

Abstract
We studied the effect of the antioxidants (AOX) ascorbic acid (2 g, I.V.) and α-tocopherol (200 mg, P.O.) on the depressant effect of subanaesthetic doses of halothane (0.11 % end-tidal concentration) on the acute isocapnic hypoxic ventilatory response (AHR), i.e. the ventilatory response upon inhalation of a hypoxic gas mixture for 3 min (leading to a haemoglobin saturation of 82 ± 1.8 %) in healthy male volunteers. In the first set of protocols, two groups of eight subjects each underwent a control hypoxic study, a halothane hypoxic study and finally a halothane hypoxic study after pretreatment with AOX (study 1) or placebo (study 2). Halothane reduced the AHR by more than 50 %, from 0.79 ± 0.31 to 0.36 ± 0.14 l min−1 %−1 in study 1 and from 0.79 ± 0.40 to 0.36 ± 0.19 l min−1 %−1 in study 2, P < 0.01 for both. Pretreatment with AOX prevented this depressant effect of halothane in the subjects of study 1 (AHR returning to 0.77 ± 0.32 l min−1 %−1, n.s. from control), whereas placebo (study 2) had no effect (AHR remaining depressed at 0.36 ± 0.27 l min−1 %−1, P < 0.01 from control). In a second set of protocols, two separate groups of eight subjects each underwent a control hypoxic study, a sham halothane hypoxic study and finally a sham halothane hypoxic study after pretreatment with AOX (study 3) or placebo (study 4). In studies 3 and 4, sham halothane did not modify the control hypoxic response, nor did AOX (study 3) or placebo (study 4). The 95 % confidence intervals for the ratio of hypoxic sensitivities, (AOX + halothane):halothane in study 1 and (AOX - sham halothane):sham halothane in study 3, were [1.7, 2.6] and [1.0, 1.2], respectively. Because the antioxidants prevented the reduction of the acute hypoxic response by halothane, we suggest that this depressant effect may be caused by reactive species produced by a reductive metabolism of halothane during hypoxia or that a change in redox state of carotid body cells by the antioxidants prevented or changed the binding of halothane to its effect site. Our findings may also suggest that reactive species have an inhibiting effect on the acute hypoxic ventilatory response.
A major defence of the mammalian body to acute hypoxia is a rapid increase in pulmonary ventilation called the acute hypoxic response (AHR). This vital chemoreflex is primarily mediated by the carotid bodies located at the bifurcations of the common carotid arteries (Gonzalez et al. 1994). During the past decade, considerable progress has been made in unravelling the cascade of events within carotid body type I cells upon exposure to a hypoxic environment, although there are still many areas of controversy (Gonzalez et al. 1994; Lopez-Barneo et al. 2001). The general picture emerging from most studies is that low oxygen decreases the open probability of potassium channels, which causes membrane depolarization and influx of Ca2+ ions. In several species, various types of potassium channels are described that may serve as an oxygen-sensing element to initiate the transduction cascade in hypoxia, for example, Kv channels in rabbits (Perez-Garcia & Lopez-Lopez, 2000; Perez-Garcia et al. 2000) and maxi-K and TASK channels in rats (Buckler et al. 2000; Riesco-Fagundo et al. 2001). Although potassium channels possess redox sensitivity and are sensitive to changes in the concentration of reactive oxygen species (ROS), it is unclear by which mechanism low oxygen is able to decrease the conductance of these channels (Kourie, 1998; Lopez-Barneo et al. 1999; Kobertz et al. 2000).
Volatile anaesthetics, e.g. halothane, can open potassium channels in various cell types, such as TASK channels in rat carotid body (Patel et al. 1999; Buckler et al. 2000; Sirois et al. 2000; Patel & Honore, 2001a,b). At the same time, volatile anaesthetics, particularly halothane, are known to depress the acute hypoxic response, an effect that may be mediated through a preferential and potent action on the carotid bodies (Knill & Clement, 1984; Dahan et al. 1994). It is unknown if the opening of potassium channels by halothane might involve the intracellular concentration of ROS or changes in the cell redox state. However, during hypoxia, halothane undergoes a reductive metabolism in the liver by which radical species are produced and lipid peroxidation is initiated; this reductive metabolism of halothane is thought to be responsible for its mild hepatotoxic effect (de Groot & Noll, 1983; de Groot & Sies, 1989; Spracklin & Kharasch, 1998; Kharasch et al. 2000). In guinea-pig liver, peroxidation of lipids following halothane administration can be inhibited by antioxidant treatment with vitamin E (Sato et al. 1992).
The above findings on the sensitivity of potassium channels to ROS, the ability of halothane to produce radical species and to open potassium channels and, finally, the role of potassium channels in the hypoxic response, all raise the question of whether halothane may reduce the hypoxic response by producing ROS and/or by influencing the redox state of the carotid body. The aim of the present study in humans, therefore, was to examine the influence of the potent antioxidants α-tocopherol and ascorbic acid on the acute hypoxic ventilatory response with and without halothane.
Methods
Subjects and apparatus
Thirty-two healthy, non-smoking, male subjects (age 20-35 years) were recruited after protocol approval by the Leiden University Medical Centre Committee on Medical Ethics. All experiments conformed to the declaration of Helsinki. None of the volunteers was taking any medication or ever had surgery under general anaesthesia. All subjects performed a series of test experiments to familiarise themselves with the apparatus and experimental procedures. The subjects were instructed not to eat or drink for at least 8 h before the study. They were not instructed about respiratory physiology, anaesthesia or the intentions of the study. All gave oral and written informed consent before their participation.
After arrival at the laboratory, an intravenous catheter was inserted in the left or right antecubital vein for drug infusion. Subsequently, electrodes for EEG monitoring (BisSensor, Aspect Medical Systems, Newton, MA, USA) were placed on the head at AT1-FP1 as specified by the manufacturer, and the subjects rested for 20-30 min before the antioxidant cocktail or placebo was administered. Next a facemask was applied over the mouth and nose. Gas flow was measured with a pneumotachograph connected to a pressure transducer and electronically integrated to yield a volume signal. This signal was calibrated with a motor-driven piston pump (stroke volume 1 l at a frequency of 20 strokes min−1). Corrections were made for the changes in gas viscosity due to changes in oxygen concentration of the inhaled gas mixtures. The pneumotachograph was connected to a T-piece. One arm of the T-piece received a gas mixture (with a flow of 50 l min−1) from a gas-mixing system consisting of three mass-flow controllers (Bronkhorst High-Tec, Veenendaal, The Netherlands). A personal computer provided control signals to the mass-flow controllers so that the composition of the inspired gas mixtures could be adjusted to force end-tidal oxygen concentration (PET,O2) to follow a specified pattern in time while the end-tidal carbon dioxide concentration (PET,CO2) was kept constant. Part of the nitrogen (5 l min−1) passed through a halothane vaporizer (Dräger 19.2, Lubeck, Germany). During the initial part of the study (control experiments), the vaporizer was kept in the ‘off’ position. Dräger Nederland BV calibrated the vaporizer before its use in this study.
The oxygen and carbon dioxide concentrations of inspired and expired gases were measured with a gas monitor (Multicap, Datex-Engstrom, Helsinki, Finland) by paramagnetic and infrared analysis. The gas monitor was calibrated with gas mixtures of known concentration delivered by a gas-mixing pump (Wösthoff, Bochum, Germany). The halothane concentration was measured at the mouth with a gas monitor (Multicap Ultima, Datex-Engstrom). This gas monitor was calibrated with a gas mixture of known concentration (Quick Cal, Datex-Engstrom). The arterial saturation of haemoglobin with O2 obtained via a finger probe (Sa,O2) was measured by pulse oximetry (Satlite Plus, Datex-Engstrom).
The EEG was recorded using an Aspect A-2000 EEG monitor (Aspect Medical Systems, Newton, MA, USA).
Study design
In the first set of studies, designed to test the effect of antioxidant pretreatment on the depression by halothane of the AHR, two separate groups of eight subjects underwent a control hypoxic study, followed by a halothane hypoxic study and finally by a halothane hypoxic study after pretreatment with a cocktail of antioxidants (study 1) or placebo (study 2). In a second set of studies, which was designed to investigate the effect of antioxidant pretreatment on the hypoxic ventilatory response in the absence of halothane, two separate groups of eight subjects underwent a control hypoxic study, followed by a sham halothane hypoxic study and then a sham halothane study after pretreatment with a cocktail of antioxidants (study 3) or placebo (study 4). While the design of the halothane administration was randomized and blinded to the subjects only, both subjects and researchers were blinded to the pretreatment with antioxidants or placebo.
After each hypoxic study blood was drawn from the capillary bed of a hyperaemic finger for the determination of blood acidity (Astrup equilibration technique, Radiometer, Copenhagen, Denmark).
Hypoxia
Hypoxia was induced with a ‘dynamic end-tidal forcing’ system (Dahan et al. 1995, 1996); steps from normoxia (PET,O2 15 kPa) into hypoxia (PET,O2 6.2 kPa obtained within 4-6 breaths) were applied. Since peak hypoxic responses occur within 3 min (Dahan et al. 1995), hypoxia was maintained for 3 min, after which hyperoxia was introduced for 5 min (fractional inspired O2, FI,O2 > 0.5). The PET,CO2 was maintained just above individual resting values.
Halothane
During the appropriate studies, the subjects inhaled halothane (Fluothane, Zeneca Ltd, Macclesfield, UK). The flow through the vaporizer was 5 l min−1. This was added to a fresh gas flow of 45 l min−1. Both flows were very precise and generated by mass-flow controllers (Bronkhorst High-Tec). The setting of the vaporizer was 1.1-1.5 % (measured at the outflow tract). After mixing, the measurements were repeated (now 0.1-0.15 %). This procedure ensured that the concentrations employed and measured were in the range as stated in Table 1. The subjects inhaled a halothane concentration that maintained the end-expiratory concentration at 0.11 % for 10 min before the hypoxic study started, resulting in a minimum alveolar concentration (MAC) equivalent of 0.13 (assuming an age-adjusted MAC of 0.84 % halothane in our young subjects; Gregory et al. 1969). Note that because of the short (10 min) exposure time to this end-tidal level of halothane, the brain concentration will be less than 0.11 %, minimizing the occurrence of significant central effects (i.e. within the central nervous system) of halothane. The subjects were under the impression that halothane was given during the sham halothane studies by manipulating an empty vaporizer.
Table 1.
Influence of antioxidant and placebo pretreatment on halothane and sham halothane induced depression of the ventilatory response to hypoxia
| Baseline ventilation (1 min−1) | End-expiratory CO2 (mmHg) | pH | Halothane (vol. %) | Hypoxic ventilatory response (l min−1%−1) | Hypoxic ventilatory response (% of control) | |
|---|---|---|---|---|---|---|
| Study 1 | ||||||
| Control | 12.1 ± 0.5 | 6.1 ± 0.4 | 7.41 ± 0.02 | 0 | 0.79 ± 0.31 | 100 |
| Halothane | 12.5 ± 3.3 | 6.1 ± 0.3 | 7.41 ± 0.02 | 0.11 | 0.36 ± 0.14 * | 46 ± 11 * |
| AOX + halothane | 4.0 ± 2.1 | 6.2 ± 0.2 | 7.42 ± 0.02 | 0.11 | 0.77 ± 0.32 | 96 ± 20 |
| Study 2 | ||||||
| Control | 12.5 ± 1.6 | 6.0 ± 0.2 | 7.40 ± 0.02 | 0 | 0.79 ± 0.40 | 100 |
| Halothane | 12.7 ± 3.2 | 6.1 ± 0.2 | 7.40 ± 0.03 | 0.11 | 0.36 ± 0.19 * | 47 ± 14 * |
| Placebo + halothane | 13.7 ± 4.1 | 6.0 ± 0.2 | 7.41 ± 0.02 | 0.12 | 0.36 ± 0.27 *† | 40 ± 15 *† |
| Study 3 | ||||||
| Control | 13.9 ± 1.9 | 5.8 ± 0.3 | 7.43 ± 0.03 | 0 | 0.89 ± 0.42 | 100 |
| Sham halothane | 14.5 ± 3.6 | 5.9 ± 0.2 | 7.44 ± 0.02 | 0 | 0.90 ± 0.44 | 102 ± 14 |
| AOX + sham halothane | 14.5 ± 2.8 | 5.8 ± 0.3 | 7.43 ± 0.03 | 0 | 1.00 ± 0.54 | 116 ± 22 |
| Study 4 | ||||||
| Control | 14.6 ± 3.3 | 5.9 ± 0.4 | 7.42 ± 0.03 | 0 | 0.83 ± 0.42 | 100 |
| Sham halothane | 16.9 ± 3.8 | 5.9 ± 0.4 | 7.42 ± 0.02 | 0 | 0.88 ± 0.45 | 104 ± 15 |
| Placebo + sham halothane | 16.1 ± 2.3 | 5.9 ± 0.4 | 7.41 ± 0.02 | 0 | 0.89 ± 0.45 | 110 ± 10 |
Values are means ± s.d.
P < 0.01 vs. control of identical study (one way ANOVA)
P < 0.01vs. AOX + halothane of study 1 (Student's paired t test). AOX, antioxidant cocktail.
Antioxidant cocktail (AOX)
The antioxidant cocktail consisted of 200 mg of oral α-tocopherol (Organon, Oss, The Netherlands) given 1 h before the start of the appropriate hypoxic study, ingested with a cup of yoghurt, and two 1 g intravenous doses of ascorbic acid (Ascorbinezuur CF, 5 ml, Centrafarm, The Netherlands) given 10 and 4 min before the appropriate hypoxic study. Placebos consisted of cellulose tablets and 0.9 % NaCl manufactured by the local pharmacy. The oral placebo was also ingested with yoghurt.
Data and statistical analysis
Analysis was performed on a blinded data set. The breath-to-breath data of the last 10 breaths of normoxia and the last 10 breaths of hypoxia were averaged. Since the relationship between ventilation and arterial oxygen saturation is found to be linear (Dahan et al. 1996), we calculated the difference between the mean inspiratory ventilation (VI) and the Sa,O2 data points and expressed the hypoxic ventilatory response or sensitivity as follows (Dahan et al. 1996):
The statistical analysis was performed using SPSS v10.0 for Windows. To detect the significance of differences among the three treatment groups of each study, a two-way analysis of variance was performed. Post hoc analysis was by least significant differences and Bonferroni correction. To assess the effect of antioxidant vs. placebo pretreatment, Student's paired t tests were performed on the appropriate treatment levels of studies 1 and 2 and studies 3 and 4. Values reported are means ± s.d. P values less than 0.05 were considered significant.
Results
All subjects completed the protocols without side effects. During all studies, PET,CO2 values were kept constant at 0.1-0.2 kPa above individual resting values, with no differences between baseline (prehypoxia) and hypoxic PET,CO2 values and pH. In all hypoxic studies, Sa,O2 values were 82 ± 2 %.
The values of baseline ventilatory parameters and the control ventilatory responses to hypoxia (Table 1) are in agreement with earlier observations (Dahan et al. 1994, 1996). We observed no effect from low-dose halothane on baseline ventilation. Similarly, antioxidant and placebo pretreatment had no significant effect on baseline parameters (Table 1). Halothane (0.11 % end-expiratory concentration) decreased the ventilatory response to hypoxia by more than 50 %. As shown in Fig. 1, this effect was completely prevented by pretreatment with the antioxidant cocktail (study 1) but not by placebo pretreatment (study 2). Sham halothane did not affect any of the ventilatory baseline and hypoxic parameters, nor did antioxidant (study 3) or placebo (study 4) pretreatment (Table 1 and Fig. 2). The 95 % confidence intervals of antioxidant effect relative to halothane or sham halothane (ratio of (AOX + halothane):halothane in study 1 and ratio of (AOX + sham halothane):sham halothane in study 3) did not overlap: [1.7, 2.6] and [1.0, 1.2] in studies 1 and 3, respectively (Fig. 3). These data do not indicate that the effect of AOX to abolish halothane's depressant effect are explained by an increase of the AHR by the antioxidants per se. However, it cannot be excluded that at higher doses AOX may increase the AHR.
Figure 1. Hypoxic ventilatory responses of individual subjects in studies 1 and 2.
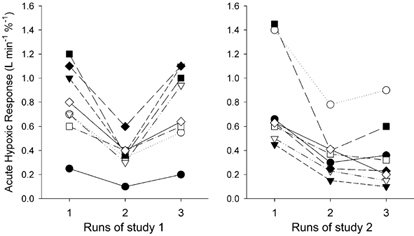
Study 1 (left panel): control and halothane hypoxic ventilatory responses and influence of AOX pretreatment on halothane-induced impairment of the hypoxic drive. Study 2 (right panel): control and halothane hypoxic responses and influence of placebo pretreatment on halothane-induced impairment of the hypoxic drive. Note the ability of antioxidant but not placebo pretreatment to prevent depression of the hypoxic response by halothane. Different symbols are used for each subject. Subjects in studies 1 and 2 were different.
Figure 2. Hypoxic ventilatory responses of individual subjects of studies 3 and 4.

Study 3 (left panel): control and sham halothane hypoxic responses and the influence of AOX pretreatment on a sham halothane response. Study 4 (right panel): control and sham halothane hypoxic responses and the influence of placebo pretreatment on a sham halothane hypoxic response. Note the absence of effect of antioxidant relative to placebo on the hypoxic response. Different symbols are used for each subject. Subjects in studies 3 and 4 were different.
Figure 3. The effect of AOX or placebo on halothane or sham halothane-induced depression of the acute hypoxic response.
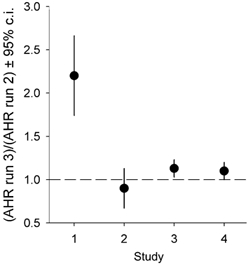
Values are the ratio of the third hypoxic run to the the second hypoxic run of studies 1-4. •, means; vertical lines depict the 95 % confidence intervals. A value of 1 indicates no effect of the AOX or placebo pretreatment on halothane's effect on the acute hypoxic ventilatory response. Note that the 95 % confidence intervals of studies 1 (AOX + halothane) and 3 (AOX + sham halothane) do not overlap.
Discussion
We have found that while an antioxidant cocktail had only a small, statistically insignificant effect on the acute hypoxic response (Fig. 3), it did reverse the large depression in the hypoxic response caused by low-dose halothane. To place this result into context, we need to discuss methodological considerations, the possible roles of ROS in the chemoreception process and the possible mechanisms by which halothane depresses the hypoxic ventilatory response.
The measurement of the hypoxic ventilatory response requires isocapnia both across drug treatments and during the hypoxic test. As seen in Table 1, the mean differences in PET,CO2 for the different treatment conditions in the four studies were closely matched and did not contribute to the changes in the measured AHR.
While we attempted to achieve blinding, the subjects were probably aware of when halothane was being inhaled. However, the depression of the AHR by halothane is large and consistent across subjects (Fig. 1), while the changes in the AHR with the sham halothane protocol are variable and similar to the variation expected with repeated hypoxic tests (Fig. 2). In testing the effects of inhalational anaesthetics, the experimental conditions are very important. We have previously shown that arousing the subject with audiovisual stimulation can reverse the depression of the AHR by isoflurane (van den Elsen et al. 1994). Therefore, possible variations in arousal level could be a confounding factor in our experiments. Since one of the researchers continuously observed the raw EEG during the studies and did not see any sign of sleep normally observed in sleep EEGs, we believe that the arousal level was similar in all studies employing halothane (i.e. halothane alone, halothane + AOX and halothane + placebo) as determined from subjective measures (all subjects opened their eyes when spoken to in a soft voice) and EEG measurements.
While we believe that the antioxidant cocktail we used was effective in altering the intracellular and/or extracellular redox state, we have no direct measurement of its efficacy in our subjects. Taking into account that the effects of halothane could be located at several sites - at the outer face of the membrane, within the membrane, in the cytosol or possibly at the mitochondrial level - we rationalized the combined use of water-soluble ascorbic acid, which is a particularly potent antioxidant in plasma and in the cytosol (Frei et al. 1989; Carr & Frei, 1999), and α-tocopherol, which, due to its lipid solubility, may be the most important free radical and lipid peroxide scavenger in membranes (Burton et al. 1983). Furthermore, it is known that the combined effectiveness of ascorbate and α-tocopherol is synergistic, with the net result that radicals originating from the membrane are removed using two different antioxidants (Packer et al. 1979; Niki, 1987). Combined administration of α-tocopherol (2000 i.u. i.m.) and ascorbic acid (2 g i.v.) has been shown to reduce lipid peroxidation in patients undergoing cardiac bypass surgery (Barta et al. 1991).
The oxygen transduction cascade in the carotid body (as in the similarly oxygen-sensitive pulmonary artery smooth muscle and the pulmonary neural epithelial cell bodies) has been subject to considerable research over the past decade and, while a much clearer picture of the process has emerged, there are many areas of considerable controversy (see Gonzalez et al. 1994 and Lopez-Barneo et al. 2001 for recent reviews). The most generally accepted model is that low oxygen decreases the open probability of potassium channels in the membrane of carotid body type I cells, which results in depolarization. This membrane depolarization opens voltage-gated calcium channels, with the resulting influx of Ca2+ causing neurotransmitter release, activating the synaptically adjacent carotid sinus nerve. Currently, much interest has focused on the oxygen-sensitive potassium channels in the carotid bodies of several species (Peers, 1997; Lewis et al. 2002). The rat and the rabbit have been most commonly studied and they appear to have different types of oxygen-sensitive potassium channels. The rat appears to have both TASK (Buckler et al. 2000) and maxi-K channels (Riesco-Fagundo et al. 2001) that are oxygen sensitive, while in the rabbit Kv channels seem to serve this role (Perez-Garcia & Lopez-Lopez, 2000; Perez-Garcia et al. 2000). However, within this general model, it is not determined how low oxygen closes the potassium channel that seems to initiate the cascade.
Several studies have indicated that potassium channels show redox sensitivity and considerable sensitivity to levels of ROS (Kourie, 1998; Lopez-Barneo et al. 1999; Kobertz et al. 2000). It is unclear whether potassium channels possess intrinsic oxygen sensitivity, or whether other elements are required or modulate the O2-sensing cascade (e.g. cytosolic, possibly membrane-associated, redox couples). Intrinsic oxygen sensitivity could exist in the form of reduction/oxidation of thiol-containing free cysteine residues in K+ channel β subunits that are required for hypoxic sensitivity (Perez-Garcia et al. 1999). One proposed redox model associated with enzymatic production of ROS that may influence potassium channel conductance is the cytochrome P450 system that utilises NAD(P)H as an electron donor. Inhibition of this enzyme system has been shown to prevent the hypoxic inhibition of potassium channels (Hatton & Peers, 1996) but this has not been found in all model systems (Roy et al. 2001).
It is clear that within this general framework of hypoxic chemoreception there is considerable variety in specific sensor elements and couplings. Particularly when channels are expressed in heterologous systems, all the elements for the in vivo cascade may not be present. In addition, there may be substantive differences between sensing elements of the cascade between the different oxygen-sensitive tissues. Thus, it has been difficult to verify the roles for ROS in carotid body chemotransduction in more physiologically intact preparations. In fact, there is considerable controversy as to whether ROS increase (Leach et al. 2001; Waypa et al. 2001) or decrease (Lahiri & Acker, 1999) with hypoxia in oxygen-sensitive cells. Experiments in which the redox state of carotid body cells was altered would seem to indicate that ROS might not provide a direct link between hypoxia and the membrane depolarization initiated by the closure of the K+ channel (Roy et al. 2001; Sanz-Alfayate et al. 2001). Exogenous reductants, however, have been shown to mimic the effect of hypoxia on O2-sensitive potassium channels in carotid body cells (Benot et al. 1993). Thus, whatever the precise mechanism, there is likely to be at least a modulating role for the redox state of the type I cell in O2 sensing.
The depressant effect of subanaesthetic doses of halothane in humans on ventilation during hypoxia may occur via a preferential and potent action on the carotid bodies (Knill & Clement, 1984; Dahan et al. 1994). The mechanism for this depression is unknown, but inhalational anaesthetics can directly open two-pore domain potassium (TASK) channels in various cell types (Patel et al. 1999; Sirois et al. 2000; Patel & Honore, 2001a,b) and in particular in the rat carotid body (Buckler et al. 2000). The action of inhalational anaesthetics on TASK channels may be located at a specific region at the junction between the final transmembrane domain and the cytoplasmic C-terminus (Patel et al. 1999; Talley & Bayliss, 2002). This site is also involved in neurotransmitter inhibition of the channel but does not contain a motif that is known to be involved in cell signalling mechanisms (Talley & Bayliss, 2002). How changes in ROS and/or redox state could alter the properties of this binding site is unknown. In the lung carcinoma cell line H146, a representative model for pulmonary oxygen-sensitive neuroepithelial body cells, halothane transiently reverses hypoxic inhibition of potassium currents, similar to the reversal caused by the reactive species H2O2 (Hartness et al. 2001).
The metabolism of halothane itself may also change the redox status of cells. In hypoxia, halothane undergoes a reductive metabolism that in the liver is catalysed by isoforms of cytochrome P450 but in other tissues possibly also by other haem-containing proteins (de Groot & Sies, 1989; Spracklin & Kharasch, 1998; Kharasch et al. 2000). Reduction of halothane yields CF3CHCl radicals able to inactivate cytochrome P450 by covalent binding or, alternatively, to remove hydrogen from polyunsaturated lipids thus initiating lipid peroxidation (de Groot & Noll, 1983; de Groot & Sies, 1989; Kharasch et al. 2000). In guinea-pigs, the hepatotoxic effect caused by this reductive metabolism of halothane can be prevented by antioxidant treatment (Sato et al. 1992). In humans, haemin induction of haem oxygenase-1, which has an antioxidant role in oxidative stress, has been shown to be effective against halothane-induced liver damage (Odaka et al. 2000).
The susceptibility of halothane's depressant effect to antioxidant treatment that we found in this study indicates that the cellular redox state influences the effect of halothane on the oxgyen-sensing mechanism. This could be explained by a modulation by ROS of the coupling of halothane to the potassium channel (or other channels). Whether or not the ROS was generated from halothane's metabolism or from other intracellular processes (Waypa et al. 2001), the reduction in ROS with antioxidant treatment could reduce the coupling of halothane to the channel and prevent it from opening the channel.
An alternative way to explain our findings would be to suggest that an increase in the concentration of ROS has an inhibitory effect on the mechanism involved in the acute hypoxic response. In this scenario, the cellular redox state or the signalling from a particular ROS would be the coupling from low oxygen to potassium channel closure. For example, an NAD(P)H oxidase has been proposed as the membrane-bound source of oxygen-sensitive ROS, implying a decrease in ROS in hypoxia (Kummer & Acker, 1995; Semenza, 1999; Jones et al. 2000; Kietzmann et al. 2000). The increase in local ROS caused by the reductive metabolism of halothane in hypoxia would thus counter the hypoxia-induced decrease in ROS and prevent the hypoxic closure of the K+ channel. This effect would be most noticeable in hypoxia since halothane's reductive metabolism is increased in hypoxia.
In animal species, the effect of halothane on the hypoxic ventilatory response is variable. In the goat, for example, an end-tidal concentration of 0.5 % halothane does not significantly depress it (Koh & Severinghaus, 1990). In the rabbit and cat, 0.5-1 % halothane reduces the hypoxic response, the effect in the latter species being larger (Davies et al. 1982; Ponte & Sadler, 1989). As shown in this and previous studies, the effect of 0.11 % halothane in man is to reduce hypoxic sensitivity by more than 50 %. These species differences could originate from the differences in the type of oxygen-sensitive potassium channel that initiates the transduction cascade (e.g. TASK vs. Kv) and their differences in anaesthetic sensitivity or in splice variants of the expressed channel. An alternative explanation could also lie in species differences in the defence against ROS. Goats produce large quantities of ascorbic acid (Chatterjee et al. 1975) and may thus be better protected against the adverse effects of free radicals produced by halothane. To a lesser degree this may also be the case for rabbits. Cats produce low quantities of ascorbic acid (Chatterjee et al. 1975) and this might explain their higher susceptibility to halothane than rabbits. Humans have lost the ability to synthesize ascorbic acid and may therefore be more vulnerable to the adverse effects of reactive species that are produced by halothane.
It is worth mentioning that in a previous study we were not able to demonstrate a clear depression of the normocapnic AHR by desflurane (Dahan et al. 1996). This volatile anaesthetic has a low metabolism, with little production of free radicals (Koblin, 1992). In another study, we found that low-dose propofol, which is known to have antioxidant properties (De La Cruz et al. 1999), depressed neither the CO2 sensitivity of the peripheral chemoreflex loop nor the fast (carotid body-mediated) component of the acute hypoxic response (Nieuwenhuijs et al. 2000, 2001). Together with the present findings, these previous data suggest that the (lack of) depressant effects of anaesthetics on the hypoxic response may be related to their pro-oxidant (antioxidant) properties, but further studies are needed to support this hypothesis.
From the data that we present in this study, we conclude that changing the cellular redox state can modulate the depressant effect of halothane on the acute hypoxic response. Furthermore, although our results do not supply direct evidence for an inhibitory role of ROS in the oxygen-sensing cascade, they could be explained by ascribing at least a modulating role to radical species in the AHR. Our observation that antioxidant pretreatment markedly reduces the depressant effect of halothane on the AHR demonstrates a specific pharmacological reversal of an anaesthetic effect. Further work is needed in both humans and animal preparations to clarify the interaction of cellular redox status, inhalational anaesthetics and oxygen-sensitive potassium channels in the carotid body.
Acknowledgments
This study was made possible by Grant MW902-19-144 from The Netherlands Organization for Pure Research (ZonMW, Zorgonderzoek Nederland, Medische wetenschappen NOW), Den Haag, The Netherlands.
References
- Barta E, Pechan I, Cornak V, Luknarova O, Rendekova V, Verchovodko P. Protective effect of alpha-tocopherol and l-ascorbic acid against the ischemic-reperfusion injury in patients during open-heart surgery. Bratislavske Lekarske Listy. 1991;92:174–183. [PubMed] [Google Scholar]
- Benot AR, Ganfornina MD, Lopez-Barneo J. Potassium channel modulated by hypoxia and the redox status in glomus cells of the carotid body. In: Weir EK, Hume JR, Reeves JT, editors. Ion Flux in Pulmonary Vascular Control. New York, NY, USA: Plenum Press; 1993. pp. 177–187. [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemorecptor cells. Journal of Physiology. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GW, Joyce A, Ingold KU. Is vitamin E the only lipid-soluble, chain-breaking antioxidant in human blood plasma and erythrocyte membranes? Archives of Biochemistry and Biophysics. 1983;221:281–290. doi: 10.1016/0003-9861(83)90145-5. [DOI] [PubMed] [Google Scholar]
- Carr A, Frei B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB Journal. 1999;13:1007–1024. doi: 10.1096/fasebj.13.9.1007. [DOI] [PubMed] [Google Scholar]
- Chatterjee IB, Majumder AK, Nandi BK, Subramanian N. Synthesis and some major functions of vitamin C in animals. Annals of the New York Academy of Sciences. 1975;258:24–47. doi: 10.1111/j.1749-6632.1975.tb29266.x. [DOI] [PubMed] [Google Scholar]
- Dahan A, Berkenbosch A, Degoede J, van den Elsen M, Olievier I, van Kleef J. Influence of hypoxic duration and posthypoxic inspired O2 concentration on short term potentiation of breathing in humans. Journal of Physiology. 1995;488:803–813. doi: 10.1113/jphysiol.1995.sp021012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan A, Sarton E, van den Elsen M, van Kleef J, Teppema L, Berkenbosch A. Ventilatory response to hypoxia in humans. Influences of subanesthetic desflurane. Anesthesiology. 1996;85:60–68. doi: 10.1097/00000542-199607000-00009. [DOI] [PubMed] [Google Scholar]
- Dahan A, van den Elsen MJLJ, Berkenbosch A, Degoede J, Olievier ICW, van Kleef J, Bovill JG. Effects of subanesthetic halothane on the ventilatory responses to hypercapnia and acute hypoxia in healthy volunteers. Anesthesiology. 1994;80:727–738. doi: 10.1097/00000542-199404000-00004. [DOI] [PubMed] [Google Scholar]
- Davies RO, Edwards MWE, Lahiri S. Halothane depresses the response of carotid body chemoreceptors to hypoxia and hypercapnia in the cat. Anesthesiology. 1982;57:153–159. doi: 10.1097/00000542-198209000-00002. [DOI] [PubMed] [Google Scholar]
- de Groot H, Noll T. Halothane hepatotoxicity: relation between metabolic activation, hypoxia, covalent binding, lipid peroxidation and liver cell damage. Hepatology. 1983;3:601–606. doi: 10.1002/hep.1840030421. [DOI] [PubMed] [Google Scholar]
- de Groot H, Sies H. Cytochrome P-450, reductive metabolism, and cell injury. Drug Metabolism Review. 1989;20:275–284. doi: 10.3109/03602538909103543. [DOI] [PubMed] [Google Scholar]
- De La Cruz JP, Zanca A, Carmona JA, de da Cuesta FS. The effect of propofol on oxidative stress in platelets from surgical patients. Anesthesia and Analgesia. 1999;89:1050–1055. doi: 10.1097/00000539-199910000-00043. [DOI] [PubMed] [Google Scholar]
- Frei B, England L, Ames BN. Ascorbate is an outstanding antioxidant in human blood plasma. Proceedings of the National Academy of Sciences of the USA. 1989;86:6377–6381. doi: 10.1073/pnas.86.16.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiological Reviews. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Gregory GA, Eger EI, Munson ES. The relationship between age and halothane requirement in man. Anesthesiology. 1969;30:488–491. doi: 10.1097/00000542-196905000-00003. [DOI] [PubMed] [Google Scholar]
- Hartness ME, Lewis A, Searle GJ, O'Kelly I, Peers C, Kemp PJ. Combined antisense and pharmacological approaches implicate hTASK as an airway O2 sensing K+ channel. Journal of Biological Chemistry. 2001;276:26499–26508. doi: 10.1074/jbc.M010357200. [DOI] [PubMed] [Google Scholar]
- Hatton CJ, Peers C. Effects of cytochrome P-450 inhibitors on ionic currents in isolated rat type I carotid body cells. American Journal of Physiology. 1996;271:C85–92. doi: 10.1152/ajpcell.1996.271.1.C85. [DOI] [PubMed] [Google Scholar]
- Jones RD, Hancock JT, Morice AH. NADPH oxidase: a universal oxygen sensor? Free Radical Biology and Medicine. 2000;29:416–424. doi: 10.1016/s0891-5849(00)00320-8. [DOI] [PubMed] [Google Scholar]
- Kharasch ED, Hankins DC, Fenstamaker K, Cox K. Human halothane metabolism, lipid peroxidation, and cytochromes P(450)2A6 and P(450)3A4. European Journal of Clinical Pharmacology. 2000;55:853–859. doi: 10.1007/s002280050707. [DOI] [PubMed] [Google Scholar]
- Kietzmann T, Fandrey J. Oxygen radicals as messengers in oxygen-dependent gene expression. News in Physiological Sciences. 2000;15:202–208. doi: 10.1152/physiologyonline.2000.15.4.202. [DOI] [PubMed] [Google Scholar]
- Knill RL, Clement JL. Site of selective action of halothane on the peripheral chemoreflex pathway in humans. Anesthesiology. 1984;61:121–126. doi: 10.1097/00000542-198408000-00002. [DOI] [PubMed] [Google Scholar]
- Kobertz WR, Williams C, Miller C. Hanging gondola structure of the T1 domain in a voltage-gated K+ channel. Biochemistry. 2000;39:10347–10352. doi: 10.1021/bi001292j. [DOI] [PubMed] [Google Scholar]
- Koblin DD. Characteristics and implications of desflurane metabolism and toxicity. Anesthesia and Analgesia. 1992;75:S10–16. [PubMed] [Google Scholar]
- Koh SO, Severinghaus JW. Effects of halothane on hypoxic and hypercapnic ventilatory responses of goats. British Journal of Anaesthesia. 1990;65:713–717. doi: 10.1093/bja/65.5.713. [DOI] [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. American Journal of Physiology. 1998;275:C1–24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Kummer W, Acker H. Immunohistochemical demonstration of four subunits of neutrophil NAD(P)H oxidase in type I cells of carotid body. Journal of Applied Physiology. 1995;78:1904–1909. doi: 10.1152/jappl.1995.78.5.1904. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Acker H. Redox-dependent binding of CO to heme protein controls PO2-sensitive chemoreceptor discharge of the rat carotid body. Respiration Physiology. 1999;115:169–177. doi: 10.1016/s0034-5687(99)00014-6. [DOI] [PubMed] [Google Scholar]
- Leach RM, Hill HM, Snetkov VA, Robertson TP, Ward JPT. Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: identity of the hypoxic sensor. Journal of Physiology. 2001;536:211–224. doi: 10.1111/j.1469-7793.2001.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A, Peers C, Ashford MLJ, Kemp PJ. Hypoxia inhibits human recombinant large conductance, Ca2+-activated K+ (maxi-K) channels by a mechanism which is membrane delimited and Ca2+ sensitive. Journal of Physiology. 2002;540:771–780. doi: 10.1113/jphysiol.2001.013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Montoro RJ, Smani T, Garcia-Hirschfeld J, Urena J. K+ and Ca2+ channel activity and cytosolic [Ca2+] in oxygen-sensing tissues. Respiration Physiology. 1999;115:215–227. doi: 10.1016/s0034-5687(99)00016-x. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanisms of oxygen sensing. Annual Review of Physiology. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuijs D, Sarton E, Teppema L, Dahan A. Propofol for monitored anesthesia care: implications on hypoxic control of cardiorespiratory responses. Anesthesiology. 2000;92:46–54. doi: 10.1097/00000542-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuijs D, Sarton E, Teppema LJ, Kruyt E, Olievier I, van Kleef J, Dahan A. Respiratory sites of action of propofol: absence of depression of peripheral chemoreflex loop by low-dose propofol. Anesthesiology. 2001;95:889–895. doi: 10.1097/00000542-200110000-00017. [DOI] [PubMed] [Google Scholar]
- Niki E. Interaction of ascorbate and alpha-tocopherol. Annals of the New York Academy of Sciences. 1987;498:186–199. doi: 10.1111/j.1749-6632.1987.tb23761.x. [DOI] [PubMed] [Google Scholar]
- Odaka Y, Takahashi T, Yamasaki A, Suzuki T, Fujiwara T, Yamada T, Hirakawa M, Fujita H, Ohmori E, Akagi R. Prevention of halothane-induced hepatotoxicity by hemin pretreatment: protective role of heme oxygenase-1 induction. Biochemical Pharmacology. 2000;59:871–880. doi: 10.1016/s0006-2952(99)00386-x. [DOI] [PubMed] [Google Scholar]
- Packer JE, Slater TF, Willson RL. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature. 1979;278:737–738. doi: 10.1038/278737a0. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E. Anesthetic-sensitive 2P domain K+ channels. Anesthesiology. 2001a;95:1013–1021. doi: 10.1097/00000542-200110000-00034. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E. Properties and modulation of mammalian 2P domain K+ channels. Trends in Neurosciences. 2001b;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nature Neuroscience. 1999;2:422–426. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- Peers C. Oxygen-sensitive ion channels. Trends in Pharmacological Sciences. 1997;18:405–408. doi: 10.1016/s0165-6147(97)01120-6. [DOI] [PubMed] [Google Scholar]
- Perez-Garcia MT, Lopez-Lopez JR. Are Kv channels the essence of O2 sensing? Circulation Research. 2000;86:490–491. doi: 10.1161/01.res.86.5.490. [DOI] [PubMed] [Google Scholar]
- Perez-Garcia MT, Lopez-Lopez JR, Gonzalez C. Kvβ1.2 subunit coexpression in HEK293 cells confers O2 sensitivity to kv4.2 but not to Shaker channels. Journal of General Physiology. 1999;113:897–907. doi: 10.1085/jgp.113.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia MT, Lopez-Lopez JR, Riesco AM, Hoppe UC, Marban E, Gonzalez C, Johns DC. Viral gene transfer of dominant-negative Kv4 construct suppresses an O2-sensitive K+ current in chemoreceptor cells. Journal of Neuroscience. 2000;20:5689–5695. doi: 10.1523/JNEUROSCI.20-15-05689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte J, Sadler CL. Effect of halothane, enflurane and isoflurane on carotid body chemoreceptor activity in the rabbit and the cat. British Journal of Anaesthesia. 1989;62:33–40. doi: 10.1093/bja/62.1.33. [DOI] [PubMed] [Google Scholar]
- Riesco-Fagundo AM, Perez-Garcia MT, Gonzalez C, Lopez-Lopez JR. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circulation Research. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- Roy A, Mokashi A, Rozanov C, Daudu PA, Lahiri S. Reduced glutathione, dithiothreitol and cytochrome P-450 inhibitors do not influence hypoxic chemosensory responses in the rat carotid body. Brain Research. 2001;889:131–137. doi: 10.1016/s0006-8993(00)03125-5. [DOI] [PubMed] [Google Scholar]
- Sanz-Alfayate G, Obeso A, Agapito MT, Gonzalez C. Reduced to oxidized glutathione ratios and oxygen sensing in calf and rabbit carotid body chemoreceptor cells. Journal of Physiology. 2001;537:209–220. doi: 10.1111/j.1469-7793.2001.0209k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Fujii K, Yuge O, Tanaka A, Morio M. Suppressive effect of vitamin E on lipid peroxidation in halothane-administered guinea pig liver. In Vivo. 1992;6:503–505. [PubMed] [Google Scholar]
- Semenza GL. Perspectives on oxygen sensing. Cell. 1999;98:281–284. doi: 10.1016/s0092-8674(00)81957-1. [DOI] [PubMed] [Google Scholar]
- Sirois JE, Lei Q, Talley EM, Lynch C, III, Bayliss DA. The TASK-1 two-pore domain K+ channel is a molecular substrate for neuronal effects of inhalation anesthetics. Journal of Neuroscience. 2000;20:6347–6354. doi: 10.1523/JNEUROSCI.20-17-06347.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklin DK, Kharasch ED. Human halothane reduction in vitro by cytochrome P450 2A6 and 3A4: identification of low and high KM isoforms. Drug Metabolism and Disposition. 1998;26:605–607. [PubMed] [Google Scholar]
- Talley EM, Bayliss DA. Modulation of TASK-1 (Kcnk3) and TASK-3 (Kcnk9) potassium channels: volatile anesthetics and neurotransmitters share a molecular site of action. Journal of Biological Chemistry. 2002;277:17733–17742. doi: 10.1074/jbc.M200502200. [DOI] [PubMed] [Google Scholar]
- van den Elsen MJLJ, Dahan A, Berkenbosch A, DeGoede J, van Kleef J, Olievier ICW. Does subanesthetic isoflurane affect the ventilatory response to acute isocapnic hypoxia in healthy volunteers? Anesthesiology. 1994;81:860–867. doi: 10.1097/00000542-199410000-00013. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circulation Research. 2001;88:1259–1266. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]