, respectively.
Figure 6. A suppressor mutation in the pH sensor (K80M) rescues Kir1.1a 351X function, restoring high open probability gating kinetics.
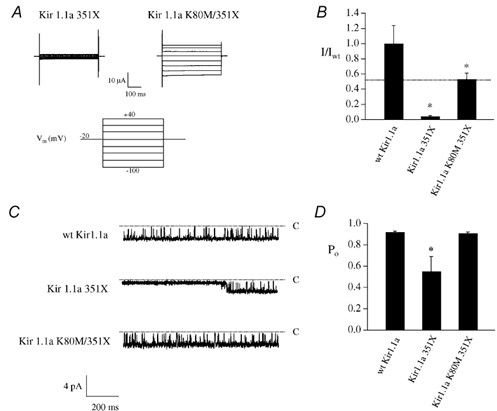
A, representative families of whole-cell currents measured in Xenopus oocytes injected with either Kir1.1a 351X or Kir1.1a 351X/K80M cRNA (250 pg). Currents were obtained using two-microelectrode voltage clamp (45 mm K+, Vhold = −20 mV, stepped in 20 mV increments from −100 to + 40 mV). B, summarized data of whole-cell potassium current relative to wild-type at −100 mV. C, single-channel records obtained in symmetrical 150 mm KCl from oocytes injected with either wild-type or mutant cRNA in the cell-attached configuration (Vm = −80 mV). Inward currents are shown and upward deflections represent channel closures. D, average open probabilities (Po) for both wild-type and mutant channels over the duration of 3 min traces (* P < 0.01).
Interdomain interactions between cytoplasmic NH2- and COOH-termini
Our model predicts that the intracellular NH2-terminal and COOH-terminal domains of Kir1.1 should physically interact with one another. To test this hypothesis, we developed an in vitro protein-protein interaction assay between the two cytoplasmic domains of Kir1.1 expressed as fusion proteins in bacteria. Recombinant NH2-terminal and COOH-terminal domains were made as glutathione S-transferase (GST) and maltose-binding protein (MBP) fusions, respectively, in E. coli and then purified to homogeneity under non-denaturing conditions using glutathione-sepharose 4B or amylose resin affinity chromatography (see Fig. 7A and B). The GST protein alone, the NH2-terminal Kir1.1-GST fusion protein or GST fusion of the NH2-terminal domain of the Shaker B channel (Li et al. 1992) were resolved by SDS-PAGE, immobilized on nitrocellulose, renatured and then probed with the MBP-Kir1.1 COOH-terminal fusion protein. The results of a typical overlay are shown in Fig. 7C. The GST protein alone or a GST fusion of the NH2-terminal domain of the Shaker B channel did not interact with the MBP fusion. In contrast, the MBP-Kir1.1 COOH-terminal fusion protein strongly bound to the GST NH2-terminal domain of Kir1.1. Specific interaction between the GST NH2-terminus and the MBP-Kir1.1 COOH-terminus was observed over a wide pH range (5.7-7.4). No interaction was observed with the MBP fusion alone. Collectively, the study provides strong evidence that the NH2- and COOH-terminal domains of Kir1.1 specifically associate with one another.
Figure 7. Cytoplasmic NH2- and COOH-terminal domains of Kir1.1 interact in vitro.
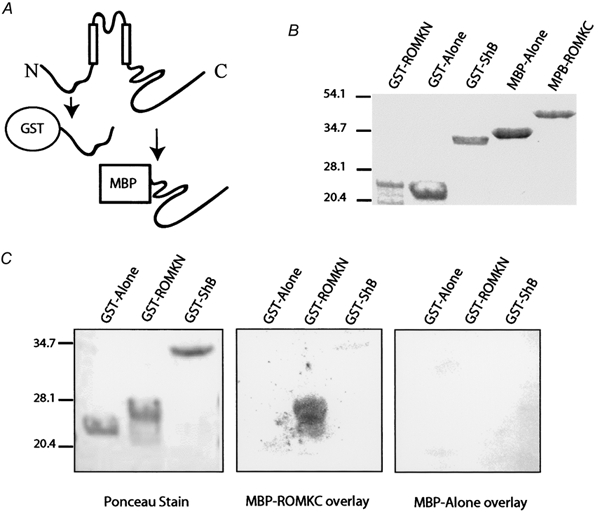
A, cartoon illustrating the cytoplasmic domains of Kir1.1 and the corresponding bacterial fusion proteins. The NH2-domain was made as a GST fusion and the COOH-domain was made as a MBP fusion. B, purified recombinant domains resolved by SDS-PAGE and visualized by Coomassie Brillant Blue staining. C, gel overlay assay: GST alone or GST fusions of the Shaker B cytoplasmic NH2-terminus (GST-ShB) or the Kir1.1 N-terminus (GST-ROMKN) were resolved by SDS-PAGE, transferred to nitrocellulose, renatured, visualized by Ponceau staining (left) and then blotted with either the MBP protein alone (right) or MBP fusion of the Kir1.1 COOH-terminal domain (MBP-ROMKC, middle). After extensive washes, bound MBP protein was detected using an anti-MBP antibody.
Delimiting amino acids responsible for regulating pH-dependent gating
To demarcate the region within the extreme COOH-terminus of the channel that is responsible for determining pH-dependent gating, pH-sensitivity of a series of truncated channels (351X, 361X and 388X) was assessed and compared with the wild-type channel. As shown in Fig. 8A, only Kir1.1a 351X was more susceptible to gating by intracellular protons (apparent pKa = 7.00 ± 0.04 in 351X compared with 6.63 ± 0.04 in wild-type). None of the other mutant channels tested exhibited an apparent pKa that was significantly different from the wild-type channel (pKa = 6.49 ± 0.04 in 361X and 6.59 ± 0.09 in 388X). These observations define a COOH-terminal domain, encoded by amino acids 332-361, that determines Kir1.1a pH-sensitivity. The region is identical to what we earlier defined (see Fig. 1) as a structural element that is essential for appropriate channel gating, effectively linking the two processes.
Figure 8. Residues 332-361 form an intrasteric pH-regulatory domain.
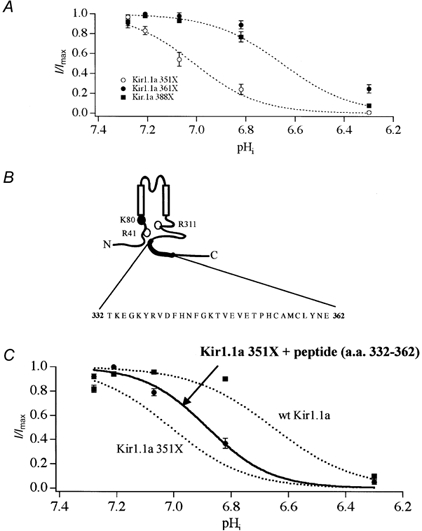
To delimit the COOH-terminal pH-gating determinant, the pH sensitivity of the COOH-terminal truncation mutants were determined. In contrast to Kir1.1a 351X, the pH sensitivities of both Kir1.1a 361X and Kir1.1a 388X were identical to wild-type channel (A). These data identify amino acids 332-361 as an essential pH-dependent gating element (B). Consistent with an intrasteric regulatory role of this domain, injection of a synthetic peptide corresponding to amino acids 332-362 partially restored the pH sensitivity (pKa = 6.88 ± 0.03, n = 15) of Kir1.1a 351X. The peptide had no effect on wild-type channel (n = 8). The pKa of wild-type channel in the presence of the peptide was 6.56 ± 0.03. •, 351X + peptide; ▪, wild-type + peptide superimposed on 351X or wild-type titration curves (dotted lines) (C).
If the COOH-terminal domain (332-361) determines pH sensitivity by protecting the pH sensor from proton attack, the domain should rescue mutant 351X channel function in trans. Accordingly, we assessed pH sensitivity of 351X and wild-type channels in oocytes injected with a synthetic peptide (50 ng) corresponding to amino acids 332-362 of Kir1.1a. As shown in Fig. 8C, the synthetic peptide partially restored the pH sensitivity of Kir1.1a 351X, shifting the apparent pKa from 7.00 to 6.88 ± 0.03 (n = 15) towards the wild-type pKa of 6.63. In contrast, the peptide had no effect on the wild-type channel (n = 8); in the presence of the peptide the pKa of the wild-type channel was 6.56 ± 0.03, no different from the wild-type channel without the peptide (6.63 ± 0.04). Interestingly, the peptide did not restore channel function in the completely inactive 331X channel, presumably because these channels enter an irreversible closed state. Nevertheless, the results with the defective 351X channel clearly demonstrate that the COOH-terminal domain functionally interacts with the NH2-terminal pH sensor to shift the pKa in an acid direction, allowing the channel to stay open at physiological intracellular pH.
DISCUSSION
A Bartter's disease mutation in the Kir1.1 channel removes the last 60 amino acids (Simon et al. 1996b) and induces an aberrant ‘locked-closed’ gating conformation (Flagg et al. 1999). In the present study, we have elucidated the molecular mechanism. Our data identify a domain within the COOH-terminus that controls pHi-dependent channel gating. Truncating the domain by disease or artificial mutation produces an alkaline shift in the current-pHi relation, causing channel inactivation at normal intracellular pH. Because the pH-dependent gating machinery remains intact, the observation indicates that the COOH-terminal domain usually co-ordinates the effective range of the pH sensor. Reinforcing this view, we found that a suppressor mutation within the pH sensor restored wild-type high open probability gating on the 351X truncated channel. Collectively, the data demonstrate that the COOH-terminal domain normally participates in pH-dependent gating, presumably interacting with, or nearby to the pH sensor.
A single lysine, K80, which is found in the cytoplasmic NH2-terminus close to the first transmembrane domain, acts as the pH sensor in the Kir1.1 channel (Fakler et al. 1996). Although protonation of the residue triggers channel closure (Fakler et al. 1996; Schulte et al. 1999), Kir1.1 are normally open at physiological pHi because titration of K80 is anomalous, occurring in the neutral pH range, far from the predicted pKa of a free lysine (pKa≈10). The response indicates that other structures in the Kir1.1 protein shield the pH sensor from proton attack. For instance, structural studies in ovotransferrin and human transferrin indicate that interdomain interactions between side chains of two lysines cause a dramatic acid shift in the pKa of the two titratable residues, important for the pH-sensitive transferrin ion binding and release mechanism (Dewan et al. 1993; Fakler et al. 1996; He et al. 1999). Similar to this model, Schulte et al. recently provided compelling evidence for two arginine residues – R41 and R311 – in distant NH2- and COOH-termini that interact in close spatial proximity with the pH sensor to form an electrostatic shield which shifts the pKa of K80 into the neutral range (Schulte et al. 1999).
Our data identify a more distant structure, encoded by amino acids 332-361, that also determines Kir1.1a pH-dependent gating by setting the range of the pH sensor. The cognate synthetic peptide partially restored pH-sensitivity in the 351X channel but had no effect on the wild-type channel, strongly suggesting that the COOH-terminal domain usually forms interdomain contacts close to the pH sensor or with other structures that stabilize electrostatic interactions of R41 and R311. Supporting this model, we found that the COOH-terminus is capable of interacting with the cytoplasmic NH2-terminus in vitro. Unlike the pH-dependent interaction between NH2- and COOH-terminal domains reported for Kir2.3 (Qu et al. 2000) channel, interaction between the cytoplasmic domains of Kir1.1 was detected over a wide pH range, perhaps more similar to what has been reported for the Kir6.2 channel (Tucker et al. 1999). Collectively, the data imply a relatively pH stable tertiary structure of the Kir1.1 channel in which the extreme COOH-terminus folds onto the NH2-terminus to form an electrostatic pocket around the pH sensor near the first transmembrane domain.
It remains to be determined whether the COOH-terminal structure contributes to physiological regulation of channel activity, acting in a matter similar to intrasteric regulatory domains in protein kinases and phosphatases (Kobe & Kemp, 1999), which control catalytic activity by reversibly and directly binding to the active site. Interestingly, the COOH-terminal domain in Kir1.1 exhibits a degree of amino acid conservation, sharing about 30 % identity with other members of the inward rectifying channel family. Structure-function studies in Kir6.2, the pore-forming subunit of β-islet cells and cardiac K+ channels, indicate that residues within both the NH2- and COOH-termini act to control ATP sensitivity (Tucker et al. 1998; Drain et al. 1998; Shyng & Nichols, 1998; Koster et al. 1999; Enkvetchakul et al. 2001). In particular, Drain et al. identified a structural determinant of ATP-dependent gating that lies within this moderately conserved domain (Drain et al. 1998). These observations suggest a widespread modulatory function of this structure in Kir channels.
In contrast to the pH sensor, the pH-dependent gating machinery has not been precisely identified. In the two-transmembrane domain potassium channel from Streptomyces lividans, KcsA, extracellular acidification induces a large conformational change in the COOH-terminal end of the second transmembrane helix, which presumably reduces the diameter of the internal vestibule of the pore to alter the conduction pathway (Perozo et al. 1998). Several studies have offered evidence for a similar mechanism in Kir1.1 channels. For instance, recent work by Schulte et al. suggests that pH gating and K+-dependent gating are coupled via structural rearrangements in the inner pore, presumably involving the P-helix (Schulte et al. 2001). Moreover, pH-dependent conformational changes in the cytoplasmic NH2- and COOH-termini have been observed by examining state-dependent chemical modification of cysteine residues with sulphydryl reagents (Schulte et al. 1998). The only cysteine residues (C49 and C308) that acquire thiol reactivity in a pH state-dependent manner lie in close proximity to the critical arginine residues of the R-K-R triad, raising the intriguing possibility that the gating machinery is directly conjoined with the neighbouring pH sensor. If such a model is accurate, structures modulating the pH sensor, such as the COOH-domain, may also directly control the efficacy of pH-dependent gating.
In summary, we have elucidated the molecular mechanism underlying the loss of channel function by a Bartter's disease mutation. Our data reveal a previously unappreciated structure within the extreme COOH-terminus that normally interacts near the pH sensor in the cytoplasmic NH2-terminus to set a physiological range of pH sensitivity. Deletion of the domain shifts the open probability-pHi relation in alkaline shift, causing channel inactivation at normal intracellular pH. Our results imply a tertiary structure of Kir1.1 in which the extreme COOH-terminus interacts with the NH2-terminus and raises the possibility that similar interdomain interactions may underscore the regulation and gating of other Kir channels.
Acknowledgments
This work was supported by grants from the NIH (DK-54231, P. A. W.), American Heart Association (P. A. W. and M. F. R.) and a HHMI institutional grant to CWRU (M. F. R.). T. F. and D. Y. were supported by pre-doctoral fellowships from an NIH Training Programme in Intregrative Membrane Biology (T32GM08181). C. M. S. was supported by a pre-doctoral fellowship (DK07678). Paul Welling is an Established Investigator of the American Heart Association. We thank W. N. Zagotta for the ShB-GST cDNA.
REFERENCES
- Bartter FC, Pronove P, Gill JR, MacCardle RC., Jr Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. American Journal of Medicine. 1962;33:811–828. doi: 10.1016/0002-9343(62)90214-0. [DOI] [PubMed] [Google Scholar]
- Chanchevalap S, Yang Z, Cui N, Qu Z, Zhu G, Liu C, Giwa LR, Abdulkadir L, Jiang C. Involvement of histidine residues in proton sensing of ROMK1 channel. Journal of Biological Chemistry. 2001;275:7811–7817. doi: 10.1074/jbc.275.11.7811. [DOI] [PubMed] [Google Scholar]
- Choe H, Zhou H, Palmer LG, Sackin H. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. American Journal of Physiology. 1997;273:F516–529. doi: 10.1152/ajprenal.1997.273.4.F516. [DOI] [PubMed] [Google Scholar]
- Derst C, Konrad M, Kockerling A, Karolyi L, Deschenes G, Daut J, Seyberth HW. Mutations in the ROMK gene in antenatal Bartter syndrome are associated with impaired K+ channel function. Biochemical and Biophysical Research Communications. 1997;230:641–645. doi: 10.1006/bbrc.1996.6024. [DOI] [PubMed] [Google Scholar]
- Derst C, Wischmeyer E, Preisig-Muller R, Spauschus A, Konrad M, Hensen P, Jeck N, Seyberth HW, Daut J, Karschin A. A hyperprostaglandin E syndrome mutation in Kir1. 1 (renal outer medullary potassium) channels reveals a crucial residue for channel function in Kir1.3 channels. Journal of Biological Chemistry. 1998;273:23884–23891. doi: 10.1074/jbc.273.37.23884. [DOI] [PubMed] [Google Scholar]
- Dewan JC, Mikami B, Hirose M, Sacchettini JC. Structural evidence for a pH-sensitive dilysine trigger in the hen ovotransferrin N-lobe: implications for transferrin iron release. Biochemistry. 1993;32:11963–11980. doi: 10.1021/bi00096a004. [DOI] [PubMed] [Google Scholar]
- Drain P, Li L, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul D, Loussouarn G, Makhina E, Shyng SL, Nichols CG. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophysical Journal. 2001;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler B, Schultz JH, Yang J, Schulte U, Brandle U, Zenner HP, Janl Y, Ruppersberg JP. Identification of a titratable lysine residue that determines sensitivity of kidney potassium channels (ROMK) to intracellular pH. EMBO Journal. 1996;15:4093–4099. [PMC free article] [PubMed] [Google Scholar]
- Fanning AS, Anderson JM. Protein-protein interactions: PDZ domain networks. Current Biology. 1996;6:1385–1388. doi: 10.1016/s0960-9822(96)00737-3. [DOI] [PubMed] [Google Scholar]
- Flagg TP, Tate M, Merot J, Welling PA. A mutation linked with Bartter's syndrome locks Kir1. 1a (ROMK1) channels in a closed state. Journal of General Physiology. 1999;114:685–700. doi: 10.1085/jgp.114.5.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebisch G. Renal potassium transport: mechanisms and regulation. American Journal of Physiology. 1998;274:F817–833. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- Guay-Woodford LM. Bartter syndrome: unraveling the pathophysiologic enigma. American Journal of Medicine. 1998;105:151–161. doi: 10.1016/s0002-9343(98)00196-x. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- He QY, Mason AB, Tam BM, MacGillivray RT, Woodworth RC. Dual role of Lys206-Lys296 interaction in human transferrin N-lobe: iron-release trigger and anion-binding site. Biochemistry. 1999;38:9704–9711. doi: 10.1021/bi990134t. [DOI] [PubMed] [Google Scholar]
- Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–886. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Jones PA, Tucker SJ, Ashcroft FM. Multiple sites of interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6. 2. FEBS Letters. 2001;508:85–89. doi: 10.1016/s0014-5793(01)03023-x. [DOI] [PubMed] [Google Scholar]
- Karaloyi L International Collaborative Study Group For Bartter-like Syndromes. Mutations in the gene encoding the inwardly-rectifying renal potassium channel, ROMK, cause the antenatal variant of Bartter syndrome: evidence for genetic heterogeneity. Human Molecular Genetics. 1997;6:17–26. doi: 10.1093/hmg/6.1.17. [DOI] [PubMed] [Google Scholar]
- Kobe B, Kemp BE. Active site-directed protein regulation. Nature. 1999;402:373–36. doi: 10.1038/46478. [DOI] [PubMed] [Google Scholar]
- Koster JC, Bentle KA, Nichols CG, Ho K. Assembly of ROMK1 (Kir1. 1a) inward rectifier K+ channel subunits involves multiple interaction sites. Biophysical Journal. 1998;74:1821–1829. doi: 10.1016/S0006-3495(98)77892-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster JC, Sha Q, Shyng S, Nichols CG. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6. 2 subunit. Journal of Physiology. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg PA, Melton DA. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Research. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipziger J, MacGregor GG, Cooper GJ, Xu J, Hebert SC, Giebisch G. PKA site mutations of ROMK2 channels shift the pH dependence to more alkaline values. American Journal of Physiology – Renal Physiology. 2001;279:F919–926. doi: 10.1152/ajprenal.2000.279.5.F919. [DOI] [PubMed] [Google Scholar]
- Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1230. doi: 10.1126/science.1519059. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Ma D, Zerangue N, Lin YF, Collins A, Yu M, Jan YN, Jan LY. Role of ER export signals in controlling surface potassium channel numbers. Science. 2001;291:316–339. doi: 10.1126/science.291.5502.316. [DOI] [PubMed] [Google Scholar]
- MacGregor GG, Xu JZ, McNicholas CM, Giebisch G, Hebert SC. Partially active channels produced by PKA site mutation of the cloned renal K+ channel, ROMK2 (kir1. 2) American Journal of Physiology. 1998;275:F415–422. doi: 10.1152/ajprenal.1998.275.3.F415. [DOI] [PubMed] [Google Scholar]
- McNicholas CM, MacGregor GG, Islas LD, Yang Y, Hebert SC, Giebisch G. pH-dependent modulation of the cloned renal K+ channel, ROMK. American Journal of Physiology. 1998;275:F972–981. doi: 10.1152/ajprenal.1998.275.6.F972. [DOI] [PubMed] [Google Scholar]
- McNicholas CM, Wang W, Ho K, Hebert SC, Giebisch G. Regulation of ROMK1 K+ channel activity involves phosphorylation processes. Proceedings of the National Academy of Sciences of the USA. 1994;91:8077–8081. doi: 10.1073/pnas.91.17.8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methfessel C, Witzemann V, Takahashi T, Mishina M, Numa S, Sakmann B. Patch clamp measurements on Xenopus laevis oocytes: currents through endogenous channels and implanted acetylcholine receptor and sodium channels. Pflügers Archiv. 1986;407:577–588. doi: 10.1007/BF00582635. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annual Review of Physiology. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Perozo E, Cortes DM, Cuello LG. Three-dimensional architecture and gating mechanism of a K+ channel studied by EPR spectroscopy. Nature Structural Biology. 1998;5:459–469. doi: 10.1038/nsb0698-459. [DOI] [PubMed] [Google Scholar]
- Qu Z, Yang Z, Cui N, Zhu G, Liu C, Xu H, Chanchevalap S, Shen W, Wu J, Li Y, Jiang C. Gating of inward rectifier K+ channels by proton-mediated interactions of N- and C-terminal domains. Journal of Biological Chemistry. 2000;275:31573–31580. doi: 10.1074/jbc.M003473200. [DOI] [PubMed] [Google Scholar]
- Romero MF, Hediger MA, Boulpaep EL, Boron WF. Expression cloning and characterization of a renal electrogenic Na+/HCO3− cotransporter. Nature. 1997;387:409–413. doi: 10.1038/387409a0. [DOI] [PubMed] [Google Scholar]
- Romero MF, Henry D, Nelson S, Harte PJ, Dillon AK, Sciortino CM. Cloning and characterization of a Na+-driven anion exchanger (NDAE1). A new bicarbonate transporter. Journal of Biological Chemistry. 2001;275:24552–24559. doi: 10.1074/jbc.M003476200. [DOI] [PubMed] [Google Scholar]
- Schulte U, Hahn H, Konrad M, Jeck N, Derst C, Wild K, Weidemann S, Ruppersberg JP, Fakler B, Ludwig J. pH gating of ROMK (K(ir)1. 1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proceedings of the National Academy of Sciences of the USA. 1999;96:15298–15303. doi: 10.1073/pnas.96.26.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Hahn H, Wiesinger H, Ruppersberg JP, Fakler B. pH-dependent gating of ROMK (Kir1. 1) channels involves conformational changes in both N and C termini. Journal of Biological Chemistry. 1998;273:34575–34579. doi: 10.1074/jbc.273.51.34575. [DOI] [PubMed] [Google Scholar]
- Schulte U, Weidemann S, Ludwig J, Ruppersberg J, Fakler B. K(+)-dependent gating of K(ir)1. 1 channels is linked to pH gating through a conformational change in the pore. Journal of Physiology. 2001;534:49–58. doi: 10.1111/j.1469-7793.2001.t01-1-00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciortino CM, Romero MF. Cation and voltage dependence of rat kidney electrogenic Na(+)-HCO(−)(3) cotransporter, rkNBC, expressed in oocytes. American Journal of Physiology. 1999;277:F611–623. doi: 10.1152/ajprenal.1999.277.4.F611. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophysical Journal. 1987;52:1047–1154. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nature Genetics. 1997;17:171–118. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- Simon DB, Karet FE, Hamdan JM, Dipietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nature Genetics. 1996a;13:183–188. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, Dipietro A, Sanjad SA, Lifton RP. Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nature Genetics. 1996b;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- Tinker A, Jan YN, Jan LY. Regions responsible for the assembly of inwardly rectifying potassium channels. Cell. 1996;87:857–868. doi: 10.1016/s0092-8674(00)81993-5. [DOI] [PubMed] [Google Scholar]
- Tsai TD, Shuck ME, Thompson DP, Bienkowski MJ, Lee KS. Intracellular H+ inhibits a cloned rat kidney outer medulla K+ channel expressed in Xenopus oocytes. American Journal of Physiology. 1995;268:C1173–1180. doi: 10.1152/ajpcell.1995.268.5.C1173. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Ashcroft FM. Mapping of the physical interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6. 2. Journal of Biological Chemistry. 1999;274:33393–33397. doi: 10.1074/jbc.274.47.33393. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of KATP channel inhibition by ATP. EMBO Journal. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Schwab A, Giebisch G. Regulation of small-conductance K channel in apical membrane of rat cortical collecting tubule. American Journal of Physiology. 1990;259:F494–502. doi: 10.1152/ajprenal.1990.259.3.F494. [DOI] [PubMed] [Google Scholar]
- Welling PA. Primary structure and functional expression of of a cortical collecting duct Kir channel. American Journal of Physiology. 1997;273:F825–836. doi: 10.1152/ajprenal.1997.273.5.F825. [DOI] [PubMed] [Google Scholar]
- Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature. 1994;371:246–249. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Sheng M. Analysis of ion channel associated proteins. Methods in Enzymology. 1999;294:371–385. doi: 10.1016/s0076-6879(99)94023-5. [DOI] [PubMed] [Google Scholar]
- Xu ZC, Yang Y, Hebert SC. Phosphorylation of the ATP-sensitive, inwardly rectifying K+ channel, ROMK, by cyclic AMP-dependent protein kinase. Journal of Biological Chemistry. 1996;271:9313–9319. doi: 10.1074/jbc.271.16.9313. [DOI] [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 1995;14:1047–1054. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]