

Abstract
This study investigated the effect of reduced acetylcarnitine availability on oxidative metabolism during the transition from rest to steady-state exercise. Eight male subjects completed two randomised exercise trials at 68 % of the peak rate of O2 uptake (V̇O2,peak). On one occasion subjects ingested 1 g (kg body mass)−1 glucose 75 min prior to exercise (CHO), whereas the other trial acted as a control (CON). Muscle samples were obtained pre- and 75 min post-ingestion, and following 1 and 10 min of exercise. Plasma glucose and insulin were elevated (P < 0.05), and plasma free fatty acids (FFA) were lower at the onset of exercise in CHO. Acetylcarnitine (CON, 4.8 ± 1.8; CHO, 1.5 ± 0.9 mmol (kg dry mass (d.m.))−1, P < 0.05) and acetyl CoA (CON, 13.2 ± 2.3; CHO, 6.3 ± 0.6 μmol (kg d.m.)−1, P < 0.05) were lower at rest, whereas pyruvate dehydrogenase activation (PDHa) was greater in CHO compared with CON (CON, 0.78 ± 0.07; CHO, 1.44 ± 0.19 mmol min−1 (kg wet mass (w.m.))−1). Respiratory exchange ratio (RER) was significantly elevated during exercise in CHO. The acetyl groups increased at similar rates at the onset of exercise (1 min) and there was no difference in substrate phosphorylation as determined from lactate accumulation and phosphocreatine degradation between trials. Subsequently, oxidative metabolism during the transition from rest to steady-state exercise was not affected by prior carbohydrate ingestion. Although exercise resulted in the rapid activation of PDH in both trials, PDHa was greater at 1 min in CHO (CON, 2.36 ± 0.22; CHO, 2.91 ± 0.18 mmol min−1 (kg w.m.)−1). No differences in muscle metabolite levels and PDHa were observed after 10 min of moderate exercise between trials. In summary, at rest, carbohydrate ingestion induced multiple metabolic changes which included decreased acetylcarnitine availability and small increases in PDHa. The prior changes in PDHa and acetylcarnitine availability had no effect on substrate phosphorylation and oxidative metabolism at the onset of exercise. These data suggest that acetylcarnitine availability is unlikely to be the site of metabolic inertia during the transition from rest to steady-state moderate intensity exercise.
During the transition from rest to steady-state exercise the mitochondria require a finite amount of time (≈1-2 min) to produce energy from oxidative phosphorylation to fully supply the required energy for muscular contractions. A subsequent mismatch between aerobic ATP provision and demand occurs (Wilson, 1994; Bangsbo et al. 2000; Evans et al. 2001), and the energy necessary to maintain the ATP turnover in the muscle and sustain force is supplied by substrate level phosphorylation (glycogenolysis/glycolysis and phosphocreatine (PCr) degradation).
The lag in oxidative metabolism has historically been thought to be due to inadequate oxygen availability to the mitochondria (Tschakovsky & Hughson, 1999). However, others have demonstrated that the delay in oxidative energy production is due to a metabolic inertia associated with delayed acetyl CoA delivery to the tricarboxylic acid (TCA) cycle secondary to (1) a lag in pyruvate dehydrogenase (PDH) activation (PDHa; Gibala & Saltin, 1999; Howlett et al. 1999a; Parolin et al. 2000; Evans et al. 2001), and/or (2) decreased delivery via the carnitine acetyltransferase reaction (acetylcarnitine + CoASH ™⇌ acetyl CoA + carnitine) (Timmons et al. 1998a; Campbell-O'Sullivan et al. 2002).
A major hypothesis forwarded by Greenhaff and collegues is that acetyl CoA from acetylcarnitine is the major determinant of the rate of oxidative phosphorylation at the onset of exercise. This interpretation is largely based on results from isolated ischaemic canine and human studies that infused dichloroacetate (DCA) prior to contraction/ exercise. DCA administration resulted in large increases in resting acetylcarnitine that were hypothesised to minimise the lag in oxidative phosphorylation by providing ready available substrate at the onset of exercise via the near-equilibruim carnitine acetyltransferase reaction. However, despite the finding of less PCr utilisation and lactate accumulation following DCA, there is little experimental evidence demonstrating acetylcarnitine utilisation at the onset of exercise (Timmons et al. 1997, 1998a,b; Howlett et al. 1999a; Campbell-O'Sullivan et al. 2002; Grassi et al. 2002). Moreover, DCA also results in the prior maximal activation of resting PDHa, and the provision of extra substrate at the onset of exercise is likely to result from greater flux through PDHa, not the accumulation of acetylcarnitine (Howlett et al. 1999b).
In contrast to the interpretation of Greenhaff and others, studies from our laboratory suggest that acetylcarnitine does not provide acetyl units at the onset of exercise and that the lag in PDH activation is the site of the metabolic inertia. We have previously shown that infusing acetate prior to exercise increases the acetylcarnitine store independently of changes in PDHa (Putman et al. 1995; Evans et al. 2001) and that this is without effect on the rate of oxidative phosphorylation at the onset of exercise (Evans et al. 2001). Thus, the primary aim of the present study was to further examine the role of acetylcarnitine availability on oxidative metabolism by lowering acetylcarnitine prior to the onset of exercise.
We tested the hypothesis that reduced pre-exercise acetylcarnitine would have no effect on oxidative metabolism during the transition from rest to steady-state exercise. To achieve this, subjects ingested carbohydrate prior to exercise, because unlike DCA, which markedly increases both acetylcarnitine and PDHa at rest, carbohydrate ingestion decreases resting acetylcarnitine and results in only modest increases in PDHa (Pehleman et al. 2000). We have incorporated both a pre- and postcarbohydrate ingestion biopsy to confirm the reduction in acetylcarnitine, and a 1 min biopsy to examine acetylcarnitine levels, PDHa and substrate level phosphorylation during the transition from rest to steady-state exercise. Rather than measuring rate of oxygen uptake (V̇O2) on-kinetics at the mouth we directly estimated substrate level phosphorylation by changes in skeletal muscle lactate and PCr. Because the energy demand of each trial was the same, we assumed that any change in oxidative metabolism would be reflected by altered substrate level phosphorylation.
Methods
Subjects
Eight recreationally active males (25 ± 2 years old; 78 ± 4 kg body weight) volunteered to participate in this study, which was approved by the University of Guelph and McMaster University Human Ethics Committees and was performed according to the Declaration of Helsinki. All experimental procedures and possible risks were explained to subjects orally and in writing, and subjects provided their written consent.
Pre-experimental protocol
Peak pulmonary oxygen uptake (V̇O2,peak) was determined during an incremental cycling test to volitional exhaustion on a Lode electromagnetically braked cycle ergometer (Excalibur, Quinton Inc., Bothell, WA, USA), and averaged 53.3 ± 1.8 ml kg−1 min−1. At least 24 h later subjects underwent a 10 min practice ride and their predetermined power output was confirmed by measuring oxygen uptake (V̇O2) on-line (Quinton Q-plex 1, Quinton Inc.).
Experimental protocol
On two separate occasions subjects completed 10 min of cycle exercise at an intensity of 68.3 ± 1.4 % V̇O2,peak. We selected this exercise intensity because it is consistent with most of the literature examining muscle metabolism during the transition from rest to steady-state exercise. On one occasion subjects ingested 1 g (kg body weight)−1 carbohydrate (CHO) 75 min prior to exercise and the other trial acted as a control (CON). Trials were randomised and separated by at least 1 week. For the day preceding all trials, subjects were instructed to maintain a normal diet and abstain from alcohol, caffeine and exercise.
On the morning of the experiments subjects arrived at the laboratory after an overnight fast (>10 hours), voided and rested quietly on a couch. An indwelling catheter was inserted into an antecubital vein for subsequent blood sampling. A resting blood sample was obtained and saline was continuously infused to maintain a patent line. Incisions were made in the skin overlying the vastus lateralis muscle under local anaesthesia (2 % lidocaine, no adrenaline) for subsequent muscle sampling. A resting muscle sample was obtained and immediately frozen in liquid nitrogen. For CHO, subjects ingested a carbohydrate solution (≈78 ml, Trutol 75, Source Medical, ON, Canada) as quickly as possible (< 2 min) and remained seated for 75 min during which time blood samples were obtained at 15 min intervals. A second resting muscle sample was obtained at 75 min post-ingestion whilst subjects remained on the couch. In the control trial, subjects remained resting with no pre-biopsy or blood samples taken. A biopsy was taken immediately before exercise.
For both trials, subjects then moved to the cycle ergometer and commenced cycling at their predetermined workload. Muscle samples were obtained at 1 and 10 min of exercise whilst the subjects remained seated on the cycle ergometer. The time lapse between cessation of exercise and freezing of muscle samples was < 10 s and at 1 min, subjects always recommenced cycling within 30 s of cessation of exercise. Blood samples were obtained at 5 and 10 min of exercise and expired pulmonary gases were collected on-line between 5 and 9 min, with the final minute being recorded. Whole-body carbohydrate oxidation rate was calculated using the non-protein respiratory quotient (Peronnet & Massicotte, 1991).
Analysis
Blood was drawn into heparinised tubes and one portion of whole blood was immediately deproteinised 1:6 with 0.6 % (w/v) perchloric acid (PCA). The PCA extract was stored at -20 °C and subsequently analysed for blood glucose and lactate (Bergmeyer, 1974). A second portion of whole blood was spun (2000 g) and 400 μl plasma was added to 100 μl NaCl and incubated at 56 °C for 30 min to inactivate lipoprotein lipase. The plasma was subsequently analysed for FFA via a colormetric method (Wako NEFA C test kit, Wako Chemicals, USA). A final portion was spun and the supernatant removed for the determination of insulin by radioimmunoassay (Coat-a-Count insulin test kit, Diagnostics Products, USA).
A small piece of muscle (8-15 mg) was chipped under liquid N2 for the determination of PDHa as described by Putman et al. (1993). The remainder of the muscle was freeze dried, dissected free from all non-muscle contaminants and powdered. An aliquot of freeze-dried muscle was extracted with 0.5 m HClO4 (1 mm EDTA) and neutralized with 2.2 m KHCO3. The extract was used for the determination of ATP, creatine, PCr, lactate and glucose 6-phosphate (G-6-P) by enzymatic spectrophotometric assays (Bergmeyer, 1974; Harris et al. 1974). Pyruvate was measured on this extract by a fluorometric assay (Passoneau & Lowry, 1993) and acetyl CoA and acetylcarnitine were determined by radiometric measures (Cederblad et al. 1990). All metabolite and PDHa measurements were normalised to the highest total creatine content from the six samples obtained for each subject to correct for non-muscle contamination.
Calculations
Free Pi was calculated by adding the estimated free phosphate (10.8 mmol (kg dry mass (d.m.))−1), Dudley et al. 1987) to the difference in PCr minus the accumulation of G-6-P between rest and 1 min. Free ADP and AMP concentrations were calculated with the assumption of equilibrium of the adenylate kinase and creatine kinase reactions (Dudley et al. 1987). Free ADP was calculated using the measured ATP, creatine and PCr values and an estimated H+ concentration (Sahlin et al. 1976) and the creatine kinase equilibrium constant of 1.66 × 109. Free AMP concentration was calculated from the estimated free ADP and measured ATP with the adenylate kinase constant of 1.05. Substrate level phosphorylation was calculated between rest and 1 min using the following equation:
where Δ is the difference between rest and 1 min values and square brackets indicate concentration (Spriet et al. 1987).
Statistics
All data are presented as means ± s.e.m. Student's paired t test was used to determine differences between pre- and post-ingestion values in the CHO trial. A two-way ANOVA (treatment × time) with repeated measures was used to determine differences between measures at 0, 1 and 10 min. Specific differences were located using the Student-Newman-Keuls post hoc test. Statistical significance was accepted at P < 0.05.
Results
Respiratory and blood measures
V̇O2 during exercise was unaffected by prior carbohydrate ingestion and averaged 68.3 ± 1.4 % V̇O2,peak (Table 1). RER and the carbohydrate oxidation rate were higher (P < 0.05) in CHO compared with CON (Table 1).
Table 1.
Respiratory data following 10 min exercise at 68% V̇O2,peak with (CHO) or without (CON) prior carbohydrate ingestion
| Measure | Condition | 10 min |
|---|---|---|
| V̇O2 (ml kg−1 min−1) | CON | 35.8 ± 1.3 |
| CHO | 36.8 ± 1.4 | |
| RER | CON | 0.91 ± 0.02 |
| CHO | 0.95 ± 0.01* | |
| CHO oxidation (g min−1) | CON | 2.67 ± 0.27 |
| CHO | 3.35 ± 0.20* |
Data are means ± s.e.m., n = 8.
Significantly different from CON (P < 0.05).
Blood glucose and plasma insulin increased during the 75 min period following CHO ingestion (Fig. 1). Blood glucose was greater (P < 0.05) in CHO compared with CON at 0 min (Fig. 1). During exercise blood glucose decreased in CHO (P < 0.05) and was constant in CON. As a result, no difference in blood glucose was evident between trials during exercise. Plasma insulin was higher (P < 0.05) in CHO compared with CON at 0 min and throughout exercise (Fig. 1). Plasma FFA concentrations were significantly reduced by 0 min following carbohydrate ingestion (Table 2). Plasma FFA levels in CHO were lower (P < 0.05) at exercise onset compared with CON and, despite an exercise-induced reduction (P < 0.05) in CON, plasma FFA levels remained lower in CHO throughout exercise (Table 2). Plasma lactate was unaffected by prior carbohydrate ingestion and increased (P < 0.05) similarly during exercise in both trials (Table 2).
Figure 1.

Blood glucose (A) and plasma insulin (B) at rest and during 10 min of exercise at 68 % V̇O2,peak with (CHO) or without (CON) prior carbohydrate ingestion. Values are means ± s.e.m., n = 8. * Significantly different from -75 min for CHO; † significantly different from corresponding time point for CON (P < 0.05).
Table 2.
Venous blood metabolite concentrations at rest and during 10 min exercise at 68% V̇O2,peak with (CHO) or without (CON) prior carbohydrate ingestion
| Measure | Condition | Pre-ingestion | 0 min | 5 min | 10 min |
|---|---|---|---|---|---|
| [FFA] | CON | — | 0.05 ± 0.10 | 0.29 ± 0.08 | 0.23 ± 0.04 |
| (mm) | CHO | 0.56 ± 0.10 | 0.10 ± 0.03*† | 0.09 ± 0.03* | 0.08 ± 0.03* |
| [Lactate] | CON | — | 0.62 ± 0.21 | 3.55 ± 0.51 | 5.43 ± 0.82 |
| (mm) | CHO | 0.63 ± 0.14 | 1.22 ± 0.43 | 3.60 ± 0.72 | 4.46 ± 0.58 |
Data are means ± s.e.m., n = 8.
Significantly different from corresponding time point for CON (P < 0.05)
significantly different from pre-ingestion (P < 0.05).
Acetyl groups
Resting acetyl CoA (pre-ingestion, 15.2 ± 2.5; post-ingestion, 6.3 ± 0.6 mmol (kg d.m.)−1) and acetylcarnitine (pre-ingestion, 3.9 ± 1.2; post-ingestion, 1.5 ± 0.9 (kg d. m.)−1) contents were both reduced (P < 0.05) following carbohydrate ingestion (Fig. 2) and were significantly lower (P < 0.05) than control at the onset of exercise. During the transition from rest to steady-state (1 min) acetyl CoA and acetylcarnitine increased to a similar extent in both trials. Accordingly, acetyl CoA and acetylcarnitine (CON, 6.8 ± 1.7; CHO, 3.1 ± 0.4 mmol (kg d. m.)−1) remained lower (P < 0.05) at 1 min of exercise in CHO (Fig. 2). These differences in acetyl group accumulation were not evident at 10 min (Fig. 2).
Figure 2.

Muscle acetylcarnitine (A) and acetyl CoA (B) at rest and during 10 min of exercise at 68 % V̇O2,peak with (CHO) or without (CON) prior carbohydrate ingestion. Values are means ± s.e.m., n = 8. * Significantly different from -75 min for CHO; † significantly different from corresponding time point for CON (P < 0.05).
PDHa
PDHa increased (P < 0.05) from a resting value of 0.76 ± 0.08 to 1.44 ± 0.19 mmol min−1 (kg wet mass (w.m.))−1 following carbohydrate ingestion and was higher (P < 0.05) than CON (0.78 ± 0.07 mmol min−1 (kg wet mass (w.m.))−1) at the onset of exercise (Fig. 3). Although PDHa increased at exercise onset in both trials, PDHa remained higher (P < 0.05) at 1 min in CHO (CON, 2.36 ± 0.22; CHO, 2.91 ± 0.18 mmol min−1 (kg wet mass (w.m.))−1). There were no differences in PDHa between conditions at 10 min (Fig. 3).
Figure 3.
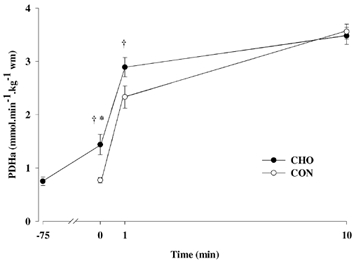
PDH activation at rest and during 10 min of exercise at 68 % V̇O2,peak with (CHO) or without (CON) prior carbohydrate ingestion. Values are means ± s.e.m., n = 8. * Significantly different from -75 min for CHO; † significantly different from corresponding time point for CON (P < 0.05).
Muscle metabolites
The muscle content of ATP was unaffected by carbohydrate ingestion or exercise (Table 3). Free ADP and AMP increased (P < 0.05) similarly at exercise onset in both trials. Free AMP was elevated (P < 0.05) at 10 min compared with 1 min in both trials and no difference was observed for free ADP. During exercise, free Pi increased but was not different between trials. Muscle G-6-P was not increased following carbohydrate ingestion and no differences were observed between trials at rest. G-6-P was higher (P < 0.05) in CON early in exercise and this effect was no longer evident by 10 min (Table 3). Muscle pyruvate was decreased (P < 0.05) by 33 % after carbohydrate ingestion at rest. Pyruvate increased (P < 0.05) during exercise in both trials and there were no differences between trials. PCr degradation (P < 0.05) and lactate accumulation at 1 and 10 min were similar between trials (Table 3). Consequently, the calculated substrate level phosphorylation between 0 and 1 min in CHO was not different from CON (CON, 58.7 ± 7.4; CHO, 55.3 ± 9.0 mmol ATP (kg d.m.)−1).
Table 3.
Muscle metabolite concentrations at rest and during 10 min exercise at 68% V̇O2,peak with (CHO) or without (CON) prior carbohydrate ingestion
| Measure | Condition | Pre-ingestion | 0 min | 1 min | 10 min |
|---|---|---|---|---|---|
| ATP | CON | — | 26.8 ± 1.1 | 26.1 ± 1.1 | 25.8 ± 0.8 |
| CHO | 25.9 ± 0.9 | 26.7 ± 1.0 | 25.8 ± 1.0 | 26.8 ± 1.3 | |
| PCr | CON | — | 89.3 ± 4.2 | 58.0 ± 4.8§ | 43.6 ± 9.1§ |
| CHO | 87.7 ± 4.3 | 88.5 ± 3.6 | 61.2 ± 4.7§ | 39.4 ± 4.9§ | |
| Creatine | CON | — | 49.1 ± 2.1 | 78.7 ± 5.0§ | 99.0 ± 6.2§ |
| CHO | 48.7 ± 2.3 | 47.8 ± 3.1 | 75.3 ± 6.8§ | 97.9 ± 5.6§ | |
| Lactate | CON | — | 5.2 ± 0.7 | 23.5 ± 3.2§ | 37.7 ± 5.7§ |
| CHO | 8.2 ± 1.7 | 5.3 ± 0.7 | 23.9 ± 3.1§ | 47.0 ± 8.3§ | |
| Pyruvate | CON | — | 0.11 ± 0.01 | 0.21 ± 0.02§ | 0.22 ± 0.01§ |
| CHO | 0.12 ± 0.01 | 0.08 ± 0.01† | 0.19 ± 0.02§ | 0.19 ± 0.01§ | |
| G-6-P | CON | — | 0.92 ± 0.12 | 3.22 ± 0.68§ | 2.04 ± 0.27§ |
| CHO | 0.99 ± 0.18 | 1.40 ± 0.34 | 2.32 ± 0.50* | 2.45 ± 0.31 | |
| Free ADP | CON | — | 97.8 ± 6.3 | 207.1 ± 25.3§ | 370.9 ± 68.7§ |
| CHO | — | 95.4 ± 6.7 | 185.9 ± 26.3§ | 319.0 ± 36.4§ | |
| Free AMP | CON | — | 0.35 ± 0.05 | 1.69 ± 0.36§ | 6.62 ± 2.29§ |
| CHO | — | 0.34 ± 0.05 | 1.43 ± 0.35§ | 4.13 ± 0.92§ | |
| Free P1 | CON | — | — | 38.8 ± 4.4 | 56.3 ± 10.4 |
| CHO | — | — | 37.9 ± 4.4 | 61.1 ± 4.2 |
Data are means ± s.e.m., n = 8. All value are expressed as mmol (kg d.m.)−1 except free ADP and free AMP which are expressed as μmol (kg d.m.)−1.
Significantly different from pre-ingestion
significantly different from corresponding time point for CON
significantly different from 0 min of same condition (P <0.05).
Discussion
In the present study, we investigated the effect of reduced pre-exercise acetylcarnitine on substrate phosphorylation and by inference oxidative phosphorylation in skeletal muscle during the onset of exercise. At rest, PDHa was increased and a paradoxical reduction in acetylcarnitine was observed. Acetylcarnitine and acetyl CoA accumulated at the same rate during the onset of exercise in both trials suggesting acetyl group availability exceeded utilisation in the TCA cycle. PDH activation was greater during the onset of exercise (1 min) in CHO; and, consistent with our hypothesis, substrate level phosphorylation was not different between trials. These data indicate that (1) reductions in pre-exercise acetylcarnitine and small increases in PDHa are without effect on oxidative metabolism at the onset of moderate exercise and (2) acetyl CoA derived from acetylcarnitine is not the factor limiting oxidative metabolism during the transition from rest to steady-state exercise.
Acetyl group availability and oxidative metabolism
The mechansims that determine the delay in oxidative metabolism at the onset of exercise have not been fully elucidated. It is thought that O2 availability at the mitochondria, a delay in the activation of PDH and TCA cycle enzymes (metabolic inertia), or a combination of these factors contributes to the delayed O2 uptake. Some lines of research suggest that this delay is a function of inadequate O2 availability at the mitochondria (Tschakovsky & Hughson, 1999). However, numerous human studies have demonstrated that the delay in oxidative energy production at ≈65 % V̇O2,peak is at least partly due to metabolic inertia associated with slow activation of PDHa and/or limited acetylcarnitine availability (Timmons et al. 1998a; Howlett et al. 1999a; Parolin et al. 2000; Evans et al. 2001; Campbell-O'Sullivan et al. 2002).
It has been hypothesised that the lag in oxidative metabolism during the transition from rest to exercise is due to a limitation in acetyl group availability (Timmons et al. 1997, 1998a; Campbell-O'Sullivan et al. 2002). This hypothesis is primarily based on studies in isolated ischaemic canine muscle (Timmons et al. 1996, 1997) and humans (Timmons et al. 1998a, b; Howlett et al. 1999a) infused with DCA prior to contraction/exercise that demonstrated increased PDHa and acetylcarnitine at rest and reduced substrate level phosphorylation during the onset of contraction/exercise. Although these data provide evidence of a metabolic inertia (lag in supplying acetyl units) they cannot distinguish the precise source of the extra acetyl units (i.e. via the PDHa or carnitine acetyltransferase reactions).
There exist a number of lines of evidence to suggest PDHa, and not acetyl group availability, limits oxidative metabolism during the transition from rest to exercise. Firstly, those studies that administered DCA have generally not observed a decrease in acetylcarnitine during the onset of exercise (Timmons et al. 1997, 1998a,b; Parolin et al. 2000; Campbell-O'Sullivan et al. 2002; Grassi et al. 2002) whereas others have actually reported an increase (Howlett et al. 1999a,b). Secondly, in the one study that clearly supports a role for acetylcarnitine, the exercise biopsy in an ischaemic canine model was obtained after 20 min of electrical contraction which is far too late to assess the rest to exercise transition (Timmons et al. 1996). Thirdly, increased acetyl group availability via acetate infusion in the absence of changes in PDHa did not accelerate oxidative phosphorylation (Evans et al. 2001). In the present study we examined the reverse situation by reducing pre-exercise acetylcarnitine availability (Fig. 2) with carbohydrate ingestion and observed no change in substrate level phosphorylation at the onset of exercise. Instead, we observed a small increase in PDHa at rest, an equal rate of PDH transformation between trials at 1 min (ΔPDHa 0-1 min: CON, 1.58 ± 0.24; CHO, 1.47 ± 0.27 mmol (kg d. m.)−1, P = 0.76) and a concomitant accumulation of acetylcarnitine that was similar in magnitude to the CON trial (CON, 2.0 ± 0.6; CHO, 1.6 ± 0.7 mmol kg−1, P = 0.62). Thus, we conclude that increasing the flux through PDH is critical for increasing acetyl CoA and NADH availability and, ultimately, oxidative metabolism during the onset of exercise.
Previous studies have suggested that the main function of acetylcarnitine is to buffer increases in acetyl CoA (Harris et al. 1987; Constantin-Teodosiu et al. 1992; Putman et al. 1993; Evans et al. 2001). Buffering increases in acetyl CoA prevents the depletion of the CoASH pool, thereby releasing CoASH for participation in the PDH and other mitochondrial reactions. In both trials of the present study, increases in mitochondrial acetyl CoA exceeded oxidation in the TCA cycle, resulting in production of acetylcarnitine via the near-equilibrium carnitine acetyltransferase reaction. Moreover, the acetyl CoA-to-acetylcarnitine ratio was not different between trials at 1 min (CON, 3.5 ± 1.3; CHO, 4.1 ± 0.9) which is consistent with an excess in acetyl CoA and equilibrium by carnitine acetyltransferase. Taken together, these data support the contention that acetylcarnitine primarily acts to buffer increases in acetyl CoA and that acetyl group availability is not limiting oxidative metabolism during the onset of exercise.
Carbohydrate ingestion and PDHa at rest
Regulation of PDH activity is an important component of fuel metabolism because PDH permits the irreversible entry of carbohydrate-derived acetyl CoA into the mitochondria for oxidation. Acute covalent modification of PDHa by phosphorylation/dephosphosphorylation on the E1 catalytic subunit is the most important regulator of this complex.
A second aim of this study was to investigate the effects of carbohydrate ingestion on PDHa at rest and during the onset of exercise. Carbohydrate ingestion in the present study resulted in a small increase in resting PDHa that may be due to increased PDH phosphatase (PDP - activates PDH) or inhibition of PDH kinase (PDK - inhibits PDH). The increase in PDHa was most probably mediated by the fourfold increase in plasma insulin (Fig. 1). PDHa was previously shown to increase during a euglycaemic hyperinsulinaemic clamp (Mandarino et al. 1987) and when hyperglycaemia was superimposed no further activation was reported (Mandarino et al. 1993). Experiments in L6 skeletal muscle cells suggest that insulin-mediated activation of PDH occurs through increased PDP activation secondary to mitochondrial translocation of protein kinase Cδ (Caruso et al. 2001). Alternatively, PDHa may have been enhanced due to inhibition of PDK via reduced acetyl CoA. A rise in intramuscular acetyl CoA-to-CoASH following a 3 day high fat diet (Putman et al. 1993) or acetate infusion (Putman et al. 1995) has been associated with a decreased resting PDHa. This is consistent with our observation of a twofold decrease in acetyl CoA and concomitant increase in PDHa (Fig. 2).
Carbohydrate ingestion and PDHa during the rest-to-exercise transition
During the transition from rest to steady-state exercise PDHa increased to the same degree in both trials; however, because PDHa was greater at rest in CHO, the difference in PDHa persisted early in exercise. Plasma insulin is a known regulator of PDHa and was elevated in CHO early in exercise. Normally, during exercise in the fasted state, where basal insulin concentrations are low, any effect of insulin is overriden by rapid alterations by the exercise allosteric regulators. However, the hyperinsulinaemia observed in the present study may have altered the balance of the phosphatase/kinase regulatory system, and the increased PDHa at rest may reflect a superimposition, or addition to the ‘normal’ activation status of PDH.
Acute changes in the other known regulators of PDHa were unlikely to account for the early (1 min) increase in PDHa. The muscle ATP-to-ADP and acetyl CoA-to-CoASH ratios were not different between trials and because the power output in each trial was identical, it is assumed that cytosolic [Ca2+] would also be similar. Although muscle [pyruvate] was not different between trials at 1 min we cannot discount the possibility that flux through the glycolytic pathway was different. Indeed, carbohydrate ingestion increased carbohydrate oxidation at rest (O'Gorman et al. 2000) and during steady-state exercise (Sparks et al. 1998) and we report greater whole-body carbohydrate oxidation at 10 min of exercise in the present study. Thus, it is reasonable to suggest that an increased pyruvate flux may have enhanced PDHa at 1 min and we were unable to detect these subtle changes.
Summary
At rest, pre-exercise carbohydrate ingestion resulted in a multitude of metabolic and hormonal changes that include hyperglycaemia, hyperinsulinaemia and reduced FFA availability. In the muscle, PDHa was increased modestly and the acetylcarnitine store was depleted. During the transition from rest to steady-state exercise, PDHa was elevated following carbohydrate ingestion and although the precise regulatory mechanism(s) remains unclear, elevated plasma insulin or an increase in pyruvate flux would seem to be the most likely regulators. Despite reduced acetylcarnitine availability following carbohydrate ingestion, there was no increase in substrate phosphorylation or decrease in oxidative metabolism at the onset of exercise. These data suggest that acetylcarnitine availability is not the site of metabolic inertia during the transition from rest to steady-state moderate intensity exercise. These results are consistent with recent work that demonstrated no effect of increased acetylcarnitine on oxidative metabolism (Evans et al. 2001). Alternatively, delayed substrate provision through PDHa seems likely to be the important metabolic factor underlying the lag in oxidative metabolism at the onset of exercise.
Acknowledgments
We thank Drs Graham Jones and Kieran Killian for their expert medical assistance. This study was supported by the Natural Sciences and Engineering Research Council of Canada (L.L.S.), the Canadian Institute of Health Research (G.J.F.) and the Australian Research Council (L.L.S. and M.H.).
References
- Bangsbo J, Krustrup P, Gonzalez-Alonso J, Boushel R, Saltin B. Muscle oxygen kinetics at onset of intense dynamic exercise in humans. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2000;279:R899–906. doi: 10.1152/ajpregu.2000.279.3.R899. [DOI] [PubMed] [Google Scholar]
- Bergmeyer HU. Methods in Enzymatic Analysis. New York: Academic Press; 1974. [Google Scholar]
- Campbell-O'Sullivan SP, Constantin-Teodosiu D, Peirce N, Greenhaff PL. Low intensity exercise in humans accelerates mitochondrial ATP production and pulmonary oxygen kinetics during subsequent more intense exercise. Journal of Physiology. 2002;538:931–939. doi: 10.1113/jphysiol.2001.013238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso M, Maitan MA, Bifulco G, Miele C, Vigliotta G, Oriente F, Formisano P, Beguinot F. Activation and mitochondrial translocation of protein kinase Cdelta are necessary for insulin stimulation of pyruvate dehydrogenase complex activity in muscle and liver cells. Journal of Biological Chemistry. 2001;276:45088–45097. doi: 10.1074/jbc.M105451200. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Carlin JI, Constantin-Teodosiu D, Harper P, Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Analytical Biochemistry. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. PDC activity and acetyl group accumulation in skeletal muscle during prolonged exercise. Journal of Applied Physiology. 1992;73:2403–2407. doi: 10.1152/jappl.1992.73.6.2403. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Tullson PC, Terjung RL. Influence of mitochondrial content on the sensitivity of respiratory control. Journal of Biological Chemistry. 1987;262:9109–9114. [PubMed] [Google Scholar]
- Evans MK, Savasi I, Heigenhauser GJ, Spriet LL. Effects of acetate infusion and hyperoxia on muscle substrate phosphorylation after onset of moderate exercise. American Journal of Physiology - Endocrinology and Metabolism. 2001;281:E1144–1150. doi: 10.1152/ajpendo.2001.281.6.E1144. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, Saltin B. PDH activation by dichloroacetate reduces TCA cycle intermediates at rest but not during exercise in humans. American Journal of Physiology. 1999;277:E33–38. doi: 10.1152/ajpendo.1999.277.1.E33. [DOI] [PubMed] [Google Scholar]
- Grassi B, Hogan MC, Greenhaff PL, Hamann JJ, Kelley KM, Aschenbach WG, Constantin-Teodosiu D, Gladden LB. Oxygen uptake on-kinetics in dog gastrocnemius in situ following activation of pyruvate dehydrogenase by dichloroacetate. Journal of Physiology. 2002;538:195–207. doi: 10.1113/jphysiol.2001.012984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RC, Foster CV, Hultman E. Acetylcarnitine formation during intense muscular contraction in humans. Journal of Applied Physiology. 1987;63:440–442. doi: 10.1152/jappl.1987.63.1.440. [DOI] [PubMed] [Google Scholar]
- Harris RC, Hultman E, Nordesjo LO. Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scandinavian Journal of Clinical and Laboratory Investigation. 1974;33:109–120. [PubMed] [Google Scholar]
- Howlett RA, Heigenhauser GJ, Hultman E, Hollidge-Horvat MG, Spriet LL. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. American Journal of Physiology. 1999a;277:E18–25. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Heigenhauser GJ, Spriet LL. Skeletal muscle metabolism during high-intensity sprint exercise is unaffected by dichloroacetate or acetate infusion. Journal of Applied Physiology. 1999b;87:1747–1751. doi: 10.1152/jappl.1999.87.5.1747. [DOI] [PubMed] [Google Scholar]
- Mandarino LJ, Consoli A, Jain A, Kelley DE. Differential regulation of intracellular glucose metabolism by glucose and insulin in human muscle. American Journal of Physiology. 1993;265:E898–905. doi: 10.1152/ajpendo.1993.265.6.E898. [DOI] [PubMed] [Google Scholar]
- Mandarino LJ, Wright KS, Verity LS, Nichols J, Bell JM, Kolterman OG, Beck-Nielsen H. Effects of insulin infusion on human skeletal muscle pyruvate dehydrogenase, phosphofructokinase, and glycogen synthase. Evidence for their role in oxidative and nonoxidative glucose metabolism. Journal of Clinical Investigation. 1987;80:655–663. doi: 10.1172/JCI113118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman DJ, Del Aguila LF, Williamson DL, Krishnan RK, Kirwan JP. Insulin and exercise differentially regulate PI3-kinase and glycogen synthase in human skeletal muscle. Journal of Applied Physiology. 2000;89:1412–1419. doi: 10.1152/jappl.2000.89.4.1412. [DOI] [PubMed] [Google Scholar]
- Parolin ML, Spriet LL, Hultman E, Matsos MP, Hollidge-Horvat MG, Jones NL, Heigenhauser GJ. Effects of PDH activation by dichloroacetate in human skeletal muscle during exercise in hypoxia. American Journal of Physiology - Endocrinology and Metabolism. 2000;279:E752–761. doi: 10.1152/ajpendo.2000.279.4.E752. [DOI] [PubMed] [Google Scholar]
- Passoneau JA, Lowry OH. Enzymatic Analysis. Totawa, NJ, USA: Humana; 1993. [Google Scholar]
- Pehleman TL, Peters SJ, Heigenhauser GF, Spriet LL. Glucose disposal into human skeletal muscle before and after a high fat/low carbohydrate diet. The Physiologist. 2000;43(330) doi: 10.1152/japplphysiol.00686.2004. [DOI] [PubMed] [Google Scholar]
- Peronnet F, Massicotte D. Table of nonprotein respiratory quotient: an update. Canadian Journal of Sport Sciences. 1991;16:23–29. [PubMed] [Google Scholar]
- Putman CT, Spriet LL, Hultman E, Dyck DJ, Heigenhauser GJ. Skeletal muscle pyruvate dehydrogenase activity during acetate infusion in humans. American Journal of Physiology. 1995;268:E1007–1017. doi: 10.1152/ajpendo.1995.268.5.E1007. [DOI] [PubMed] [Google Scholar]
- Putman CT, Spriet LL, Hultman E, Lindinger MI, Lands LC, McKelvie RS, Cederblad G, Jones NL, Heigenhauser GJ. Pyruvate dehydrogenase activity and acetyl group accumulation during exercise after different diets. American Journal of Physiology. 1993;265:E752–760. doi: 10.1152/ajpendo.1993.265.5.E752. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Harris RC, Nylind B, Hultman E. Lactate content and pH in muscle obtained after dynamic exercise. Pflügers Archiv. 1976;367:143–149. doi: 10.1007/BF00585150. [DOI] [PubMed] [Google Scholar]
- Sparks MJ, Selig SS, Febbraio MA. Pre-exercise carbohydrate ingestion: effect of the glycemic index on endurance exercise performance. Medicine and Science in Sports and Exercise. 1998;30:844–849. doi: 10.1097/00005768-199806000-00011. [DOI] [PubMed] [Google Scholar]
- Spriet LL, Soderlund K, Bergstrom M, Hultman E. Anaerobic energy release in skeletal muscle during electrical stimulation in men. Journal of Applied Physiology. 1987;62:611–615. doi: 10.1152/jappl.1987.62.2.611. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. American Journal of Physiology. 1998a;274:E377–380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Hultman E, Kaijser L, Chwalbinska-Moneta J, Constantin-Teodosiu D, Macdonald IA, Greenhaff PL. Substrate availability limits human skeletal muscle oxidative ATP regeneration at the onset of ischemic exercise. Journal of Clinical Investigation. 1998b;101:79–85. doi: 10.1172/JCI1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Poucher SM, Constantin-Teodosiu D, Macdonald IA, Greenhaff PL. Metabolic responses from rest to steady state determine contractile function in ischemic skeletal muscle. American Journal of Physiology. 1997;273:E233–238. doi: 10.1152/ajpendo.1997.273.2.E233. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Poucher SM, Constantin-Teodosiu D, Worrall V, Macdonald IA, Greenhaff PL. Increased acetyl group availability enhances contractile function of canine skeletal muscle during ischemia. Journal of Clinical Investigation. 1996;97:879–883. doi: 10.1172/JCI118490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschakovsky ME, Hughson RL. Interaction of factors determining oxygen uptake at the onset of exercise. Journal of Applied Physiology. 1999;86:1101–1113. doi: 10.1152/jappl.1999.86.4.1101. [DOI] [PubMed] [Google Scholar]
- Wilson DF. Factors affecting the rate and energetics of mitochondrial oxidative phosphorylation. Medicine and Science in Sports and Exercise. 1994;26:37–43. [PubMed] [Google Scholar]