

Abstract
Following contraction-induced damage of skeletal muscle there is a loss of calcium homeostasis. Attenuating the damage-induced rise in myocellular calcium concentration may reduce proteolytic activation and attenuate other indices of damage; calcium channel blockers have been shown to be effective in this regard. The effect of administration of a calcium channel blocker (CCB), amlodipine, on indices of muscle damage following a unilateral ‘damage protocol’, during which subjects performed 300 maximal isokinetic (0.52 rad s−1) eccentric contractions with the knee extensors was investigated. The design was a randomized, double-blind crossover. On one occasion, prior to the damage protocol, subjects consumed CCB for 7 days prior to and for 7 days following the damage protocol. Biopsies were taken from the vastus lateralis prior to (baseline) and following the damage protocol at 4 h and 24 h post-damage. Isometric peak knee extensor torque was reduced (P < 0.05) immediately post-, 24 h post- and 48 h post-damage protocol compared to pre-exercise values with no effect of treatment. Desmin disruption was attenuated (P < 0.05) with CCB versus placebo at 4 h post-damage. Z-band streaming was significantly (P < 0.05) elevated compared to baseline at both times post-damage, but was lower with CCB at 4 h (P < 0.05). Damage resulted in increased inflammatory cell (macrophage) infiltration into skeletal muscle at both 4 h and 24 h post-damage, with no effect of CCB. Neutrophil number was elevated by the damage protocol, but was higher at 24 h post-damage in the CCB condition (P < 0.05). Creatine kinase (CK) activity was higher (P < 0.05) at 24 h and 48 h following the damage protocol compared to baseline, with no effect of treatment. In conclusion, the reduction in desmin disruption and Z-band streaming indicates that CCB attenuated, or delayed, the contraction-induced damage to sarcomeric proteins.
It is known that eccentric contractions cause damage to skeletal muscle that results in disruption of the normal muscle protein structure. Eccentric contraction-induced muscle damage is characterized by a number of metabolic events including infiltration of inflammatory cells (Duncan, 1987), increased resting intracellular calcium concentration (Duan et al. 1990; Warren et al. 1993; Lowe et al. 1994), muscle enzyme release (Clarkson et al. 1992; Sayers et al. 2000), muscle soreness and a profound loss of both voluntary and involuntary force (Gibala et al. 1995).
Muscle tissue samples obtained via needle biopsies provide direct evidence of the damage, thought to be due to the high forces per active muscle fibre, induced by eccentric contractions (Friden & Lieber 1992; Gibala et al. 1995; Gibala et al. 2000). Subsequent increases in intracellular calcium concentration following the initial contraction-induced injury have been suggested to contribute to the progression of muscle damage (Duan et al. 1990; Armstrong et al. 1993; Warren et al. 1993; Lowe et al. 1994; Lynch et al. 1997; Belcastro et al. 1998). Increases in intracellular calcium may contribute to the progression of muscle damage by stimulating calcium-activated neutral proteases (CANP) such as calpain (high Ca2+ affinity μ-and low Ca2+ affinity m-forms). These proteases may initiate proteolysis by cleaving ‘susceptible’ Z-line-associated proteins such as desmin and α-actinin (Belcastro, 1993; Belcastro et al. 1998). Exposure of muscle to other treatments, such as calcium ionophores, also elevates myocellular calcium with the treated muscle showing similar morphological and ultrastructural changes (Duncan, 1987) to those observed in eccentrically damaged muscle (Duan et al. 1990; Armstrong et al. 1993; Warren et al. 1993; Lowe et al. 1994; Lynch et al. 1997).
How contraction-induced injury brings about an elevation in calcium within the muscle cell remains unknown. However, one or a combination of the following are plausible reasons: a loss of sarcoplasmic reticulum (SR) membrane integrity; ruptures in the sarcolemmal membrane; opening of stretch-responsive channels; or alterations in triad and t-tubule orientation resulting in calcium entry via voltage-sensitive channels, such as the dihydropyridine receptor (DHPR; Duan et al. 1990; Armstrong et al. 1993; Warren et al. 1993; Lowe et al. 1994; Lynch et al. 1997; Belcastro et al. 1998). Calcium channel blockers (CCB) and other calcium-chelating agents reduce or prevent the contraction-induced rise in intracellular calcium levels and subsequent injury following muscle damage in rodents (Soza et al. 1986; Duan et al. 1990; Duarte et al. 1992; Armstrong et al. 1993). The ability of CCB to prevent contraction-induced increases in cytosolic and mitochondrial calcium concentrations, and the subsequent histological evidence of injury, indicates that the disruption of the normal action of the CCB-sensitive calcium channels is at least partially responsible for allowing calcium entry into the muscle cell (Soza et al. 1986; Duan et al. 1990; Duarte et al. 1992).
The effect of CCB administration on indices of muscle damage after eccentric contractions in humans has not been examined. Given the reported ability of CCB administration to attenuate contraction-induced muscle damage (Soza et al. 1986; Duan et al. 1990; Duarte et al. 1992; Armstrong et al. 1993;), the purpose of this study was to investigate whether CCB treatment would attenuate the extent of muscle damage, as indicated by direct and indirect indices of muscle damage, after a bout of 300 maximal isokinetic eccentric contractions, in healthy human males. It was hypothesized that CCB treatment compared to placebo would attenuate the extent of inflammatory cell infiltration, the extent of protein (dystrophin and desmin) disruption and the elevation of plasma CK activity.
Methods
Subjects
Nine healthy males (age 23.8 ± 2.5 years, height 178.3 ± 4.7 cm, weight 76.5 ± 6.8 kg) who had ‘recreational’ exercise experience (i.e. no weight training and no more than two exercise bouts per week), were recruited for the study. Subjects were required to complete a routine medical screening and health questionnaire. Individuals, based on their questionnaire responses, were deemed healthy and none were smokers. All subjects were advised of the purposes of the study and associated risks, and all subjects gave written informed consent. The project was approved by the Research Ethics Board of the Hamilton Health Sciences. All of the procedures regarding the treatment of human subjects conformed to the Helsinki declaration on the use of human subjects in research.
Design
The design was a randomized double-blind placebo crossover design in which treatment (CCB or placebo) as well as leg dominance (based on strength) was randomized in a counter-balanced manner.
Exercise protocol
Prior to initiating any testing, subjects had a baseline muscle biopsy taken from the mid portion of their vastus lateralis from both the right and left leg as described previously (Stupka et al. 2001). One week following the biopsy, subjects attended a familiarization session on the testing apparatus (Biodex-System 3, Biodex Medical Systems, Inc., NY, USA) and underwent two preliminary leg strength tests, prior to beginning the study protocol, to determine peak isometric torque on both legs (see Strength measurements). All strength tests were performed on the Biodex and were applied to each leg individually (i.e. unilateral strength for each leg). Following the strength tests, the subjects (n = 9) were randomized in a double-blind manner to commence taking either a placebo or CCB. One subject withdrew after completing only one arm of the study, for reasons unrelated to the study.
Subjects consumed 2.5 mg on day one, 5.0 mg on day two and 10 mg (2 × 5.0 mg) of CCB per day for the remaining 14 days of the protocol, commencing consumption 7 days prior to the exercise protocol (Fig. 1). Compliance with the drug regime (Fig. 1) was not checked via external measures (i.e. urinary or blood levels of the drug); however, we had frequent contact with the subjects, who were financially remunerated, checked empty pill containers and have no reason to suspect that subjects did not follow our prescribed drug or placebo regime. We chose to use amlodipine as opposed to other calcium channel antagonists that have been used in animals, such as nifedipine and diltiazem, due to amlodipine's long half-life, high degree of specificity for L-type calcium channels and low incidence of side effects (Scholz, 1997). Further, this drug has been used in exercise situations and has been shown to have no effect on haemodynamic responses (Stankovic et al. 1999). On day 7 of the protocol (Fig. 1), subjects reported to the testing centre to have their resting blood pressure (BP) measured, a venous blood sample taken and to complete a battery of pre-damage protocol strength tests on one leg. Following the strength tests subjects performed an eccentrically biased isokinetic protocol designed to induce muscle damage. This ‘damage protocol’ was adapted from a protocol previously described by MacIntyre et al. (1996). Briefly, the protocol comprised 30 sets (10 repetitions per set) of knee flexion and extension on the Biodex, with 1 min of recovery between sets, at a velocity of 0.52 rad s−1. Subjects were seated throughout the protocol and had their shoulders strapped to the chair so as to isolate the torque production by the knee extensors during the activity. The contractions were performed over a range of motion of 1.04 rad (2.08-1.04 rad of flexion, where 3.14 rad is full extension). The subjects were instructed to extend their knee to 2.08 rad as the lever arm of the Biodex was raised upwards with no resistance, except the mass of their leg, during the concentric movement. Subsequently, subjects were verbally encouraged to maximally resist the lowering lever arm of the Biodex during the eccentric movement, which pushed their leg to 1.04 rad of knee flexion. To further encourage maximal effort, a computer screen provided subjects with a visual feedback of their force production throughout the exercise movement. To monitor strength throughout the protocol and between the two study conditions, peak and average torque (N m) were collected for each set throughout the protocol.
Figure 1. Outline of study design.
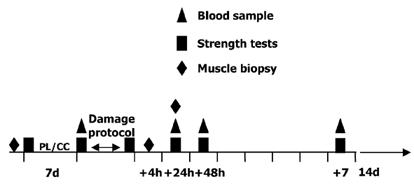
PL, placebo; CC, calcium channel blocker.
Following the damage protocol, muscle biopsies were taken from the exercised leg at two different times: 4 h and 24 h after the damage protocol. Subjects reported back to the testing centre to have venous blood samples taken and strength evaluated at 24 h, 48 h and 7 days following the exercise protocol. At 7 days after the contraction-induced damage, the subjects commenced a 14 day washout period, whereby they discontinued consumption of the CCB or placebo. Upon completion of the washout period, subjects began taking the CCB or placebo, and recommenced the study as previously outlined (Fig. 1), using their contralateral leg to perform the damage protocol.
Strength measurements
To observe the time course of changes in maximal force generating capacity, isometric knee extensor torque was assessed using the Biodex at the following times: baseline (pre-treatment), immediately pre-exercise, immediately following the damage protocol, and at 24 h, 48 h and 7 days after inducing the muscle damage. Subjects performed three repetitions while being verbally encouraged to sustain an isometric maximal voluntary contraction (MVC). Each contraction was 5 s in duration. A 60 s rest was given between each repetition, and all repetitions were performed with the subject's knee at an angle of 1.57 rad, where 3.14 rad is full extension. The peak torque exerted was recorded for all three repetitions with the highest value being considered the MVC torque.
Blood
Venous blood samples were collected into heparinized tubes from the antecubital vein of all subjects at the following times: immediately pre-exercise, 24 h, 48 h, and 7 days after the muscle damage. Blood samples were centrifuged immediately at 4 °C for 10 min, to separate out the blood plasma. Plasma samples were stored at -80 °C prior to measurement (in duplicate) of plasma CK activity using an enzymatic kit (Procedure No. 47-UV, Sigma Diagnostics, St Louis, MO, USA). The intra-assay coefficient of variation for quadruplicate samples, using this procedure was < 5 %.
Muscle biopsies
Needle biopsy samples were obtained from each subject under local anaesthesia (2 % lignocaine (lidocaine)) using a modified 5 mm Bergstrom biopsy needle with manual suction. Briefly, three biopsies were taken from each leg with one biopsy being taken from each leg at least 14 days prior to participating in the damage protocol to establish a baseline. Incisions were made on the lateral portion of the vastus lateralis starting ≈15 cm from the midline of the knee. The biopsy needle was inserted to a depth of ≈4 cm, dependent on the depth of subcutaneous fat, and once it was felt to be ≈2 cm beyond the fascia, the needle was advanced proximally along the muscle to ensure that the opening of the needle was essentially parallel with the longitudinal axis of the muscle fibres. A single clip with the biopsy needle was used to obtain a sample of muscle. Two needle biopsies were then taken from each of the exercised legs at 4 h and 24 h following the damage protocol as described above. These biopsies were taken from incisions that were made ≈8 cm proximal to the first (i.e. baseline biopsy) incision. Hence with each biopsy, the needle would be sampling from a site in the vastus lateralis muscle that was anatomically distinct from the previous site. We have recently confirmed that successive biopsies taken in this manner from resting (i.e. non-damaged) muscle do not induce damage in subsequent biopsies (S. M. Phillips, unpublished observations).
Each sample was immediately dissected free of fat and connective tissue, and partitioned into two portions. One portion was placed in a chilled (4 °C) fixative (2 % gluteraldehyde buffered with 0.1 % sodium cacodylate) for staining with toluidine blue, while another portion was placed in optimum cutting temperature embedding medium (OCT; Tissue-Tek) with its fibres perpendicular to the horizontal plane. The muscle portions placed in OCT were then quick frozen in isopentane cooled by liquid nitrogen, and stored at -50 °C until subsequent analysis.
Light microscopy
After initial fixation, the tissue samples were post-fixed in osmium tetroxide, dehydrated in graded baths of ethyl alcohol, and embedded in an epoxy resin (Spurr's) with the fibres orientated longitudinally. Each block was then sectioned (0.5 μm) and stained with toluidine blue. Individual fibres from each longitudinal muscle section were studied under 1000× magnification and examined for moderate (3-10 continuous and/or adjacent Z-bands showing obvious disruption) and extreme (10 or more continuous and/or adjacent Z-bands showing obvious disruption) Z-band streaming (Gibala et al. 1995; Stupka et al. 2001). Sample areas were calculated and the extent of Z-band streaming was expressed per mm2 of muscle analysed. The numbers of fibres examined in each biopsy varied from 106-372, mean = 184 ± 45. All muscle sections were scored by two independent investigators and viewed blind as to the treatment and subject. Agreement between two investigators on what constituted Z-band streaming was excellent (Pearson correlation coefficient r = 0.97). Using this method of identifying Z-band disruption it has been shown that randomly selected sections identified as being disrupted were confirmed to be damaged using electron microscopy (Stupka et al. 2000; Stupka et al. 2001). Further, since we had baseline tissue samples with which to compare our post-damage samples, the Z-band streaming we observed was not artefactual (i.e. due to the biopsy procedure itself).
Cross sections (10 μm thick) taken from the tissue blocks were cut on a cryostat (Microm, Heidelberg, Germany) at -20 °C. Serial sections were stained for individual fibre myosin ATPase activity using previously described procedures (Staron et al. 2000). Briefly, transverse serial sections were incubated in one of three different pH solutions (pH 4.3, 4.6, or 10.5). After preincubating at a specific pH, slides were incubated in a solution (pH 9.4) containing ATP (0.003 mm; Sigma) and Ca2+ (0.06 mm) for 45 min at 37 °C. The slides were then placed in a solution of 1 % CaCl2 for 3 min, followed by a solution of 2 % CoCl2 for 3 min, and finally a solution of 1 % ammonium sulphide for 1 min, all at room temperature. The tissue sections were prepared for examination under the microscope (BX-60, Olympus) at 200× magnification by dehydrating and clearing the tissue, and subsequently placing a coverslip over the stained muscle section. Sections were analysed using SPOT software (V2.2; Diagnostic instruments, Inc., Sterling Heights, MI, USA) and Image-Pro Plus (V4; Media Cybernetics, Silver Springs, MD, USA). Muscle fibres were classified (preincubation pH 4.6) as type I, IIa, IIax or IIx. Classification of fibre types was based on both visual and optical density criteria. Optical density measurements were used as an aid in grouping fibre types. Hence, we did not visually identify every fibre of a given type once optical density limits had been set, but instead used the analysis software, which allowed all visible fibres in the field of view to be classified. Discrete bins of optical ranges, specific to each slide, were used to group type I, IIa, IIax, and IIx, respectively. This analysis was only carried out for all six biopsies (n = 3 subjects). Fibre type composition at each time point was determined from 296 ± 62 fibres for each subject.
Cross sections (8 μm thick) taken from the tissue blocks, were cut on a cryostat at -20 °C. Sections were stained with haematoxylin and eosin for routine histological analysis and to ensure completely perpendicular fibre orientation.
Immunohistochemistry
Frozen muscle was serially cross sectioned to 8 μm thickness on a cryostat and dried overnight. Muscle cross sections were on average 1.6 ± 0.5 mm2 in size and ranged from 1.2-3.1 mm2 (126-644 muscle fibres). A negative control serial section (with no primary antibody) was included in all immunological analysis. The slides were post-fixed in cold acetone (-20 °C) for 15 min. After being washed with 0.05 m TBS, peroxidase was added for 15 min using liquid diaminobenzidine (DAB) substrate kit (00-2014, Zymed Laboratories, Inc., San Francisco, CA, USA). Following another wash, all slides were blocked with 1 % goat serum for 15 min. Monoclonal primary antibodies (Mab) to macrophage (CD68, DAKO, Denmark), myeloperoxidase (MPO; MPO-7, DAKO) and neutrophil elastase (NP57, DAKO) were diluted in 1 % goat serum (anti-macrophage: 1:75; anti-MPO: 1:300; and anti- elastase: 1:200), added individually onto the positive slides and allowed to incubate for 30 min. MPO is a neutrophil-specific enzyme and was used within this study for the identification of neutrophils (Raj et al. 1998; Stupka et al. 2001). Slides were then washed and incubated in secondary goat anti-mouse antibody (Histostain-SP Kit 95-6543-B, Zymed,) for 20 min. Aminoethyl carbazole substrate (AEC Kit 00-2007, Zymed) was used for colour (red) development.
Sections were analysed under a light microscope (BX-60, Olympus), using SPOT software (V2.2, Diagnostic Instruments, Inc.) and Image-Pro Plus (V4; Media Cybernetics). The number of macrophages and MPO-positive cells, all of which were external to the muscle cells, were counted in the total cross sectional area and expressed per mm2 of muscle. Only MPO-positive cells within the muscle and interstitial fluid were counted; these cells represented the majority of cells that were visible. The numbers of positive cells (MPO- or CD68-positive) were counted by two independent and blinded (to condition) individuals (inter-investigator r = 0.95).
Immunofluorescence
Primary Mab for desmin(DER11, Novocastra Laboratories Ltd, UK), dystrophin (DYS1, Novocastra Laboratories Ltd), and anti-α-actinin (Sarcomeric-clone no. EA-53; A7811, Sigma-Aldrich, St Louis, MO, USA) were used to identify possible disruption to the muscle cytoskeleton. Frozen muscle was cut as described for immunohistochemistry. A negative control serial section (with no primary antibody) was included in all analyses. Slides for α-actinin were post-fixed in cold acetone (-20 °C) for 15 min, while desmin and dystrophin slides were washed in 0.05 m TBS. After washing with 0.05 m TBS, all slides were blocked with 1 % goat serum for 15 min. Mabs were diluted in 1 % goat serum (anti-desmin: 1:50; anti-dystrophin: 1:20; and anti-α-actinin: 1:200) and allowed to incubate (anti-desmin: 1 h; anti-dystrophin: 24 h; and anti-α-actinin: 1 h). Slides were then washed and incubated in the dark with a Cy3-conjugated goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 1 h. The slides were washed and mounted with Vectashield mounting medium (no. H-1000, Vector Laboratories, Burlingame, CA, USA).
Muscle sections were analysed under a light microscope using SPOT software and Image-Pro Plus, as described above. The fibres that exhibited discontinuous, or completely absent intermyofibrillar staining of the desmin network were counted as ‘desmin-negative’ (Fig. 6). The number of discontinuous stains was counted for dystrophin and expressed as a percentage of the total number of fibres counted. The total number of fibres counted for these analyses was > 160 (range 161-340) for both dystrophin and desmin. The number of fibres exhibiting the aforementioned staining patterns was counted by two independent and blinded (to condition) individuals (inter-investigator agreement, r = 0.97, P < 0.001).
Figure 6. Muscle biopsy cross sections stained for desmin.

See Methods for details. A, baseline muscle samples (400× magnification) showed no discontinous, disrupted or desmin-negative staining in any fibres. At 4 h (B, 400× magnification) and at 24 h (C, 200× magnification) following the damage protocol, evidence of desmin-negative fibres was apparent. Arrows indicate some of the fibres identified as desmin negative or disrupted, other such fibres (not labelled) are also clearly visible.
Statistical analysis
Strength, plasma CK, and immunohistochemistry/fluorescence data were analysed using a two-factor repeated measures analysis of variance (ANOVA), with treatment (CCB or placebo) and time (hours) as the within factors. All data were checked for assumptions of normality and data that were found not to be normally distributed were transformed to ensure a normal distribution. Due to the lack of desmin, dystrophin and Z-band disruption at baseline, data for these variables were analysed without the baseline data (i.e. only two levels - 4 h and 24 h - for time). In addition, a t test was used to analyse whether data at 4 h and 24 h for these variables were significantly different from zero. For all other measurements, baseline data were included within the ANOVA. Pearson correlation coefficients were calculated to determine inter-individual agreement for blinded analyses and intra-class correlations for fibre types. Post hoc statistical power for main effects and interactions was also calculated. Statistical significance for all analyses was accepted as P ≤ 0.05. Significant effects identified by ANOVA were further analysed using Tukey's post hoc test. Values presented are means ± s.d.
Results
Strength measurements
Eccentric peak torque and total work were progressively reduced throughout the exercise protocol and were determined to be statistically lower than set 1 at sets 10, 15, 20, 25 and 30 (post hoc tests were only run for every 5th set). More importantly, however, there were no differences between treatments (P = 0.95 for treatment; P < 0.05, main effect for time; Fig. 2). As an index of fatigue, the drop in peak and average torque over the course of the 30 sets (i.e. set 1 versus set 30) was not different between the treatment conditions.
Figure 2. Peak isokinetic (0.52 rad s−1) torque (N m) throughout damage protocol.

* Significantly different (P < 0.05) from set 1. Values are means ± s.d.
Isometric peak torque was reduced (P < 0.05; Fig. 3) compared to pre-exercise (CCB: 220 ± 50 N m; placebo: 214 ± 61 N m; P = 0.92) values at time points immediately post-damage (expressed as a percentage of pre-exercise peak torque: CCB: 53 ± 17 %; placebo: 54 ± 15 %), 24 h post-damage (CCB: 74 ± 25 %; placebo: 75 ± 20 %), and 48 h post-damage protocol (CCB: 69 ± 24 %; placebo: 71 ± 29 %); however, there was no effect of treatment. Pre-exercise values (i.e. after treatment with placebo or CCB) were found not to be significantly different from baseline (i.e. before treatment) values (CCB: 237 ± 37 N m; placebo: 246 ± 58 N m; P = 0.93).
Figure 3. Isometric peak torque (N m) at specified times in the experimental protocol.

Pre and post, before and immediately post-damage protocol, respectively. 24 h, 48 h and 7 d, interval post-exercise (see Methods for details). * Significantly different (P < 0.05) compared to pre-protocol. Values are means ± s.d.
Fibre type
Type I fibres accounted for 44 ± 6 %; type IIa fibres for 34 ± 5 %; IIax fibres for 3 ± 3 %; and IIx fibres for 20 ± 6 % of all observable fibres. Intra-class correlation coefficients within subjects (6 biopsies total) varied from 0.68-0.93. Hence, while the fibre types seen in each biopsy within a subject varied, overall all biopsies contained fibre type distributions that were within the range of what has been reported previously (Staron et al. 2000).
Muscle damage
Evidence of either moderate or extreme Z-band streaming was undetectable in baseline samples; hence, baseline data do not appear in the figure (Fig. 4A and B). The extent of moderate Z-band streaming was greater (P < 0.05) than zero at 4 h (CCB: 4 ± 3 per mm2 muscle; placebo: 10 ± 8 per mm2 muscle; Fig. 4A) and 24 h post-damage protocol (CCB: 6 ± 5 per mm2 muscle; placebo: 7 ± 4 per mm2 muscle; Fig. 4A). However, no significant effect for treatment (interaction, P = 0.14; main effect, P = 0.42) was observed. Post hoc power analysis revealed an observed power (threshold P value (α) = 0.05) for a main (treatment, i.e. CCB versus placebo) effect of 0.68, for a time effect of 0.60 and for an interaction of 0.52.
Figure 4. Change (from baseline) in moderate and extreme Z-band streaming.

See Methods for details; results are shown relative to the baseline at 4 h and 24 h post-damage. * Significantly different (P < 0.05) versus baseline values. + Significantly different from CCB treatment. Values are means ± s.d.
Extreme Z-band streaming was observed to be greater (P < 0.05) at 4 h post-damage protocol following placebo than with CCB treatment (Fig. 4B). The extent of extreme Z-band streaming was observed to be greater (P < 0.05; Fig. 4B) at 24 h post-damage protocol compared to baseline for both treatments. Extreme Z-band streaming was not elevated above baseline at 4 h post-damage protocol following treatment with CCB (Fig. 4B). Post hoc power analysis revealed an observed power (α = 0.05) for a main (treatment) effect of 0.71, for a time effect of 0.64 and for an interaction of 0.61.
Examination of dystrophin staining revealed no cells that exhibited discontinuous or disrupted fluorescence patterns in the baseline biopsies. The number of discontinuous stains (expressed as percentage of total fibres) observed at 4 h (CCB: 0.6 ± 0.7 %; placebo: 0.6 ± 1.2 %) and 24 h (CCB: 0.4 ± 0.9 %; placebo: 0.5 ± 0.6 %) following the damage protocol were elevated (P < 0.05) compared to baseline. There was no interaction or main effect of CCB treatment. Post hoc power analysis revealed an observed power (α = 0.05) for a main (treatment) effect of 0.51, for a time effect of 0.48 and for an interaction of 0.43.
Examination of α-actinin staining revealed no cells that exhibited discontinuous or disrupted fluorescence patterns in the baseline or the post-damage biopsies. Further analysis of the α-actinin staining was not undertaken.
The damage protocol induced a significant increase in the number of fibres that had disrupted or absent desmin staining, regardless of condition (P < 0.05) at 4 h (CCB: 0.4 ± 0.5 %; placebo: 2.6 ± 1.6 %) and 24 h (CCB: 0.4 ± 0.6 %; placebo: 1.0 ± 0.8 %) compared to zero (no desmin-negative fibres found in baseline biopsies; Fig. 6). However, there was a significant interaction effect between time and condition (P < 0.05); there were fewer (P < 0.05; Fig. 5) desmin-negative fibres at both 4 h and 24 h post-damage protocol in the CCB condition. Post hoc power analysis revealed an observed power (α = 0.05) for a main (treatment) effect of 0.71, for a time effect of 0.70 and for an interaction of 0.60.
Figure 5. Change (from baseline) in number of muscle fibres exhibiting patterns of disrupted or negative desmin.

See Methods for details; results are shown for 4 h and 24 h post-damage protocol. * Significantly different (P < 0.05) compared to baseline. + Significantly different from CCB treatment. Values are means ± s.d.
Plasma CK activity was elevated (P < 0.05) at 24 h post-damage (CCB: 327 ± 156 U l−1; placebo: 253 ± 117 U l−1) and 48 h post-damage protocol (CCB: 298 ± 102 U l−1; placebo: 301 ± 82 U l−1) versus pre-exercise (CCB: 61 ± 37 U l−1; placebo: 50 ± 20 U l−1). CK activity at 7 days post-damage protocol (CCB: 223 ± 94 U l−1; placebo: 231 ± 101 U l−1) was not different from pre-exercise, nor were there any differences found between treatments.
The number of neutrophils at 24 h post-damage protocol for CCB treatment was higher (P < 0.05; Fig. 7A.) than baseline (CCB: 1.0 ± 0.9 per mm2 muscle; placebo: 0.9 ± 0.5 per mm2 muscle), 4 h (CCB: 2.9 ± 2.5 per mm2 muscle; placebo: 2.4 ± 1.0 per mm2 muscle) and 24 h post-exercise (CCB: 6.3 ± 3.7 per mm2 muscle; placebo: 1.9 ± 1.1 per mm2 muscle). There was also a greater (P < 0.05) number of neutrophils at 24 h post-damage protocol for CCB treatment versus placebo. The number of neutrophils observed using an alternative antibody thought to be specific for neutrophils (elastase) was not different from myeloperoxidase (MPO) for either treatment or time (data not shown). Post hoc power analysis revealed an observed power (α = 0.05) for a main (treatment) effect of 0.74, for a time effect of 0.68 and for an interaction of 0.56.
Figure 7. Number of macrophages and neutrophils observed in muscle following the damage protocol.

A, macrophages (CD68+ cells) and B, neutrophils (MPO+ cells) observed per mm2 muscle post-damage. * Significantly different compared to baseline. # Significantly different from 4 h post-damage protocol. + Significantly different from placebo. Values are means ± s.d.
A higher (P < 0.05; Fig. 7B.) number of macrophages was found at 4 h (CCB: 5.5 ± 4.7 per mm2 muscle; placebo: 7.3 ± 5.1 per mm2 muscle) and 24 h post-damage protocol (CCB: 9.2 ± 3.2 per mm2 muscle; placebo: 7.8 ± 3.1 per mm2 muscle) versus baseline (CCB: 2.7 ± 1.1 per mm2 muscle; placebo: 2.9 ± 1.4 per mm2 muscle) in both treatments. No differences in macrophage number were found for CCB and placebo treatment at any time point. Post hoc power analysis revealed an observed power (α = 0.05) for a main (treatment) effect of 0.68, for a time effect of 0.63 and for an interaction of 0.48.
Discussion
The results from the present investigation indicate that treatment with CCB in healthy males selectively attenuated damage to some sarcomeric proteins, as indicated by the reduced number of desmin-negative stains with CCB treatment and a reduction in the extent of extreme Z-band streaming at 4 h post-damage. However, CCB administration was not found to affect disruption of either dystrophin or α-actinin, muscle CK release, inflammatory cell infiltration, or the contraction-induced force deficit following 300 maximal isokinetic (0.52 rad s−1) muscle actions. This is the first study of its kind to investigate muscle damage in humans following CCB administration and to use muscle biopsies to examine the time course of the damage response.
An increase in intracellular calcium concentration has been hypothesized to act as a trigger for calcium-dependent neutral protease (calpain) activity in the myoplasm (Belcastro, 1993; Belcastro et al. 1998; Arthur et al. 1999). In vitro, calpain has been found to preferentially cleave a variety of proteins including structural and Z-line-associated proteins such as α-actinin, fodrin, desmin, titin, and vimentin (Belcastro, 1993; Huang & Forsberg 1998; Arthur et al. 1999) and is activated at calcium concentrations as low as 1 μm (Belcastro, 1993). In the present study, we investigated the effect of CCB treatment on two known calpain substrates, α-actinin and desmin. Disruption to desmin, indicated by cells negative for antibody staining was evident at both 4 h and 24 h following the damage protocol. It is important to emphasize that independent and blinded investigators saw no disruption or desmin-negative fibres in baseline samples. The extent of desmin disruption observed post-exercise was similar to that observed by Komulainen et al. (1999), who reported desmin protein loss in rats after a downhill running protocol. A greater percentage of disrupted fibres was observed by Lieber et al. (1994, 1996) in rabbit hindlimb muscles, peaking at 30 % of fibres exhibiting desmin loss at 3 days following cyclic eccentric contractions (Lieber et al. 1994). Consistent with our hypothesis we observed that desmin disruption was significantly attenuated with CCB treatment. Moreover, given that we did not observe any disruption of either α-actinin or dystrophin in the face of increased desmin disruption, we believe that this is evidence of a rather specific (i.e. calcium-activated calpain mechanism) and not a ‘global’ response to muscle damage.
In the present study, we did not have direct measures of intracellular calcium concentration or calpain activity. We propose, however, that the reduction in desmin disruption may have been due to CCB blocking entry of calcium via CCB-sensitive calcium channels (i.e. the dihydropyridine receptor) and thus preventing the rise in intracellular calcium concentration (Duan et al. 1990). It is possible that other mechanisms such as mitochondrial calcium loading or activation of other calcium pumps could have buffered the rise in intracellular calcium. However, there is ample evidence showing that preventing the damage-induced rise in calcium, by CCB administration, does attenuate indices of damage (Armstrong et al. 1993). Reducing the rise in myoplasmic calcium concentration would then reduce the subsequent increase in calpain activity, preventing the selective cleavage of proteins such as desmin (Belcastro, 1993; Belcastro et al. 1998; Arthur et al. 1999). Measures of calpain mRNA expression, and calpain activity would be useful in determining if the hypothesized mechanism behind the CCB-induced attenuation of desmin disruption is sound.
It is unclear why α-actinin was not seen to be disrupted in this protocol, since it is a known substrate of calpain (Belcastro, 1993). The pattern of staining obtained for α-actinin revealed that the antibody used to label α-actinin may not have been specific enough to reveal any real disruption to the protein network, had it occurred. It is clear (Fig. 6) that the intermyofibrillar desmin lattice was disrupted and this was seen to be the case by two independent blinded investigators. The results from labelling of α-actinin showed no disruption, a result that was also seen by the same two blinded investigators. Hence, we cannot discount the possibility that disruption of α-actinin did in fact occur, but that it may not have been detected.
Antibody staining against the cytoskeletal protein dystrophin was also used within this study as a marker of muscle cell membrane damage (Komulainen et al. 1999). Discontinuous antibody staining around the periphery of muscle cells was observed at both 4 h and 24 h after the exercise protocol. In addition, treatment with CCB was found to have no significant effect in preventing disruption to dystrophin at either time point following the exercise protocol. Significant disruption and proteolysis of dystrophin following eccentric contraction protocols in humans has to our knowledge not been previously reported; however, Komulainen et al. (1999) reported disruption to dystrophin after eccentrically biased downhill running in rats immediately post- and up to 96 h post-damage. It is unclear why the potentially proteolytically mediated degradation of proteins such as desmin was affected by CCB treatment, whereas dystrophin and α-actinin disruption and proteolysis was not. However, if it is the damage-induced rise in myoplasmic calcium that is triggering increased calpain activity (Belcastro, 1993, 1998), resulting in proteolysis of susceptible (i.e. calpain-sensitive) proteins, it may be that dystrophin and/or α-actinin are simply not susceptible ‘substrates’ for calpain-based proteolysis, at least not in this model of damage.
Disruption of Z-bands was reduced by CCB administration at 4 h post-exercise (Fig. 4). Duarte et al. (1992) reported that nifedipine significantly attenuated the number of damaged fibres following downhill running in rats; in particular this was demonstrated by alteration in muscle striation pattern, the presence of central nuclei, and the extent of necrosis. It is unknown how CCB might reduce Z-band streaming, but it could be the result of reduced calcium-mediated calpain-induced proteolysis of Z-band-associated proteins such as the calpain-sensitive protein titin (Belcastro, 1993; Belcastro et al. 1998); such a thesis will obviously need to be tested in future experiments. That neither Z-band streaming nor desmin disruption were reduced with CCB at 24 h versus placebo post-damage suggests that the blocker may have been ineffective in attenuating calcium influx at this time and that other means of calcium entry into the cell, or other proteolytic pathways (Farges et al. 2002), may have begun to predominate. That agents such as verapamil have been shown to attenuate the elevation of mitochondrial calcium (Armstrong et al. 1993), but not to the same extent as calcium chelators (Duan et al. 1990), suggests that the primary source of calcium entering the cell following contraction-induced damage may not be CCB-sensitive membrane calcium channels.
Muscle function was significantly compromised upon completion of the damage protocol, and in the days following the protocol. Consistent with our hypothesis, treatment with CCB had no effect in reducing the extent of post-exercise torque deficit. This hypothesis was based on a number of studies that have pointed to a disruption of the excitation-contraction (EC) coupling process that is not related to the actual physical damage per se (for a review see Warren et al. 2001). Recently, Takekura et al. (2001) found that downhill running in rats caused changes in the orientation of t-tubules (longitudinal orientation opposed to transverse orientation) and a change in the orientation and properties of the triads. In addition, the binding properties of the t-tubular voltage sensor for the dihydropyridine receptor (DHPR) have been found to increase immediately after and 3 days after injury, indicating that muscle injury may alter the functional properties of the voltage sensors (Ingalls et al. 1999; Warren et al. 2001). These findings (Ingalls et al. 1999; Takekura et al. 2001) would seem to indicate that the changes to t-tubule and triad orientation and voltage sensor properties may play a role in EC coupling failure. It is also plausible that alterations in the DHPR following contraction-induced damage could increase its opening frequency and allow it to act as a calcium channel in addition to its normal role in voltage sensing. Another possibility is that the damage seen in the biopsy does not relate to whole-limb function (i.e. the force-generating capacity of the entire knee extensor muscle group). The damage we observed as Z-band streaming in the vastus lateralis may not have been representative of damage in other muscles that are used in force generation during knee extension, which might explain the discrepancy between physical muscle damage and the torque deficit. The variability in the Z-band damage that we have observed probably means that it would be difficult to assess low cellular levels of damage and mechanical responses (i.e. force production) from whole-limb functional tests.
Following the initial muscle damage, inflammatory cells such as neutrophils and macrophages infiltrate into the damaged muscle to remove damaged tissue and initiate muscle repair (Cannon et al. 1990; Tidball, 1995; Tidball et al. 1999). In this study neutrophils, as indicated by MPO activity, were observed at both 4 h and 24 h post-protocol. An unexpected finding was the pronounced elevation in neutrophil number at 24 h following CCB treatment. Following exercise-induced muscle damage in rats, calpain-like activity has been found to be associated with a greater presence of intramuscular neutrophils, as indicated by increased myeloperoxidase (MPO) activity (Raj et al. 1998). Moreover, inhibition of calpain with a cysteine protease inhibitor reduced the activity of both calpain and MPO (Raj et al. 1998). One would expect therefore, that preventing the rise in intracellular calcium concentrations would attenuate the activation of calpain and, given the findings of Raj et al. (1998), reduce the extent of neutrophil infiltration. The rise in MPO activity post-exercise with CCB treatment observed within the present study, however, may indicate that CCB was unable to attenuate a later (24 h) rise in intracellular calcium concentrations. It is also possible that CCB, through its action on the vascular and smooth muscle tone (Burges & Moisey, 1994), may have facilitated the migration of neutrophils into the muscle, accounting for the greater rise in MPO activity post-exercise with CCB treatment than with placebo. To our knowledge, the effect of a CCB on MPO activity within skeletal muscle has not been investigated prior to this study.
In contrast to neutrophils, it is thought that macrophages are primarily responsible for removal of cellular debris (Tidball, 1995). In this study, however, we found that the number of macrophages per mm2 of muscle was significantly greater at 4 h post- and 24 h post-exercise than at baseline, regardless of treatment. Following eccentric contraction-induced damage, significant elevations in intramuscular macrophage numbers have been reported in humans at 24 h (M. A. Tarnopolsky, unpublished results), 48 h (Malm et al. 2000), and 4 days (Malm et al. 2000). Intracellular macrophage infiltration into muscle following an eccentric contraction protocol has not been investigated earlier than 24 h post-exercise in humans. Elevated levels of circulating monocytes in men have been reported by Malm et al. (1999) as early as 6 h following eccentric stair stepping.
An outstanding issue with this study is the representative nature of a single muscle biopsy from the vastus lateralis muscle in determining indices of muscle damage. Naturally, the proportion of the muscle sampled in a biopsy is small and hence, our findings may not be reflective of those events occurring in the muscle as a whole. In fact, we have data indicating a large intra-site variability in macrophage number and Z-band streaming following contraction-induced damage (L. J. Beaton & S. M. Phillips, unpublished observations). However, even given the variability in measures of muscle damage in human biopsies we were able to detect significant differences in the current study, with reasonable statistical power. Hence, the positive findings of this study are robust; moreover, due to the high variability, we may have been unable to detect some differences that occurred. In addition, of importance is whether successive biopsies induced damage that could have contaminated the results. Several lines of evidence suggest otherwise. Firstly, data from our lab indicates that Z-line streaming and other indices of damage including desmin disruption remain unchanged in biopsies taken from non-exercised individuals from the same leg on three successive days (S. M. Phillips, unpublished observations). Secondly, our data are supported by previous research that has reported on the effect of successive biopsies on ultrastructural damage in humans (Staron et al. 1992). While the previous report (Staron et al. 1992) concluded that an initial biopsy could result in evidence of damage in a subsequent biopsy, the damage was only focal (i.e. encompassing only one sarcomere), and was not substantial when expressed as a percentage of the fibres exhibiting damage. Hence, we believe that the 2-fold (extreme damage: 10 or more adjacent sarcomeres) and 10-fold (moderate: 3-10 adjacent sarcomeres) increases in areas of damaged muscle are a result of the damage protocol and not of the previous biopsy. In addition, tremendous care was taken to sample from an anatomically distinct biopsy site with each subsequent biopsy, which we believe means that the chance of seeing biopsy-induced damage is extremely low. While it is certain that areas of focal damage are present in muscle biopsies from normal persons (Meltzer et al. 1976; Staron et al. 1992), large areas of ‘extensive’ damage are extremely rare (Meltzer et al. 1976) or are never seen (Staron et al. 1992). In fact, we have never, in more than 80 biopsies spanning past (Gibala et al. 1995; Gibala et al. 2000) and present results, seen areas of extensive damage in resting biopsies from biceps (Gibala et al. 1995; Gibala et al. 2000) or vastus lateralis (present results and unpublished observations) muscles. Finally, based on our analysis of fibre types within biopsies from a given subject, the data showed that ranges of fibre types were not biased toward fast twitch fibres, and hence we have sampled from a relatively homogeneous portion of the vastus lateralis. More importantly, we did not see in any biopsy a fibre type distribution that suggested a much higher proportion of fast twitch fibres, which are more susceptible to damage (Lieber et al. 1996).
In conclusion, the reduction in the number of desmin-negative fibres and attenuation of the extent of Z-band streaming following the eccentric protocol with CCB treatment as compared to placebo, indicate that CCB administration appears to have been effective in attenuating eccentric contraction-induced disruption of sarcomeric proteins. However, CCB administration was not found to have a post-damage effect on the other indices of muscle damage. Further research is warranted to quantify the effect of CCB treatment on intracellular calcium concentrations in human skeletal muscle both before and following a bout of eccentric exercise. Furthermore, other likely sites of calcium entry into the cell need to be investigated to determine their contribution to post-exercise intracellular calcium concentrations and subsequent muscle protein breakdown.
Acknowledgments
Thank you to the subjects for their compliance and patience during the study. This study was supported by NSERC (S.M.P.). S.M.P. and M.A.T. are supported by Premier's Research Excellence Awards and acknowledge these awards in allowing the completion of this work. S.M.P. is supported by a Canadian Institute of Health Research New Investigator award and acknowledges that source of funding in allowing the completion of this work. The expert technical assistance of Dr K. Chorneyko and Dr J. Bourgeois (Department of Pathology) and the help of staff of the electron microscope facility (E. Spitzer, M. Moore and C. Nguyen) at McMaster University in analysis of the Z-bands is also greatly appreciated.
References
- Armstrong RB, Duan C, Delp MD, Hayes DA, Glenn GM, Allen GD. Elevations in rat soleus muscle [Ca2+] with passive stretch. Journal of Applied Physiology. 1993;74:2990–2997. doi: 10.1152/jappl.1993.74.6.2990. [DOI] [PubMed] [Google Scholar]
- Arthur GD, Booker TS, Belcastro AN. Exercise promotes a subcellular redistribution of calcium-stimulated protease activity in striated muscle. Canadian Journal of Physiology andPharmacology. 1999;77:42–47. doi: 10.1139/cjpp-77-1-42. [DOI] [PubMed] [Google Scholar]
- Belcastro AN. Skeletal muscle calcium-activated neutral protease (calpain). with exercise. Journal of Applied Physiology. 1993;74:1381–1386. doi: 10.1152/jappl.1993.74.3.1381. [DOI] [PubMed] [Google Scholar]
- Belcastro AN, Shewchuk LD, Raj DA. Exercise-induced muscle injury: a calpain hypothesis. Molecular and Cellular Biochemistry. 1998;179:135–145. doi: 10.1023/a:1006816123601. [DOI] [PubMed] [Google Scholar]
- Burges R, Moisey D. Unique pharmacologic properties of amlodipine. American Journal of Cardiology. 1994;73:2–9A. doi: 10.1016/0002-9149(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Cannon JG, Orencole SF, Fielding RA, Meydani M, Meydani SN, Fiatarone MA, Blumberg JB, Evans WJ. Acute phase response in exercise: interaction of age and vitamin E on neutrophils and muscle enzyme release. American Journal of Physiology. 1990;259:R1214–1219. doi: 10.1152/ajpregu.1990.259.6.R1214. [DOI] [PubMed] [Google Scholar]
- Clarkson PM, Nosaka K, Braun B. Muscle function after exercise-induced muscle damage and rapid adaptation. Medicine and Science in Sports and Exercise. 1992;24:512–520. [PubMed] [Google Scholar]
- Duan C, Delp MD, Hayes DA, Delp PD, Armstrong RB. Rat skeletal muscle mitochondrial [Ca2+] and injury from downhill running. Journal of Applied Physiology. 1990;68:1241–1251. doi: 10.1152/jappl.1990.68.3.1241. [DOI] [PubMed] [Google Scholar]
- Duarte JA, Soares JM, Appell HJ. Nifedipine diminishes exercise-induced muscle damage in mouse. International Journal of Sports Medicine. 1992;13:274–277. doi: 10.1055/s-2007-1021266. [DOI] [PubMed] [Google Scholar]
- Duncan CJ. Role of calcium in triggering rapid ultrastructural damage in muscle: a study with chemically skinned fibres. Journal of Cell Science. 1987;87:581–594. doi: 10.1242/jcs.87.4.581. [DOI] [PubMed] [Google Scholar]
- Farges MC, Balcerzak D, Fisher BD, Attaix D, Bechet D, Ferrara M, Baracos VE. Increased muscle proteolysis after local trauma mainly reflects macrophage-associated lysosomal proteolysis. American Journal of Physiology - Endocrinology and Metabolism. 2002;282:E326–335. doi: 10.1152/ajpendo.00345.2001. [DOI] [PubMed] [Google Scholar]
- Friden J, Lieber RL. Structural and mechanical basis of exercise-induced muscle injury. Medicine and Science in Sports Exercise. 1992;24:521–530. [PubMed] [Google Scholar]
- Gibala MJ, Interisano SA, Tarnopolsky MA, Roy BD, MacDonald JR, Yarasheski KE, MacDougall JD. Myofibrillar disruption following acute concentric and eccentric resistance exercise in strength-trained men. Canadian Journal of Physiology and Pharmacology. 2000;78:656–661. [PubMed] [Google Scholar]
- Gibala MJ, MacDougall JD, Tarnopolsky MA, Stauber WT, Elorriaga A. Changes in human skeletal muscle ultrastructure and force production after acute resistance exercise. Journal of Applied Physiology. 1995;78:702–708. doi: 10.1152/jappl.1995.78.2.702. [DOI] [PubMed] [Google Scholar]
- Huang J, Forsberg NE. Role of calpain in skeletal-muscle protein degradation. Proceedings of theNational Academy of Sciences of the USA. 1998;95:12100–12105. doi: 10.1073/pnas.95.21.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingalls CP, Warren GL, Hamilton SL, Armstrong RB. Dihydropyridine and ryanodine receptor binding after eccentric contractions in mouse skeletal muscle. Medicine and Science in Sports and Exercise. 1999;31:S72. doi: 10.1152/japplphysiol.00084.2003. [DOI] [PubMed] [Google Scholar]
- Komulainen J, Koskinen SO, Kalliokoski R, Takala TE, Vihko V. Gender differences in skeletal muscle fibre damage after eccentrically biased downhill running in rats. Acta Physiologica Scandanavica. 1999;165:57–63. doi: 10.1046/j.1365-201x.1999.00481.x. [DOI] [PubMed] [Google Scholar]
- Lieber RL, Schmitz MC, Mishra DK, Friden J. Contractile and cellular remodelling in rabbit skeletal muscle after cyclic eccentric contractions. Journal of Applied Physiology. 1994;77:1926–1934. doi: 10.1152/jappl.1994.77.4.1926. [DOI] [PubMed] [Google Scholar]
- Lieber RL, Thornell LE, Friden J. Muscle cytoskeletal disruption occurs within the first 15 min of cyclic eccentric contraction. Journal of Applied Physiology. 1996;80:278–284. doi: 10.1152/jappl.1996.80.1.278. [DOI] [PubMed] [Google Scholar]
- Lowe DA, Warren GL, Hayes DA, Farmer MA, Armstrong RB. Eccentric contraction-induced injury of mouse soleus muscle: effect of varying [Ca2+]o. Journal of Applied Physiology. 1994;76:1445–1453. doi: 10.1152/jappl.1994.76.4.1445. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Fary CJ, Williams DA. Quantitative measurement of resting skeletal muscle [Ca2+]i following acute and long-term downhill running exercise in mice. Cell Calcium. 1997;22:373–383. doi: 10.1016/s0143-4160(97)90022-1. [DOI] [PubMed] [Google Scholar]
- MacIntyre DL, Reid WD, Lyster DM, Szasz IJ, Mckenzie DC. Presence of WBC, decreased strength, and delayed soreness in muscle after eccentric exercise. Journal of Applied Physiology. 1996;80:1006–1013. doi: 10.1152/jappl.1996.80.3.1006. [DOI] [PubMed] [Google Scholar]
- Malm C, Lenkei R, Sjodin B. Effects of eccentric exercise on the immune system in men. Journal of Applied Physiology. 1999;86:461–468. doi: 10.1152/jappl.1999.86.2.461. [DOI] [PubMed] [Google Scholar]
- Malm C, Nyberg P, Engstrom M, Sjodin B, Lenkei R, Ekblom B, Lundberg I. Immunological changes in human skeletal muscle and blood after eccentric exercise and multiple biopsies. Journal of Physiology. 2000;529:243–262. doi: 10.1111/j.1469-7793.2000.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer HY, Kuncl RW, Yang V. Incidence of Z band streaming and myofibrillar disruptions in skeletal muscle from healthy young people. Neurology. 1976;26:853–857. doi: 10.1212/wnl.26.9.853. [DOI] [PubMed] [Google Scholar]
- Raj DA, Booker TS, Belcastro AN. Striated muscle calcium-stimulated cysteine protease (calpain-like) activity promotes myeloperoxidase activity with exercise. Pflügers Archiv. 1998;435:804–809. doi: 10.1007/s004240050587. [DOI] [PubMed] [Google Scholar]
- Sayers SP, Clarkson PM, Lee J. Activity and immobilization after eccentric exercise: II. Serum CK. Medicine and Science in Sports and Exercise. 2000;32:1593–1597. doi: 10.1097/00005768-200009000-00011. [DOI] [PubMed] [Google Scholar]
- Scholz H. Pharmacological aspects of calcium channel blockers. Cardiovascular Drugs and Therapeutics. 1997;10:869–872. doi: 10.1007/BF00051613. [DOI] [PubMed] [Google Scholar]
- Soza M, Karpati G, Carpenter S, Prescott S. Calcium-induced damage of skeletal muscle fibers is markedly reduced by calcium channel blockers. Acta Neuropathologica. 1986;71:70–75. doi: 10.1007/BF00687964. [DOI] [PubMed] [Google Scholar]
- Stankovic S, Panz V, Klug E, Di Nicola G, Joffe BI. Amlodipine and physiological responses to brisk exercise in healthy subjects. Cardiovascular Drugs and Therapeutics. 1999;13:513–517. doi: 10.1023/a:1007875603969. [DOI] [PubMed] [Google Scholar]
- Staron RS, Hagerman FC, Hikida RS, Murray TF, Hostler DP, Crill MT, Ragg KE, Toma K. Fiber type composition of the vastus lateralis muscle of young men and women. Journal of Histochemistry and Cytochemistry. 2000;48:623–629. doi: 10.1177/002215540004800506. [DOI] [PubMed] [Google Scholar]
- Staron RS, Hikida RS, Murray TF, Nelson MM, Johnson P, Hagerman F. Assessment of skeletal muscle damage in successive biopsies from strength-trained and untrained men and women. European Journal of Applied Physiology. 1992;65:258–264. doi: 10.1007/BF00705091. [DOI] [PubMed] [Google Scholar]
- Stupka N, Lowther S, Chorneyko K, Bourgeois JM, Hogben C, Tarnopolsky MA. Gender differences in muscle inflammation after eccentric exercise. Journal of Applied Physiology. 2000;89:2325–2332. doi: 10.1152/jappl.2000.89.6.2325. [DOI] [PubMed] [Google Scholar]
- Stupka N, Tarnopolsky MA, Yardley NJ, Phillips SM. Cellular adaptation to repeated eccentric exercise-induced muscle damage. Journal of Applied Physiology. 2001;91:1669–1678. doi: 10.1152/jappl.2001.91.4.1669. [DOI] [PubMed] [Google Scholar]
- Takekura H, Fujinami N, Nishizawa T, Ogasawara H, Kasuga N. Eccentric exercise-induced morphological changes in the membrane systems involved in excitation-contraction coupling in rat skeletal muscle. Journal of Physiology. 2001;533:571–583. doi: 10.1111/j.1469-7793.2001.0571a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG. Inflammatory cell response to acute muscle injury. Medicine and Science in Sports and Exercise. 1995;27:1022–1032. doi: 10.1249/00005768-199507000-00011. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Berchenko E, Frenette J. Macrophage invasion does not contribute to muscle membrane injury during inflammation. Journal of Leukocyte Biology. 1999;65:492–498. [PubMed] [Google Scholar]
- Warren GL, Hayes DA, Lowe DA, Armstrong RB. Mechanical factors in the initiation of eccentric contraction-induced injury in rat soleus muscle. Journal of Physiology. 1993;464:457–475. doi: 10.1113/jphysiol.1993.sp019645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren GL, Ingalls CP, Lowe DA, Armstrong RB. Excitation-contraction uncoupling: major role in contraction-induced muscle injury. Exercise and Sports Science Reviews. 2001;29:82–87. doi: 10.1097/00003677-200104000-00008. [DOI] [PubMed] [Google Scholar]