

Abstract
The effects of changes in the mean (Sm) and pulsatile (Sp) components of arterial wall shear stress on arterial dilatation of the iliac artery of the anaesthetized dog were examined in the absence and presence of the endothelin receptor antagonist tezosentan (10 mg kg−1 I.V.; Ro 61-0612; [5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(2-1H-tetrazol-5-yl-pyridin-4-yl)-pyrimidin-4-ylamide]). Changes in shear stress were brought about by varying local peripheral resistance and stroke volume using a distal infusion of acetylcholine and stimulation of the left ansa subclavia. An increase in Sm from 1.81 ± 0.3 to 7.29 ± 0.7 N m−2 (means ± S.E.M.) before tezosentan caused an endothelium-dependent arterial dilatation which was unaffected by administration of tezosentan for a similar increase in Sm from 1.34 ± 0.6 to 5.76 ± 1.4 N m−2 (means ± S.E.M.). In contrast, increasing the Sp from 7.1 ± 0.8 to a maximum of 11.5 ± 1.1 N m−2 (means ± S.E.M.) before tezosentan reduced arterial diameter significantly. Importantly, after administration of tezosentan subsequent increases in Sp caused arterial dilatation for the same increase in Sp achieved prior to tezosentan, increasing from a baseline of 4.23 ± 0.4 to a maximum of 9.03 ± 0.9 N m−2 (means ± S.E.M.; P < 0.001). In conclusion, the results of this study provide the first in vivo evidence that pulsatile shear stress is a stimulus for the release of endothelin from the vascular endothelium.
The ability of the arterial endothelium to transduce mechanical into biochemical events is now well accepted (Davies, 1995). Arterial wall shear stress (a function of blood flow, viscosity and geometry of the artery) is a primary mechanical stimulus to the endothelium resulting in the release of mediators of vasodilatation and constriction such as nitric oxide (NO) and endothelin, respectively. Arterial wall shear stress may be regarded as having two components mean (Sm) and pulsatile (Sp) corresponding to the mean and pulsatile components of blood flow. Previously we have shown that dilatation of the iliac artery of the anaesthetized dog, by NO released from the endothelium, was caused by increases in Sm and that, in contrast, increases in Sp caused arterial constriction (Snow et al. 1994, 2001). Evidence obtained from in vitro studies, using cultured endothelial cells, has corroborated our findings by showing that pulsatile shear stress downregulates constitutive NO synthase and increases the expression of the endothelin gene (Ziegler et al. 1998). In a similar series of experiments in which the rates of production of NO, prostacyclin and endothelin were measured, it was concluded that mean shear stress stimulates the highest production of vasodilators whereas pulsatile shear stress stimulates the highest production of vasoconstrictors (Qui & Tarbell, 2000). Therefore it is possible that the reduction in arterial diameter in response to pulsatile shear stress observed in our previous experiments in the iliac artery of the dog (Snow et al. 2001) is caused by the production of endothelin. Our aim in this study was to test this hypothesis in vivo using tezosentan, a potent endothelin A and B (ETA/ETB) receptor antagonist (Clozel et al. 1999), in an anaesthetized dog preparation (Snow et al. 1994, 2001).
Methods
General methods
This investigation was carried out under licences issued by the Department of Health, Ireland as directed by the Cruelty to Animals Act, Ireland and EU Statutory Instructions. Experiments were carried out in 18 dogs weighing 21 kg (mean; range 14.8-30 kg). The dogs were sedated with morphine sulphate (10 mg i.m.) and 30 min later were anaesthetized with pentobarbitone (induction 30 mg kg−1i.v.; maintenance averaged 3 mg kg−1i.v. every 30 min) through a cannula inserted into the long saphenous vein under local anaesthesia. Depth of anaesthesia was assessed by measuring the increase in heart rate in response to pinching of the nasal septum. At the end of the experimental procedures animals were killed using a lethal intravenous injection of pentobarbitone.
Ventilation was delivered via a tracheotomy with a mixture of 40 % oxygen in room air supplied by a Starling Ideal pump. Samples of arterial blood were withdrawn at regular intervals throughout each experiment and the pH, PCO2 and PO2 was measured using an IL pH/blood gas analyser (IL, Milan, Italy). End tidal PCO2 was measured using an infra CO2 analyser (P. K. Morgan Ltd, Gillingham, Kent). The arterial PCO2 was kept within the limits of 34-42 mmHg and the arterial pH between 7.33-7.43 either by adjustment of the ventilation or by intravenous injection of 1.0 m NaHCO3. Rectal temperature was recorded and maintained at 37 ± 1°C by heating lamps and a thermostatically controlled blanket.
The ECG lead II was recorded and used to drive a cardiotachometer. Systemic arterial pressure was recorded through a short nylon cannula inserted into the abdominal aorta via the right femoral artery using a strain gauge pressure manometer (Grass Instruments, Quincy, USA) attached to a carrier amplifier (A6WP, Astro-Med, Slough, UK). Blood flow was measured using an ultrasound flowmeter (Transonic Inc., Ithaca, NY, USA). The outside diameter of the iliac artery was measured using disc-shaped piezoelectric crystals (VD5-2NP) attached to a Sonomicrometer 120.0 (Triton Technology Inc., San Diego, CA, USA). This apparatus is capable of measuring distances within the range 2-20 mm with a resolution of ±0.005 mm (Angus et al. 1983). The ECG, heart rate, arterial pressure, blood flow and diameter measurements were recorded using a thermal array recorder (K2G, AstroMed).
Surgical procedures
A diagram of the preparation is shown in Fig. 1. The aorta and external iliac arteries were exposed through a retroperitoneal left flank incision. The right iliac artery was tied off around the cannula used for measurement of pressure. An arterial embolectomy catheter (Fogarty No.3, Baxter, Santa Anna, CA, USA) was inserted a distance of 1 cm into the distal portion of the left iliac artery via the deep femoral artery. The balloon of the catheter was distended with saline until it caused about 10 % increase in the diameter of the artery and then moved backwards and forwards over a distance of 1 cm to remove the endothelium. There is histological evidence (Lamping et al. 1985) that this technique removes the endothelium without damaging the underlying smooth muscle, since the muscle still responds to the application of catecholamines and sodium nitroprusside (Snow et al. 1994). The catheter was then replaced by a polythene cannula whose tip lay within the deep femoral artery. Two pairs of piezoelectric crystals were then placed on diametrically opposite sides of the iliac artery, one pair on the section with endothelium removed (D1) and a second pair 4-5 cm upstream on a section of artery with endothelium intact (D2). An ultrasonic blood flow transducer was placed around the artery between D1 and D2. The chest was opened in the third intercostal space, the left stellate ganglion was crushed and stimulating electrodes were placed around the two branches of the ansa subclavia. The nerves were stimulated supramaximally at frequencies in the range 1 to 10 Hz using a Grass S88 stimulator (Grass Instruments).
Figure 1. Schematic diagram of preparation.
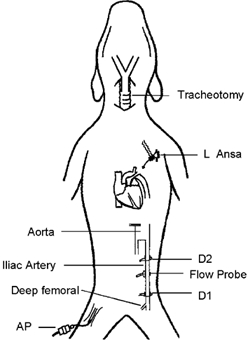
A cannula was inserted through the right femoral artery for measurement of arterial pressure (AP). Pairs of piezoelectric crystals were used to measure iliac artery diameter at D2 endothelium intact and D1 endothelium removed. An ultrasonic flow probe was used to measure iliac artery blood flow.
Experimental protocol
Changes in mean blood flow in the iliac artery were brought about by varying both peripheral resistance in the vascular bed fed by the left iliac artery and in the stroke volume of the heart before and within 1 h (half-life of tezosentan) after injection of tezosentan 10 mg kg−1i.v. (Clozel et al. 1999). Changes in peripheral resistance were produced by infusing acetylcholine downstream to the sites of diameter measurement in the iliac artery, through the cannula inserted into the deep femoral artery; the doses were 1, 2 and 4 μg min−1, a range of infusion rates at which acetylcholine does not recirculate and cause release of nitric oxide in the iliac artery (Snow et al. 1994). Changes in stroke volume and therefore pulsatile shear stress in the iliac artery were brought about by stimulation of the left ansa subclavia (Fig. 2) and subsequent inotropic effect on the left ventricle with little or no change in heart rate (Furnival et al. 1973). Measurements were taken when the artery diameter reached a steady state, about 30 s after start of dilatation. The internal diameter (D) of the two sections of the iliac artery were calculated from the distance between the piezoelectric crystals, corrected for lens effect to obtain the external diameter (Triton Technology Inc., USA), and the wall thickness of the iliac artery, measured at postmortem using a micrometer gauge.
Figure 2. Records showing the effect of an increase in mean and amplitude of blood flow on the external diameter of the iliac artery with endothelium intact (D2) and removed (D1).
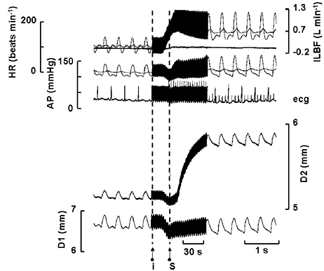
ILBF, iliac artery blood flow; AP, arterial blood pressure; HR, heart rate. Both mean and amplitude of blood flow were increased by an infusion of acetylcholine (4 μg min−1) downstream to the sites of diameter measurement at I and stimulation of the sympathetic nerves to the heart at S (left ansa subclavia, supra maximal 4 Hz). The mean diameter of the artery at D2 initially follows blood pressure along with that at D1, but after about 25 s begins to dilate in response to the maintained increase in blood flow.
Calculation of wall shear stress
The mean (Sm) and pulsatile (Sp) components of wall shear stress (N m−2) were calculated from internal diameter (D), the mean (Vm) and pulsatile component (Va) of blood velocity averaged across the artery, the fundamental frequency of oscillation (heart rate) and blood viscosity (μ, assumed to be 4 mN s m−2). To obtain changes in diameter which were endothelium only dependent, changes in the diameter in the section of artery without endothelium were subtracted from the changes in diameter of artery with endothelium. Mean (Sm) and pulsatile (Sp) components of shear stress were calculated as previously described (Snow et al. 2001).
Statistical analysis
Linear regression and correlation analysis and Student's t tests were carried out using MS Excel version 8 (Sandyford, Dublin, Ireland), P < 0.05 was taken to be statistically significant.
Results
When recording began about 2 h after the induction of anaesthesia the heart rate (HR) was 138 ± 25 beats min−1 (mean ± s.d.), the mean arterial blood pressure was 117 ± 14 mmHg (mean ± s.d.). The average mean blood flow in the left iliac artery was 100 (range 43-179 ml min−1) and the amplitude of blood flow was 467 (range 310-932 ml min−1). The mean internal diameter of the section of artery from which the endothelium had been removed (D1) was 4.02 ± 0.78 mm (mean ± s.d.) and of the section of artery with intact endothelium (D2) was 3.58 ± 0.61 mm (mean ± s.d.), this difference in diameter was significant (P < 0.05, Student's paired t test).
Increases in both the mean and amplitude of blood flow in the iliac artery were brought about by peripheral vasodilatation and inotropic stimulation of the heart. An example of records obtained in one dog is shown in Fig. 2. Blood flow was increased by a combination of a downstream (to avoid a direct action of acetylcholine on the endothelium of the iliac artery) infusion of acetylcholine (4 μg min−1) and stimulation of the cardiac sympathetic nerves (left ansa subclavia, supramaximal at 4 Hz). Both the mean and amplitude of blood flow were increased, 111 to 622 ml min−1 and 533 to 932 ml min−1, respectively; these changes in blood flow were accompanied by small and transient changes in mean arterial blood pressure. About 5 s after the start of the infusion of acetylcholine, blood pressure fell and both sections of artery decrease in diameter, blood flow increases and is further increased by sympathetic nerve stimulation which also caused arterial mean pressure to return to control levels. The external diameter of the section of artery with no endothelium (D1) followed, without delay, the changes in blood pressure. The section of artery with intact endothelium (D2) initially followed blood pressure, and only 30 s after the blood flow had increased did the artery begin to dilate and the increased blood flow had to be maintained for about 1 min before the maximum response was obtained, with, on this occasion an increase in the internal diameter from 3.75 to 4.23 mm, an effect previously shown to be mediated by nitric oxide (Snow et al. 1994, 2001). In contrast, the internal diameter of the section of artery without endothelium (D1) fell from a control of 4.75 to 4.7 mm. Figure 3 shows that tezosentan (10 mg kg−1i.v.) had no effect on heart rate, but mean arterial blood pressure (MABP), iliac artery blood flow (ILBF) and the diameter of both sections of artery were reduced by small but significant amounts; the results are summarized in Table 1.
Figure 3. An example of records showing the effect of tezosentan on the baseline parameters measured.
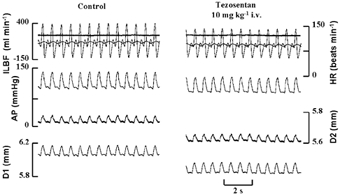
ILBF, iliac artery blood flow; AP, arterial blood pressure; HR, heart rate; D1, external diameter of section of iliac artery without endothelium; D2, external diameter of section of iliac with endothelium; tezosentan (10 mg kg−1i.v.) reduces the diameter of both sections of the iliac artery equally subsequent to the fall in arterial blood pressure.
Table 1.
Summary of results obtained showing the effects of tezosentan on the cardiovascular parameters measured
| Control | Tezosentan 10 mg kg−1 | |
|---|---|---|
| HR(beats min−1) | 134 ± 6 | 137 ± 7 |
| MABP (mmHg) | 111 ± 9 | 105 ± 10* |
| MILBF (ml min−1) | 112 ± 13 | 90 ± 14* |
| D2 (mm) | 4.3 ± 0.16 | 4.22 ± 0.15* |
| D1 (mm) | 4.7 ± 0.17 | 4.6 ± 0.17* |
Values are means ± s.e.m.; HR, heart rate; MABP, mean arterial blood pressure; MILBF, mean iliac artery blood flow; D2 section of artery with intact endothelium; D1 section with no endothelium.
P < 0.05 paired t test.
A variety of combinations of peripheral vasodilatation and cardiac stimulation were carried out in each dog before and after tezosentan (10 mg kg−1i.v.), and the effects of changes in both mean (Sm) and the pulsatile component (Sp) of wall shear stress on the internal diameter of the section of artery with intact endothelium (D2) are shown in Fig. 4 and Fig. 5, respectively. Endothelium-dependent arterial dilatation is positively correlated with an increase in Sm, further, the magnitude of the increase in arterial diameter was not altered by endothelin receptor antagonism (Fig. 4). In Fig. 4A the Sm increased from a control of 1.81 ± 0.3 to 7.29 ± 0.7 N m−2 (means ± s.e.m.), before tezosentan, and from 1.34 ± 0.6 to 5.76 ± 1.4 N m−2 (means ± s.e.m.) after tezosentan (Fig. 4B). These values were not significantly different (Student's unpaired t test). The slope of the relationship between Sm and arterial diameter was also unaffected by tezosentan, 0.052 ± 0.0045 (mean ± s.e.m.) compared to 0.05 ± 0.0095 mm N−1 m2 (mean ± s.e.m.). Thus blockade of the effects of endothelin appears to have no effect on the relationship between increases in mean shear stress and increases in the diameter of the iliac artery with intact endothelium (D2).
Figure 4. Pooled results showing the effects of changes in mean shear stress (Sm) before and after tezosentan on the change in diameter of artery with intact endothelium.
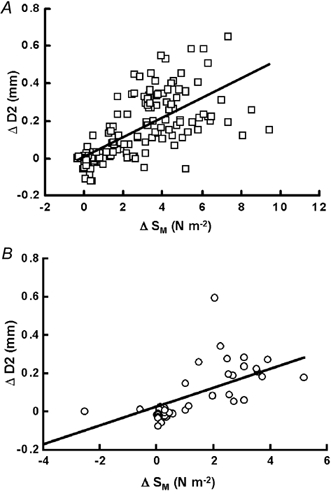
Before (A) and after (B) tezosentan (10 mg kg−1i.v.) administration. Sm shows a significant positive correlation with the change in diameter (A; correlation coefficient (r) = 0.46, P < 0.001) which is unaffected by tezosentan (B; r = 0.39, P < 0.001).
Figure 5. Pooled results showing the effects of changes in pulsatile shear stress (Sp) before and after tezosentan on the change in diameter of artery with intact endothelium.
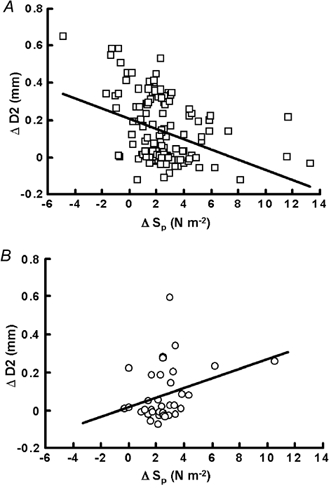
Before (A) and after (B) tezosentan (10 mg kg−1i.v.) administration. Sp shows a significant negative correlation with the change in diameter before tezosentan (A; r = -0.13, P < 0.001) which is completely reversed following the administration of tezosentan (B; r = 0.08, P < 0.05).
In contrast, before tezosentan there is a significant negative effect of pulsatile shear stress on arterial diameter (Fig. 5A). Pulsatile shear stress (Sp) increased from 7.1 ± 0.8 to a maximum of 11.5 ± 1.1 N m−2 (means ± s.e.m.) and the slope of the regression line was -0.028 ± 0.006 mm N−1 m2 (mean ± s.e.m.; P < 0.001). After tezosentan (10 mg kg−1i.v.) Sp increased from a baseline of 4.23 ± 0.4 to a maximum of 9.03 ± 0.9 N m−2 (means ± s.e.m.; P < 0.001), and importantly, the negative relationship between the increase in pulsatile shear stress was now positive (Fig. 5B) with a slope of 0.0257 ± 0.012 006 mm N−1 m2 (mean ± s.e.m.; P < 0.05).
Therefore the decrease in arterial diameter due to the increase in pulsatile shear stress is mediated by endothelin.
Discussion
The results from this study show that the arterial constriction that follows an increase in the pulsatile component of shear stress described previously (Snow et al. 2001) is prevented by tezosentan, a dual endothelin A/B (ETA/ETB) receptor antagonist (Clozel et al. 1999). Further, this effect is dependent on an intact vascular endothelium. Taken together the results are consistent with the idea that the vascular endothelium releases endothelin in response to a maintained increase in pulsatile shear stress. In these experiments, in vivo, pulsatile shear stress and pulsatile pressure are always associated and it is not possible to distinguish between their effects as possible mediators of the release of vasoconstrictor or vasodilator agents by the endothelium. However, experiments on cultured endothelial cells have shown that increases in mean shear stress favour the production of vasodilators whereas increases in pulsatile shear stress increase the production of endothelin and reduce the production of NO, and that the addition of circumferential strain to that of pulsatile shear stress does not alter the production of endothelin (Ziegler et al. 1998; Qui & Tarbell 2000). Therefore it is likely that the stimulus for the production of endothelin in our experiments is an increase in pulsatile shear stress and not circumferential strain caused by increases in pulse pressure.
When released into the bloodstream NO is rapidly deactivated by combining with haemoglobin, hence it acts as a local autocrine and paracrine hormone. For example, smooth muscle in the region of the iliac artery without endothelium does not respond to NO released from intact endothelium upstream, even though it is capable of responding to vasoconstrictors and dilators (Snow et al. 1994). Whether or not endothelin acts as both a local and systemic hormone in the normal animal is unclear, though there is evidence for a systemic action of endothelin in congestive heart failure in man (Gray & Webb 1996; Moreau 1998). Our results indicate that the action of endothelin on arterial smooth muscle is local and restricted to areas where the endothelium is intact. In all our experiments on the iliac artery (Snow et al. 1994, 2001 and those reported here), careful removal of the endothelium by a balloon embolectomy catheter at resting blood flows invariably caused the artery to dilate e.g. Table 1. We conjectured that, under our experimental conditions at resting blood flows, the artery was under a net vasoconstrictor influence from the endothelium mediated by endothelin. The fact that antagonism of endothelin receptors and the associated fall in mean arterial pressure reduces the diameter of both sections of artery with and without endothelium by a similar amount argues against the involvement of endothelin in the dilation caused by removal of the endothelium. It would appear that only when the amplitude of shear stress is sufficiently large to cause production of endothelin do the two sections of artery behave differently, the section of artery with endothelium intact constricting in response to an increase in pulsatile shear stress. Parent & Lavallee (2000) showed that shear-dependent NO release inhibited endothelin vasoconstriction in large epicardial coronary arteries. They made no attempt to differentiate between the effects of mean and pulsatile components of shear stress. In our experiments no evidence for an effect of endothelin on arterial dilatation caused by increases in mean shear stress were observed, the only effect was on the response to an increase in pulsatile shear stress.
The functional significance of the interaction between the mean and amplitude of arterial wall shear stress and the release and actions of the two hormones NO and endothelin is not fully understood. Based on the evidence known so far it is possible to speculate that the endothelial cell membrane senses the forces acting on it and responds so as to protect against cell adhesion (e.g. platelets), the formation of atheroma and the development of atherosclerosis. It must be remembered that the arguments set out here apply to the large conduit arteries only, where the resistance to blood flow and wall shear stresses are relatively low. The formation of atheroma throughout the arterial system is markedly uneven but correlates with areas of low mean and high pulsatile shear stresses (Caro et al. 1971, Moore et al. 1994) and therefore areas where the stimulus to NO production is low and that to endothelin is high. The arteries mostly affected are the central large conduit arteries, in particular the abdominal aorta, the iliac, coronary and carotid arteries. When arteries bend and divide, local areas of low mean shear stress occur and these are also sites predisposed to the formation of atheroma. There is evidence showing that both atheroma and atherosclerosis are prevented by NO through inhibition of cell adhesion and modification of lipid transport across the endothelium (For review see Snow 2002). In areas where the pulsatile component of shear stress is much higher than the mean (e.g. in the abdominal aorta where the ratio maybe 20:1) release of endothelin is increased and arterial constriction takes place, increasing mean shear stress and stimulating the production of NO.
In conclusion, a maintained increase in the pulsatile component of shear stress causes a reduction in arterial diameter that is completely reversed following administration of tezosentan, an endothelin receptor antagonist. This result suggests that pulsatile shear stress is a mechanical stimulus for the release of endothelin from the vascular endothelial cell. The findings from this study coupled with previous work carried out in our laboratory provide the first in vivo evidence of the contrasting effects of the components of shear stress, mean and pulsatile, on arterial diameter and of their interacting effects on the release of NO and endothelin from the endothelium.
Acknowledgments
We would like to thank Dr Martine Clozel (Actelion Pharmaceuticals Ltd, Allschwil, Switzerland) for her kind gift of tezosentan, and Mr Stephen Dineen and Mr Kieran McDonnell for their technical assistance.
References
- Angus JA, Campbell GR, Cocks TM, Manderson JA. Vasodilatation by acetylcholine is endothelium-dependent: a study by sonomicrometry in canine femoral artery in vivo. Journal of Physiology. 1983;344:209–222. doi: 10.1113/jphysiol.1983.sp014934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro CG, Fitzgerald JM, Schroter RC. Atheroma and arterial wall shear. Observations, correlation and proposal of shear dependent mass transfer mechanism for atherogenesis. Proceedings of the Royal Society B. 1971;177:109–159. doi: 10.1098/rspb.1971.0019. [DOI] [PubMed] [Google Scholar]
- Clozel M, Ramuz H, Clozel JP, Breu V, Hess P, Loffler BM, Coassolo P, Roux S. Pharmacology of tezosentan, new endothelin receptor antagonist designed for parenteral use. Journal of Pharmacology and Experimental Therapeutics. 1999;290:840–846. [PubMed] [Google Scholar]
- Davies PF. Flow-mediated endothelial mechanotransduction. Physiological Reviews. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnival CM, Linden RJ, Snow HM. Chronotropic and inotropic effects on the dog heart of stimulating the efferent cardiac sympathetic nerves. Journal of Physiology. 1973;230:137–153. doi: 10.1113/jphysiol.1973.sp010179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray GA, Webb DJ. The endothelin system and its potential as a therapeutic target in cardiovascular disease. Pharmacological Therapeutics. 1996;72:109–148. doi: 10.1016/s0163-7258(96)00101-5. [DOI] [PubMed] [Google Scholar]
- Lamping KG, Marcus ML, Dole WP. Removal of the endothelium potentiates canine large coronary artery constrictor responses to 5-hydroxytryptamine in vivo. Circulation Research. 1985;57:46–54. doi: 10.1161/01.res.57.1.46. [DOI] [PubMed] [Google Scholar]
- Moore JE, Xu C, Glagov S, Zarius CK, Ku DN. Fluid wall shear stress measurements in a model of the human abdominal aorta: oscillatory behaviour and relationship to atherosclerosis. Atherosclerosis. 1994;110:225–240. doi: 10.1016/0021-9150(94)90207-0. [DOI] [PubMed] [Google Scholar]
- Moreau P. Endothelin in hypertension: a role for receptor antagonists? Cardiovascular Research. 1998;39:534–542. doi: 10.1016/s0008-6363(98)00154-0. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Tarbell JM. Interaction between wall shear stress and circumferential strain affects endothelial cell biochemical production. Journal of Vascular Research. 2000;37:147–157. doi: 10.1159/000025726. [DOI] [PubMed] [Google Scholar]
- Snow HM. Atheroma and the mechanics of blood flow in arteries. Irish Journal of Medical Science. 2002;171:46–50. doi: 10.1007/BF03170508. [DOI] [PubMed] [Google Scholar]
- Snow HM, McAuliffe SJ, Moors JA, Brownlie R. The relationship between blood flow and diameter in the iliac artery of the anaesthetized dog: the role of endothelium-derived relaxing factor and shear stress. Experimental Physiology. 1994;79:635–645. doi: 10.1113/expphysiol.1994.sp003796. [DOI] [PubMed] [Google Scholar]
- Snow HM, Markos F, O'regan D, Pollock K. Characteristics of arterial wall shear stress which cause endothelium-dependent vasodilatation in the anaesthetized dog. Journal of Physiology. 2001;531:843–848. doi: 10.1111/j.1469-7793.2001.0843h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler T, Bouzourene K, Harrison VJ, Brunner HR, Hayoz D. Influence of oscillatory and unidirectional flow environments on the expression of endothelin and nitric oxide synthase in cultured endothelial cells. Arteriosclerosis Thrombosis and Vascular Biology. 1998;18:686–692. doi: 10.1161/01.atv.18.5.686. [DOI] [PubMed] [Google Scholar]