

Abstract
Puff-application of hypertonic saline (sucrose added to external saline) causes a transient increase in the frequency of spontaneous miniature synaptic currents (mSCs) at the neuromuscular junctions of Drosophila embryos. The frequency gradually returns to pre-application levels. External Ca2+ is not needed for this response, but it may modify it. At 50 mm added sucrose, for example, enhanced spontaneous release was observed only in the presence of external Ca2+, suggesting that Ca2+ augments the response. In a high-K+ solution, in which the basal mSC frequency was elevated, higher sucrose concentrations produced an increase in mSC frequency that was followed (during and after the hypertonic exposure) by depression, with the magnitude of both effects increasing with hypertonicity between 100 and 500 mm. Evoked release by nerve stimulation showed only depression in response to hypertonicity. We do not believe that the depression of spontaneous or evoked release can be explained by the depletion of releasable quanta, however, since the frequency of quantal release did not reach levels compatible with this explanation and the enhancement and depression could be obtained independent of one another. In a mutant lacking neuronal synaptobrevin, only the depression of mSC frequency was induced by hypertonicity. Conversely, only the enhancing effect was observed in wild-type embryos when the mSC frequency was elevated with forskolin in Ca2+-free saline. In cultured embryonic Drosophila neurons, Ca2+ signals that were induced by high K+ and detected by Fura-2, were reduced by hypertonicity, suggesting that the depressing response is due to a direct effect of hypertonicity on Ca2+ influx.
Quantal neurotransmitter release occurs when synaptic vesicles fuse with the presynaptic membrane at specialized release sites. This process is currently the subject of intense investigation in several systems. The embryonic Drosophila neuromuscular junction is uniquely valuable for such investigation because of the existence of a number of mutants with defects in key molecules involved in vesicle docking and/or fusion. Although these are lethal mutations, embryos or larvae are viable and can be studied electrophysiologically (Broadie & Bate, 1993a, b; Broadie et al. 1994; Deitcher et al. 1998; Aravamudan et al. 1999; Yoshihara et al. 1999, 2000). In many of these mutants, nerve stimulation does not evoke synaptic currents, so it is desirable to have other tools with which to trigger vesicle fusion. In this preparation, as in many others, vesicle fusion can be greatly enhanced by application of hypertonic solution in the absence of external Ca2+ (hypertonicity response).
In neuronal synapses, a comparable hypertonicity response can be induced by high concentrations of sucrose, even when the internal Ca2+ concentration ([Ca2+]i) was strongly buffered with BAPTA (Rosenmund & Stevens, 1996; Mochida et al. 1998). On the other hand, it has been shown that cleaving of neuronal synaptobrevin (n-syb), SNAP-25 or syntaxin by clostridial neurotoxins blocks the response to hypertonicity (Capogna et al. 1997). Similarly, at embryonic Drosophila neuromuscular junctions, the hypertonicity response was not evoked or heavily reduced in the absence of n-syb, syntaxin or Unc-13 (Aravamudan et al. 1999). Thus, hypertonicity seems to bypass the Ca2+-sensing step, but shares major elements of the basic Ca2+-triggered vesicle fusion mechanism.
To characterize this Ca2+-independent vesicle fusion process, in a previous study we examined the quantal transmitter release induced by hypertonicity at embryonic Drosophila neuromuscular junctions in the absence of external Ca2+ (Suzuki et al. 2002). The frequency of spontaneous quantal transmitter release increased upon puff-application of hypertonic sucrose solutions. Measurements of the total number of quanta released in response to various concentrations of sucrose showed a dose-response curve that reached a peak at 420 mm sucrose (420 mm sucrose added to Ca2+-free external saline) and was lower at higher osmolarities. Thus, the total number of quantal synaptic events that can be induced by hypertonicity seems to have a ceiling. This ceiling has been interpreted as the size of a readily releasable vesicle pool (Stevens & Tsujimoto, 1995; Rosenmund & Stevens, 1996). However, this interpretation is inconsistent with the observation that forskolin, an activator of adenylyl cyclase, increased and maintained a higher background frequency of spontaneous quantal release in Ca2+-free saline, even after application of 420 mm sucrose. The hypertonicity response to this treatment was as large as in the absence of forskolin, yet had no apparent effect on the subsequent occurrence of spontaneous quantal events. If the pool had been emptied by the 420 mm sucrose, quantal release should have either decreased or ceased transiently after the response. Furthermore, although the dose-response curve apparently hit a ceiling at 420 mm, the response at 600 mm was significantly smaller than at 420 mm, suggesting that at very high concentrations of sucrose another effect of hypertonicity reveals itself and suppresses vesicle release. Thus, the hypothesis that the maximum hypertonicity response represents the size of the readily releasable pool does not fit the data obtained at the embryonic Drosophila neuromuscular junction. Hypertonicity does not seem to deplete the readily releasable pool (Suzuki et al. 2002).
We have proposed an alternative hypothesis: that hypertonicity facilitates vesicle fusion as well as recruitment of vesicles for release, independent of Ca2+. The total number of quantal events that occur during the hypertonicity response may be determined principally by factors such as the extent and time course of enhancement of vesicle fusion by hypertonicity and the recruitment rate of vesicles for release (Suzuki et al. 2002). The second of these factors, the recruitment of vesicles for release, includes the translocation of vesicles to the release site and docking/ priming. In other systems, the steps involved in the recruitment of synaptic vesicles are strongly influenced by Ca2+. In adrenal chromaffin cells, for example, the recovery of secretory responsiveness was accelerated by a moderate increase of internal Ca2+ (von Ruden & Neher, 1993), and at the calyx of Held in the mouse brainstem, Ca2+ accelerates replenishment of the releasable pool (Wang & Kaczmarek, 1998). An acceleration by Ca2+ of vesicle replenishment at the retinal bipolar-cell synapse of goldfish has also been suggested (von Gersdorff & Matthews, 1997). Thus one might expect the hypertonicity response to be sensitive to Ca2+. This prediction seemingly contradicts the previous reports that the hypertonicity response is independent of Ca2+ (Rosenmund & Stevens, 1996; Mochida, et al. 1998).
To resolve this issue we have examined the hypertonicity response at the embryonic Drosophila neuromuscular junction in the presence of external Ca2+. We found that the hypertonicity response induced by a relatively low concentration of sucrose is enhanced by external Ca2+. Unexpectedly, we also observed that hypertonic solutions depressed nerve-evoked neurotransmitter release with a prolonged time course, and that the elevation in frequency of miniature synaptic currents (mSCs) induced by high K+ in the presence of Ca2+ was also depressed by hypertonic solutions. Thus, release triggered by Ca2+ influx at active zones was depressed at the same time that spontaneous quantal release was enhanced. We further found that the Ca2+ signals induced by high K+ and detected by Fura-2 in cultured embryonic Drosophila neurons were reduced by hypertonicity. Taken together, we suggest that the depressing response to hypertonicity is the result of inhibition of Ca2+ influx through voltage-gated Ca2+ channels by hypertonicity.
METHODS
Fly stocks
The following mutants were used in this study: a null allele of myosin heavy chain gene, Mhc1 (Mhc, Mogami et al. 1986, balanced with CyO, [yellow+]) and a double mutant Mhc1 n-sybF33B (Mhc n-syb). n-sybF33B is a null allele of the n-syb gene isolated by Deitcher et al. (1998) and balanced with TM6, [yellow+]. For cell cultures embryos of a wild-type strain, Canton S, was used.
Preparations
Embryos of mutants (17-19 h after fertilization) were prepared for electrophysiological experiments as described previously (Kidokoro & Nishikawa, 1994; Nishikawa & Kidokoro, 1995). The egg case was removed by treatment with 30 % hypochlorous acid for 3 min. After washing in saline we selected homozygous mutant embryos with the aid of a stereomicroscope, based upon the marker of [yellow+] and dissected them in Ca2+-free saline (see below for ionic composition). The dissected preparation was treated with collagenase (1 mg ml−1) for 30 s to 1 min, and electrical recordings of synaptic currents were carried out with the patch-clamp technique in the whole-cell configuration. The membrane potential was always held at −60 mV. The internal solution contained Cs+ (see below for ionic composition), and the junction potential of the electrode filled with the Cs+ internal solution was −5 mV. Thus, the true holding potential was −65 mV.
Hypertonic solutions were prepared by adding sucrose to the external solution and were applied by the puff method. Basically, when 340 mm sucrose was added to external saline, we refer to it as a 340 mm sucrose solution. The puff pipette had a tip diameter of 2–5 μm, and the tip was placed within ≈20 μm of the junctional area. The gas pressure used to puff hypertonic solutions was 0.5 kg cm−2. The quantal synaptic events were counted individually within every 1 s interval.
For nerve stimulation, the tip of a microelectrode, with a resistance of 10–20 MΩ after filling with 4 m potassium acetate was placed in the ventral nerve cord, and rectangular pulses of 2 ms duration and about 2 μA intensity were delivered.
For application of RGD peptides, the preparation was kept for 15 min in a low-Ca2+ (50 μm), low-Mg2+ (50 μm) solution containing the peptide at 0.2 mm. The low divalent solution destabilizes integrin bonds to native ligands, and RGD binds in their place (Gailit & Rouslahti, 1988; Kirchhofer et al. 1991; Chen & Grinnell, 1995, 1997). The recording bath solution and the hypertonic solution in the puff pipette also contained the same concentration of the peptide. To test the effect of forskolin (200 μm) on the hypertonicity response, the preparation was kept for at least 20 min in external saline containing forskolin, and the physiological experiment was carried out in the same medium.
All experiments were carried out at room temperature (18-27 °C).
Solutions
Electrophysiological experiments were carried out either in the Ca2+-containing solution or in Ca2+-free solution. The ionic composition of Ca2+-free saline was (mm): NaCl 140, KCl 2, MgCl2 6, Hepes-NaOH 5 (pH 7.1). For nerve stimulation to evoke synaptic currents, 0.1 mm Ca2+ or 0.3 mm Ca2+ was added to Ca2+-free saline, replacing the same amount of Mg2+. The ionic composition of the high-K+ saline was (mm): NaCl 80, KCl 62, MgCl2 5.9, CaCl2 0.1, Hepes-NaOH 5 (pH 7.1). The internal solution in the patch pipette contained (mm): CsCl 158, EGTA 5, Hepes-NaOH 10, ATP 2 (pH 7.1).
For Ca2+-imaging experiments with cultured embryonic neurons, HL3 medium was used (Stewart et al. 1994). This medium contained the following ingredients (mm): NaCl 70, KCl 5, CaCl2 1.5, MgCl2 20, NaHCO3 10, trehalose 5, sucrose 115, Hepes-NaOH 5 (pH 7.2). A high-K+ HL3 solution was prepared by replacing all NaCl with KCl. Thus, the total K+ concentration was 75 mm. A hypertonic, high-K+ HL3 solution was prepared by adding 420 mm sucrose to the high-K+ HL3 solution.
Cell cultures
Wild-type embryos were collected on agar plates for 1 h after egg laying and incubated for 3.5 h at 25 °C. Embryos were at the early gastrula stage (stage 7-8). To remove the chorionic membrane, embryos were treated with a mixture of antiformin (about 12 % sodium hypochlorite solution) and 90 % ethanol (1:1 by volume) for 1 min and washed with 70 % ethanol. Embryos were subsequently washed with sterile water, and egg shells were ruptured with a pair of forceps in modified Schneider medium supplemented with fetal bovine serum (20 %), insulin (200 ng ml−1), penicillin (50 U ml−1), streptomycin (50 μg ml−1) and cytochalasin B (2 μg ml−1). The treatment with cytochalasin B prevents cell division and produces giant neurons with multiple nuclei (Wu et al. 1990; Saito & Wu, 1991). To remove debris, dissociated cells were washed in the same medium and were plated on 22 mm diameter glass coverslips coated with poly-l-lysine. Cultures were kept in humidified chambers at 25 °C for 5–7 days before experiments.
Fura-2 loading
Free [Ca2+]i was measured using the Ca2+-sensitive dye, Fura-2/AM. Fura-2/AM was stored as a stock solution (1 mm) in dimethylsulphoxide at −20 °C and was diluted in the HL3 solution to 2 μm prior to use. Before Fura-2/AM loading, cells were washed with the HL3 medium three times to remove fetal bovine serum and then incubated in the Fura-2/AM solution for 30 min at 25 °C. After loading, the cells were washed twice with the HL3 medium.
Measurement of [Ca2+]i
Using silicon vacuum grease, a plastic frame was pasted on the cover slip on which cells were growing, to form a 200 μl chamber. The Fura-2 fluorescence at 510 nm elicited by excitation at 340 or 380 nm was measured using an inverted Diaphot microscope (Nikon, Tokyo, Japan) equipped with a cooled CCD camera (Hamamatsu Photonics, Hamamatsu, Japan). The excitation wavelength was switched between 340 and 380 nm by a computer-controlled filter changer. Images were recorded at 5 s intervals and were analysed with the Aquacosmos Ca2+-imaging processing system (Hamamatsu Photonics). The F340/F380 ratio was measured to estimate [Ca2+]i. The area of measurement was approximately 5 μm × 5 μm. The bath was perfused continuously at a rate of 1 ml min−1 during the experiment. Cells were stimulated with a high-K+ solution for 1 min and subsequently washed with the HL3 medium for 5 min. Only 30–50 % of cultured cells responded well with high-K+ stimulation. In those responding cells, high-K+ stimulation was repeated three times with an interval of 5 min. The first stimulation was done with the high-K+ solution alone for control conditions, the second was with the high-K+ solution plus Cd2+ (200 μm) or PLTXII (0.2 μm) or sucrose (420 mm), and the third was again with the high-K+ (control) solution to observe the recovery from the treatment.
Calibration of the Fura-2 signal
An in vitro calibration was performed using Ca2+ solutions of known concentration based on the following equation: [Ca2+]i = Kdβ(R - Rmin)/(Rmax - R), where R is the ratio of fluorescence emission intensities at 340 nm and 380 nm excitations, Rmin is the same ratio under a minimal Ca2+ condition, Rmax is the ratio at a maximal Ca2+ concentration, Kd is the dissociation constant of the Ca2+-Fura-2 complex (Kd = 135 nm at 20 °C) and β is the ratio of fluorescence intensity of the free dye to that of the bound dye measured at a 380 nm excitation wavelength. The value of Rmin was measured using a 200 μl HL3 solution containing zero Ca2+, 10 mm EGTA and 2 μm Fura-2. The value of Rmax was measured using a 200 μl HL3 solution containing 2 mm Ca2+, zero EGTA and 2 μm Fura-2. We used the following values: Rmin = 0.5667, Rmax = 4.0151 and β = 2.915 (Grynkiewicz et al. 1985).
Chemicals
The peptide Gly-Arg-Gly-Asp-Ser-Pro (GRGDSP) was obtained from Peninsula Laboratories (Belmont, CA, USA). Forskolin, tetrodotoxin (TTX) and collagenase were purchased from Sigma (St Louis, MO, USA). For stock solutions, forskolin was dissolved in ethanol at 10 mm. Ethanol does not have any effect on synaptic transmission at the concentration used in this experiment (Yoshihara et al. 2000). A synthetic spider toxin, PLTXII (Body et al. 1995), was purchased from the Peptide Institute (Osaka, Japan). Schneider's Drosophila culture medium and bovine serum were purchased from Gibco (Grand Island, NY, USA), antiformin and streptomycin were from Wako (Osaka, Japan) and Fura-2/AM from Dojindo (Kumamoto, Japan). Penicillin and insulin were purchased from Sigma.
Statistical analyses
For comparison among multiple groups, ANOVA was used with Scheffé‘s test. Student's t test was used for comparison of two groups. All data are expressed as means ± s.d., unless indicated otherwise.
RESULTS
Hypertonicity caused a reduction in nerve-evoked synaptic transmission, while spontaneous quantal transmitter release was enhanced
Nerve stimulation in wild-type embryos caused muscle contraction that often stretched nerves and adversely affected terminals. Under these conditions, asynchronous quantal release was increased, and nerve-evoked synaptic transmission was often enhanced. To avoid this instability, throughout this study we used a non-contracting muscle mutant, Mhc (Mogami et al. 1986). The basic characteristics of synaptic transmission at the neuromuscular junction in Mhc embryos are indistinguishable from those in wild-type embryos (Yoshihara et al. 2000).
When the nerve was stimulated at 1 Hz in saline containing 0.3 mm Ca2+, the time course and amplitude of synaptic currents were variable (Fig. 1A, before). Because of the variability in the time course, the amount of transmitter release was quantified by integrating the synaptic current to determine the total charge transfer. The average charge transfer before puff-application of hypertonic saline was 1.7 ± 1.4 pC stimulus−1 (n = 5). Upon application of the 340 mm sucrose solution, the charge transfer evoked by each stimulus declined immediately, and synaptic transmission was depressed for up to ≈30 s after the end of the puff pulse (Fig. 1A, during; Fig. 1C). This hypertonicity-induced depression of nerve-evoked synaptic transmission could be due to an effect on the postsynaptic receptors. However, the failure rate during the 30 s period after the end of the hypertonic puff pulse was 0.86 ± 0.14 (n = 8), which is significantly higher than the rate before application of the hypertonic solution (0.50 ± 0.20, n = 8, P < 0.01, Fig. 1B). This indicates that the depression is at least partially due to a reduction in transmitter release from the presynaptic terminal.
Figure 1. The effects of hypertonicity on nerve-evoked synaptic currents and spontaneous synaptic currents.
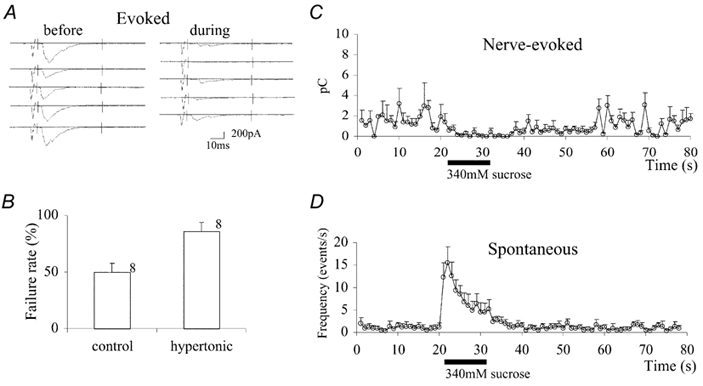
A, embryonic muscle cells from a non-contracting mutant, Mhc, were voltage clamped at −65 mV. Synaptic currents were elicited by stimulation at 1 Hz with a microelectrode placed in the ventral nerve cord. Sample records of nerve-evoked synaptic currents before (left) and during (right) the hypertonicity response are shown. A solution containing 340 mm sucrose (added to the external saline) was puff-applied to the neuromuscular junction while the postsynaptic muscle cell was voltage clamped with a patch electrode in the whole-cell configuration. The external solution contained 0.3 mm Ca2+. B, bar graph showing the failure rate before (left, n = 8) and during (right, n = 8) the hypertonicity response. The vertical bar is the standard error of the mean and numbers are the number of cells examined. C, charge transfer during synaptic currents plotted against time. The synaptic current was integrated between the two vertical bars shown in A and plotted against time. The puff pulse was applied during the time indicated by the horizontal bar below the abscissa. Data from different cells were superimposed by aligning the onsets of the puff pulse. The bars attached to each data point are the standard error of the mean. D, the frequency of spontaneous synaptic events was counted every 1 s and is expressed on the ordinate as the number of events per second (n = 4).
It is also possible that failures of synaptic transmission during the hypertonicity-induced depression were due to failure of presynaptic nerve conduction rather than in synaptic transmission. However, when the charge transfer during successful transmission, excluding failures, was compared before and after application of hypertonicity, the ratio was 0.49 ± 0.20 (n = 5), which is significantly smaller than 1 (P < 0.01). Thus the depression is not likely to be due to failures of nerve conduction, because transmission was less even when nerve conduction occurred. We conclude, therefore, that this depression is due to an effect of hypertonicity on synaptic transmission. That this depression is due to the effect of hypertonicity on the presynaptic terminal is in accord with our previous observation that the mean amplitude of quantal synaptic currents was only slightly smaller during the enhancing hypertonicity response in the absence of external Ca2+, suggesting that the postsynaptic effect of hypertonicity is minimal (Suzuki et al. 2002). The time course of changes in charge transfer during evoked synaptic currents is plotted against time in Fig. 1C. Synaptic depression was more prolonged than enhancement of the frequency of mSCs (see below and Fig. 1D) and started immediately after the onset of the pulse of hypertonic solution.
With 0.3 mm Ca2+ in the external solution, mSCs were observed at a mean frequency of 1.1 ± 1.5 s−1 (n = 5). These were mostly mSCs, as synaptic events with a similar frequency were also observed in the presence of 3 μm TTX. When a 340 mm sucrose solution was puff-applied for 11 s to the synaptic site, the mSC frequency increased to a peak of 17.2 ± 6.8 s−1 (n = 5) and declined quickly during the puff pulse (Fig. 1D). This peak frequency was similar to that observed in the absence of external Ca2+ (20.6 ± 6.7 s−1, n = 5). The mSC frequency returned to the prepuff level within 20 s after termination of the pulse. The total number of events during the period of 31 s after the onset of the puff pulse was 127 ± 73 (n = 5), which was not different from in the absence of external Ca2+ (156 ± 61, n = 5, P > 0.05).
The mSC frequency in a high-K+ solution containing Ca2+ was modulated biphasically by hypertonicity
We have shown here that the depression of charge transfer during nerve-evoked synaptic currents was due to reduced release, while the frequency of mSCs was elevated by hypertonicity. A plausible explanation for this biphasic effect is that the influx of Ca2+ during an action potential is reduced, while the fusion rate is enhanced by a mechanism that is independent of the Ca2+ level. To test this possibility, we next examined the effect of hypertonicity on mSC frequency already elevated in a high-K+ solution containing Ca2+. In a high-K+ saline (62 mm K+) containing 0.1 mm Ca2+ and 3 μm TTX, the mSC frequency was 14.5 ± 7.6 s−1 before application of hypertonic solutions (n = 31). The high frequency of mSCs under this experimental condition is most like the result of elevated internal Ca2+ concentration due to opening of voltage-gated Ca2+ channels by high-K+-induced depolarization. If hypertonicity inhibits Ca2+ influx through voltage-gated Ca2+ channels, the frequency of mSCs would be expected to decrease.
When the 340 mm sucrose solution was puff-applied, the frequency increased transiently to a peak of about 35 s−1 (+20 s−1 above the prepuff level), followed by a prolonged depression, which lasted more than 30 s after the end of the puff pulse (Fig. 2A). At 60 s after the end of the puff pulse, the frequency returned to approximately 60 % of that before the application of hypertonicity (Fig. 2B). The peak mSC rate and the extent of depression (the trough amplitude) increased with the sucrose concentration between 200 and 500 mm (Fig. 3A), but neither of the effects had saturated at 500 mm sucrose. At 50 mm, there was enhancement of mSC frequency without a subsequent depression (Fig. 3B). The enhancing effect of hypertonicity on mSC frequency in the absence of external Ca2+ was virtually absent with 50 mm sucrose (Fig. 3C). This result indicates that with 50 mm sucrose, the enhancing hypertonicity response is strongly augmented by the presence of external Ca2+. At the higher concentrations of sucrose tested, the effect of external Ca2+ on the enhancing hypertonicity response was usually not evident due to the superimposed depressing hypertonicity response.
Figure 2. Biphasic effects of hypertonicity on miniature synaptic currents (mSCs) in high-K+ saline.
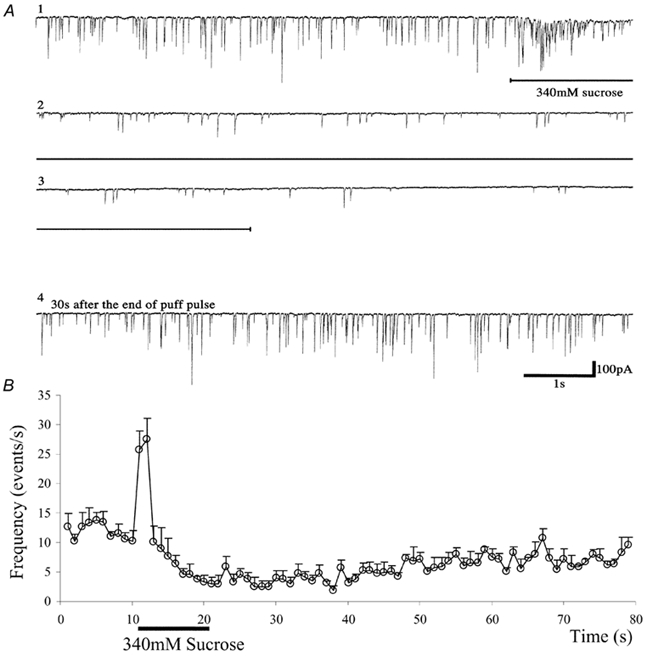
A, current records before and during the hypertonicity response induced by 340 mm sucrose in an abdominal muscle cell in an Mhc embryo. The upper three traces (1-3) are a consecutive record. The fourth trace starts 30 s after the end of the puff pulse, showing the recovery of quantal transmitter release. The hypertonic solution was applied during the horizontal bar shown below the upper three traces. Downward deflections indicate inward currents. The external solution contained 62 mm K+ and 0.1 mm Ca2+. B, the mean frequency of mSCs in five cells is plotted against time. Sucrose (340 mm) was puff-applied at the horizontal bar. Vertical bars attached to each data point are the standard error of the mean. The external solution contained 62 mm K+ and 0.1 mm Ca2+.
Figure 3. Dose-response curves for the enhancing and depressing effects of hypertonicity and the effect of Ca2+ on the enhancing hypertonicity response.
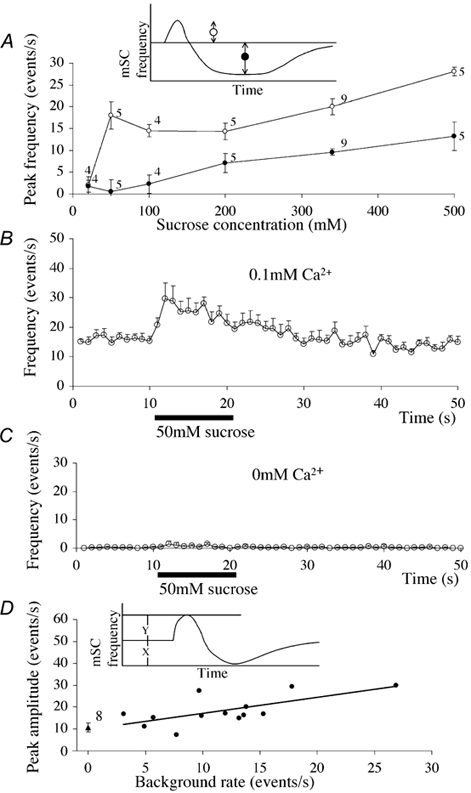
A, dose-response curves for the enhancing and depressing effects induced by hypertonicity. The magnitudes of the enhancing and depressing effects were measured as indicated in the inset, and the average of those parameters are plotted against the sucrose concentration. The trough level was measured by averaging 10 points during the period including the lowest frequency after the puff pulse. Vertical bars attached to each data point are the standard error of the mean and numbers are the number of cells examined. The bath solution contained 62 mm K+, 0.1 mm Ca2+ and 3 μm TTX. B, the enhancing effect at 50 mm sucrose in the presence of Ca2+. The bath solution contained 62 mm K+, 0.1 mm Ca2+ and 3 μm TTX. Sucrose (50 mm) was added to the bath solution. The 50 mm sucrose solution was puffed at the time shown by a horizontal bar. Vertical bars are standard error of the mean. Some bars were smaller than the radius of the symbol (n = 5). C, the enhancing effect at 50 mm sucrose in the absence of external Ca2+. The bath solution contained 62 mm K+ and 3 μm TTX. Sucrose (50 mm) was added to the bath solution. The 50 mm sucrose solution was puffed at the time shown by a horizontal bar. Vertical bars are standard error of the mean. Some bars were smaller than the radius of the symbol (n = 5). D, correlation between the peak amplitude of the enhancing hypertonicity response and the background mSC frequency. These data were obtained following puff-application of a 340 mm sucrose solution. The inset indicates the two parameters measured (i.e. the peak magnitude of the enhancing effect, Y, and the background mSC frequency, X. The regression line has a correlation coefficient of 0.67, which is significant at P < 0.02 (two-tail t test). The triangle at the left is the average of the mSC frequency in Ca2+-free saline. The number is the number of cells examined, and the vertical bar attached is the standard error of the mean.
In high-K+ saline with 0.1 mm Ca2+, the spontaneous mSC frequency was variable, probably reflecting variable [Ca2+]i in the presynaptic terminal. When the peak mSC frequency during the enhancing hypertonicity response (induced with the 340 mm sucrose solution) was plotted against the spontaneous frequency before puff application (background rate), a positive correlation was found (r = 0.67, significant at P < 0.02, Fig. 3D). In high-K+ saline in the absence of Ca2+, the spontaneous mSC frequency was close to zero and the peak rate of the hypertonicity response was smaller than in the presence of Ca2+ (Fig. 3D, the mean is indicated by a triangle). The peak response in the absence of Ca2+ was a mean of 10.8 ± 6.9 s−1 (n = 8), which is significantly smaller than that recorded in the presence of Ca2+ (18.3 ± 6.8 s−1, n = 13) (P < 0.05). These results again suggest that the enhancing effect of hypertonicity is augmented by internal Ca2+.
The mSC frequency in n-syb-null embryos was depressed without enhancement by hypertonicity
In the mutant n-sybF33B embryos, nerve-evoked synaptic currents are completely lacking, while mSCs are observed at lower frequencies than in wild-type embryos. The frequency of mSCs in n-sybF33B was increased when the external Ca2+ concentration was raised in high-K+ saline (Deitcher et al. 1998; Yoshihara et al. 1999). In these mutant embryos, no hypertonicity response was detected in the absence of external Ca2+ (data not shown). Similar results were obtained in the presence of external Ca2+ in tetanus-toxin-expressing transgenic Drosophila embryos, in which there is no functional n-syb (Aravamudan et al. 1999). Thus, n-syb is indispensable for the stimulation of transmitter release by hypertonic solution. If the depressing effect of hypertonicity is independent of the enhancing effect, we might be able to demonstrate the depressing effect in isolation in n-sybF33B embryos.
Consistent with these earlier observations, we found that in double-mutant embryos (Mhc n-syb) the mSC frequency was higher in high-K+ saline (62 mm K+ 0.1 mm Ca2+, 3 μm TTX) than in normal-K+ saline. The mean frequency was 5.0 ± 3.3 s−1 (n = 5) before application of the hypertonic solution, which is about one-third of the frequency reported here in Mhc, in accord with the previous observation (Yoshihara et al. 1999). With puff application of the 420 mm sucrose solution, the mSC frequency was depressed for a prolonged time without an initial enhancement (Fig. 4A). Thus, the depression induced by hypertonicity is not necessarily coupled to a preceding enhancement of release.
Figure 4. The effects of neural synaptobrevin gene (n-syb) mutation (A), forskolin (B) and an RGD peptide (C) on the hypertonicity response.
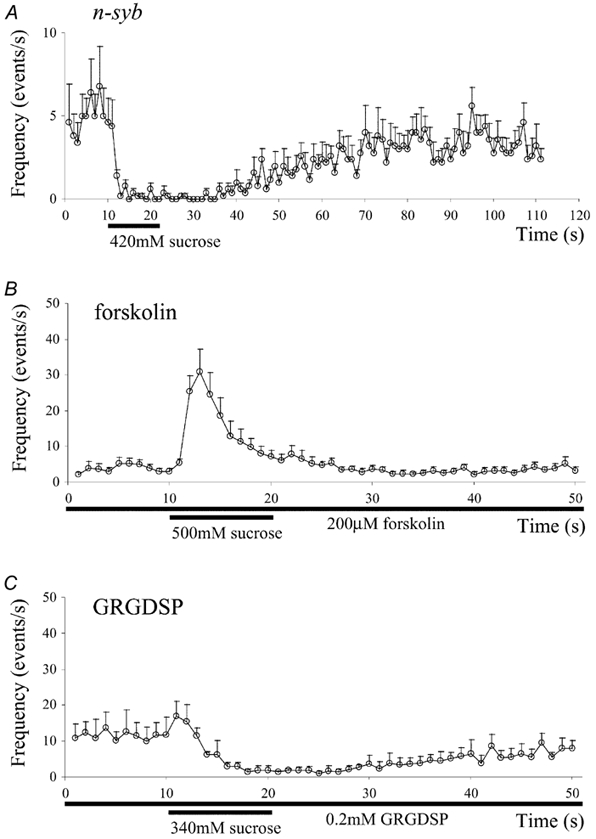
A, the hypertonicity response in Mhc n-sybF33B embryos. The bath solution contained 62 mm K+, 0.1 mm Ca2+ and 3 μm TTX. For the puff solution, 420 mm sucrose was added to the bath solution. These data were obtained in five cells and were superimposed after aligning the onsets of the puff pulse. Vertical bars represent the standard error of the mean. B, the effect of forskolin on the hypertonicity response in Ca2+-free saline. The bath contained 200 μm forskolin, and the 500 mm sucrose solution was puff-applied at the horizontal bar. The vertical bars indicate the standard error of the mean (n = 6). C, the effect of an RGD peptide in high-K+ saline. The experiment was carried out in high-K+ saline (62 mm K+, 0.1 mm Ca2+ and 3 μm TTX) and a 340 mm sucrose solution was puff-applied during the horizontal bar. These data were obtained in five cells and averaged. Vertical bars represent the standard error of the mean (n = 5).
The mSC frequency in the presence of forskolin was enhanced by hypertonicity without depression
The depression of mSC frequency by hypertonicity could be due to an inhibitory effect on Ca2+ influx through voltage-gated Ca2+ channels, as we postulated above. Alternatively, it could be due to a direct effect of hypertonicity on the vesicle fusion mechanism. It has been demonstrated that forskolin, an activator of adenylyl cyclase, increases the mSC frequency in wild-type and Mhc embryos in the absence of external Ca2+ (Yoshihara et al. 1999, 2000; Zhang et al. 1999). If hypertonicity depresses quantal transmitter release by acting directly on the vesicle fusion process, we would expect to observe a depressing effect in the absence of Ca2+ after elevating the mSC frequency by forskolin.
In Ca2+-free saline containing 200 μm forskolin, the mSC frequency in Mhc embryos was 3.9 ± 2.6 s−1 (n = 6). When a 500 mm sucrose solution was puff-applied, the mSC frequency increased to a peak of 38.7 ± 10.4 s−1 (n = 6) and declined to the prepulse level during the puff without exhibiting any depression (Fig. 4B). This result is in accord with the result reported previously in wild-type Drosophila embryos (Suzuki et al. 2002). The absence of a decrease in release suggests that the depression of release observed in high-K+ saline containing Ca2+ is not due to a direct effect of hypertonicity on the vesicle fusion process, but rather to inhibition of voltage-gated Ca2+ channels by hypertonicity.
An RGD peptide did not affect the depressing hypertonicity response, but reduced the enhancement
We have shown previously that over 50 % of the hypertonicity response in the absence of external Ca2+ is blockable with peptides containing the RGD (arginine-glycine-aspartic acid) sequence. Peptides that have this sequence bind to the ligand-binding sites of integrins and block integrin-mediated cell adhesion (Pierschbacher & Ruoslahti, 1987). Since the effects of integrins are mediated by the cAMP-protein kinase A (PKA) cascade (Suzuki et al. 2002) and cAMP facilitates synaptic transmission at Drosophila neuromuscular junctions (Zhang et al. 1999; Yoshihara et al. 2000), it is unlikely that position-specific (PS) integrins are involved in the depressing effect. To confirm this prediction we tested the effect of an RGD peptide on the depressing hypertonicity response.
After dissection, Mhc embryos were treated with 0.2 mm GRGDSP for 20 min in low-Ca2+ and low-Mg2+ saline, and the following experiments were carried out in the presence of the peptide. In high-K+ saline (62 mm K+, 0.1 mm Ca2+, 3 μm TTX), the 340 mm sucrose solution was puff-applied. The mSC frequency increased slightly and then decreased for a prolonged period of time (Fig. 4C). The peak rate (+8.2 ± 2.2 s−1, n = 5) was significantly smaller (P < 0.05) than that observed without the RGD treatment (+17.7 ± 6.0 s−1, n = 13, P < 0.05; Fig. 3A). This effect of the peptide on the enhancing hypertonicity response is in accord with our previous results in the absence of external Ca2+ (Suzuki et al. 2002). In contrast, the depressing effect (-7.8 ± 5.1 s−1, n = 5) was not different from without the peptide treatment (-8.0 ± 3.3 s−1, n = 13), suggesting that PS integrins are not involved in the depressing hypertonicity response. These results also provide further evidence that an increase in transmitter release is not required for the depression of release by hypertonic solutions.
Hypertonicity depresses the elevation of [Ca2+]i induced by a high-K+ solution in cultured embryonic neurons
Voltage-gated Ca2+ currents measured at the cell body are reduced by hypertonicity in cultured hippocampal neurons (Rosenmund & Stevens, 1996) and in anterior pituitary cells (Matzner et al. 1996). Hence, it is possible that the depressing hypertonicity response we observed is due to a reduction of Ca2+ currents in the presynaptic terminal. Since it is technically difficult to measure [Ca2+]i in the presynaptic nerve terminal of Drosophila embryos, we used cultured embryonic neurons to investigate this possibility.
Cultured neurons were loaded with Fura-2 and stimulated with a high-K+ solution (75 mm K+ HL3 solution). In the HL3 medium, the resting [Ca2+]i was 62 ± 10 nm (n = 5) in the neurite and 52 ± 7 nm (n = 8) in the cell body (Fig. 5A, Fig. 6A, leftmost pair of columns). Upon application of the high-K+ solution, [Ca2+]i increased to 453 ± 33 nm (n = 5) in the neurite and 148 ± 9 nm (n = 8) in the cell body (Fig. 5). The difference in the surface-to-volume ratio between the neurite and cell body may be a major factor in the greater elevation of [Ca2+]i in the neurite (Fig. 6A). When application of high K+ saline was repeated three times at intervals of 5 min, the extent of [Ca2+]i elevation declined only slightly (Fig. 6A). The same data are replotted in Fig. 6B as the percentage change compared to the first challenge. The elevation of [Ca2+]i induced by high-K+ saline was completely blocked when 0.2 mm Cd2+ was included in the high-K+ saline (Fig. 6C, middle columns). This blocking effect of Cd2+ was reversible (Fig. 6C, right columns). PLTX II (0.2 μm) also blocked the high-K+-induced elevation of [Ca2+]i, but this blocking effect was irreversible (Fig. 6D). These results are in accord with the previous reports that neuromuscular transmission in Drosophila larvae was irreversibly blocked (Branton et al. 1987), and that voltage-gated inward Ca2+ currents in cultured Drosophila neurons are suppressed by Plectreurys toxin (Leung et al. 1989). Thus, it is most likely that the elevation of [Ca2+]i observed in these experiments is due to Ca2+ influx through voltage-gated Ca2+ channels in the surface membrane.
Figure 5. Ca2+ signals measured with Fura-2 in cultured embryonic Drosophila neurons.
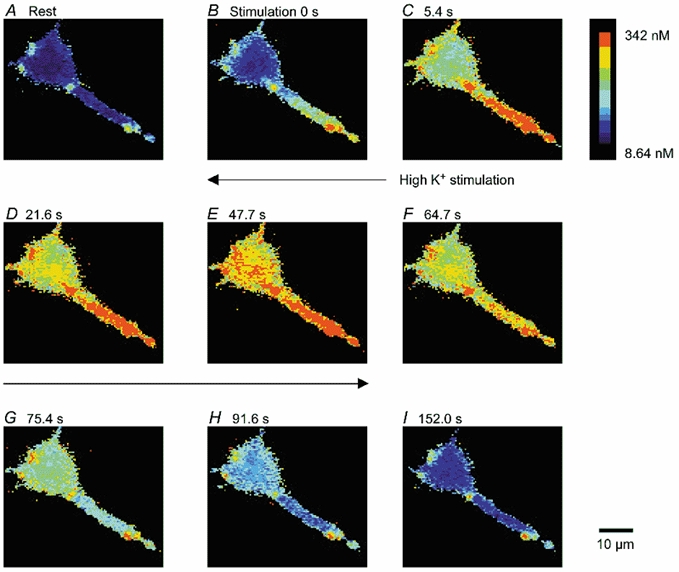
Pseudocolour demonstration of [Ca2+]i before (A), during (B-E) and after (F-I) stimulation with high-K+ saline (75 mm K+ HL3 saline). The cultured cell had been loaded with Fura-2/AM. The [Ca2+]i at rest was 52 ± 8 nm (n = 8) at the cell body and 62 ± 10 nm (n = 5) at the neurite. The [Ca2+]i increased to 148 ± 9 nm at the cell body and 453 ± 33 nm at the neurite during the first high-K+ stimulation, and returned to the resting level during a 5 min perfusion with HL3 saline.
Figure 6. Effects of Cd2+, PLTXII and hypertonicity on the high-K+-induced elevation of [Ca2+]i.
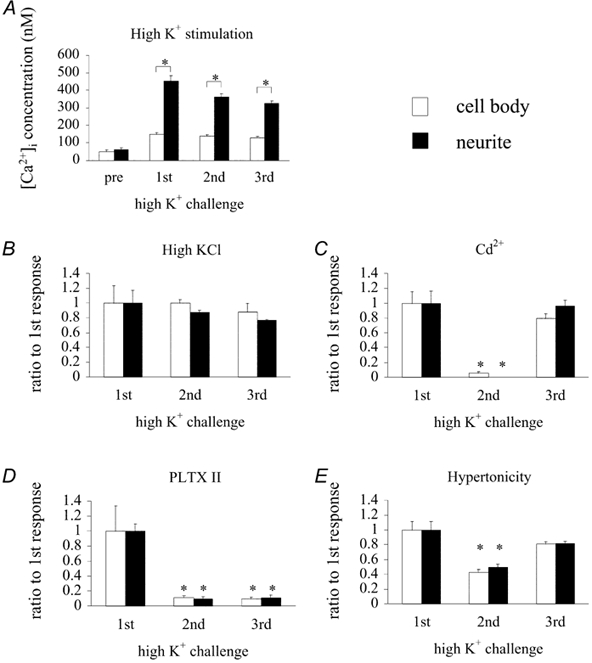
A, control: the cells were stimulated with high-K+ HL3 saline three times for 1 min at intervals of 5 min, during which the HL3 solution was continuously perfused. The ordinate indicates the Ca2+ concentration in nanomolar. Open columns are the data obtained at the cell body and filled columns are those obtained at the neurite. The height of each column represents the mean response in eight cell bodies and in five neurites. Vertical bars at the top of each column are the standard error of the mean. The pair of columns at the left represents the resting state (pre). The three pairs to the right are for three consecutive challenges with high-K+ HL3 saline. *Significant difference between the cell body and neurite (P < 0.001). B, the same data as in A, but the high-K+ responses are expressed as a percentage of the first response. C, the effect of Cd2+; 200 μm Cd2+ was added to the high-K+ solution at the second challenge. No response was detected. *Significant difference (P < 0.01) between the column with the symbol and the corresponding one in B. The response had fully recovered at the third challenge. The number of cells examined at the cell body was six, and that for the neurite was also six. D, the effect of PLTXII. PLTXII (0.2 μm) was added to the high-K+ solution at the second challenge. Recovery of the response was not observed at the third challenge. *Significant difference (P < 0.01) between the column with the symbol and the corresponding one in B. The number of cells examined at the cell body was five, and that at the neurite was also five. E, the effect of hypertonicity. Sucrose (420 mm) was added to the high-K+ solution at the second challenge. *Significant difference (P < 0.01) between the column with the symbol and the corresponding one in B. The number of cells examined at the cell body was 12, and that at the neurite was 10.
We then tested the effect of hypertonicity on Ca2+ influx in the cultured neurons. When 420 mm sucrose was added to the high-K+ saline, the elevation of [Ca2+]i was significantly smaller than that induced by high K+ alone (Fig. 6E). With the first application of high K+ saline, [Ca2+]i increased to 373 ± 33 nm (n = 7) at the neurite and 190 ± 28 nm (n = 7) at the cell body. With the second application of high K+ plus sucrose saline, [Ca2+]i increased to 50 ± 4 % (n = 10) of the first response at the neurite and 43 ± 4 % (n = 12) at the cell body. These values are significantly smaller than the corresponding values shown in Fig. 6B for the second high-K+ challenge (88 ± 3 % at the neurite, n = 5, and 100 ± 5 % at the cell body, n = 8, P < 0.01). At the third stimulation they were 82 ± 13 % at the neurite and 81 ± 3 % at the cell body, values that are not different from the corresponding values shown in Fig. 6B (77 ± 1 % at the neurite and 88 ± 11 % at the cell body; P > 0.05). Thus, the effect of hypertonicity on high-K+-induced Ca2+ influx was reversible.
We conclude that hypertonicity reduces the Ca2+ influx induced by high-K+ stimulation in cultured embryonic Drosophila neurons.
DISCUSSION
Hypertonic solutions increase spontaneous quantal transmitter release at embryonic Drosophila neuromuscular junctions in the absence of external Ca2+ (Suzuki et al. 2002), and we have now shown that this response can be amplified in the presence of external Ca2+ (Fig. 3B). However, with external Ca2+, where it is possible to test effects on evoked release by nerve stimulation, we found that nerve-evoked synaptic currents were depressed by hypertonicity (Fig. 1A and C). This is at least in part a presynaptic depression, since the failure rate also increased during this period (Fig. 1B). The depression of evoked release was seen at the same time the spontaneous release was elevated (Fig. 1D). Hypertonicity had a similar depressing effect on mSC frequency when the baseline frequency was elevated in high K+ (Fig. 2 and Fig. 3). These observations might suggest that the elevated spontaneous release depletes the population of readily releasable vesicles, thus suppressing evoked release. However, we feel that this is not the case, since forskolin increased the background mSC frequency in Ca2+-free saline and augmented the response to hypertonicity without evidence of depression (Fig. 4B). Furthermore, in n-sybF33B mutant embryos there was no enhancement of mSC frequency, but the depression remained (Fig. 4A). Thus, the enhancing and depressing effects of hypertonicity are separable. These observations lead us to conclude that the depressing effect is not coupled to the enhancement, and that these two effects are the result of two separate processes induced by hypertonicity.
The effect of hypertonic solutions on nerve-evoked synaptic currents has been studied in several preparations. Low levels of hypertonicity (up to 100 mosmol) increased evoked release either moderately or not at all (Barton et al. 1983; Tanabe & Kijima, 1988; Kashani et al. 2001), while larger changes in osmolarity decreased evoked release (Thesleff, 1959; Hubbard et al. 1968; Kita & Van der Kloot, 1977). These results are in accord with our findings in the embryonic Drosophila neuromuscular junction. Although the predominant effect of hypertonicity on nerve-evoked release at ≥ 200 mm sucrose was depression, we did observe the enhancement of spontaneous release at a low sucrose concentration (50 mm sucrose; Fig. 3B).
In cultured hippocampal synapses, the amplitude of nerve-evoked synaptic currents was reduced shortly after puff application of hypertonic solutions, and recovered as the interval between the puff pulse and nerve stimulation increased. The evoked current amplitude recovered with a time constant of about 8 s. Based upon the assumption that the application of hypertonicity depleted vesicles from the readily releasable pool, this time constant was interpreted as the rate of refilling of the pool (Rosenmund & Stevens, 1996). However, this interpretation does not seem to be valid, at least in our preparation, for the following reasons. First, the maximum hypertonicity response does not deplete the readily releasable pool (Fig. 4B and see Discussion in Suzuki et al. 2002). Second, the depressing effect of hypertonicity on voltage-gated Ca2+ channels (Fig. 5 and Fig. 6) should reduce the amplitude of nerve-evoked synaptic currents upon application of a hypertonic solution. Third, the depression of quantal release can occur without enhancement of quantal release (Fig. 4A).
We suggest that the depressing effect of hypertonicity on vesicle fusion results from the inhibitory effect of hypertonicity on voltage-gated Ca2+ channels. Depression of synaptic transmission was observed only when voltage-gated Ca2+ channels were activated either with electrical nerve stimulation or with high-K+-induced depolarization, and was not detected when the mSC frequency was elevated with forskolin in Ca2+-free saline. Moreover, the mSC frequency was not depressed in normal saline (Fig. 1D), in which voltage-gated Ca2+ channels were presumably not activated. Furthermore, in cultured embryonic Drosophila neurons, the Ca2+ signals induced by a high K+ solution were inhibited by hypertonicity (Fig. 6E). Our interpretation is in accord with the reports on pituitary cells (Matzner et al. 1996) and in cultured hippocampal neurons (Rosenmund & Stevens, 1996), in which hypertonicity depresses voltage-gated Ca2+ currents. However, in the latter case the inhibition was observed only during application of a hypertonic solution (300 mm sucrose), and the current recovered immediately after the puff (Fig. 5D in Rosenmund & Stevens, 1996). Thus, it is not clear whether the prolonged inhibition of synaptic currents after application of hypertonicity, which Rosenmund & Stevens (1996) observed and interpreted as the refilling time course of the readily releasable pool, can be explained by the mechanism we postulated.
In the study presented here, we found that 50 mm sucrose produced a clear enhancement of mSC frequency only in the presence of external Ca2+, and that the response was greater in cells in which the background mSC frequency was higher, and presumably [Ca2+]i was higher (Fig. 3D). These findings suggest that the hypertonicity-induced increase in mSC frequency is augmented by Ca2+. On the other hand, the enhancement induced by high concentrations of sucrose remained even when the presynaptic neuron was injected with high concentrations of BAPTA, which blocked nerve-induced synaptic currents (Rosenmund & Stevens, 1996; Mochida et al. 1998). Thus, at least a part of the enhancing hypertonicity response is independent of Ca2+.
In all those studies in which BAPTA was used to reduce [Ca2+]i, the hypertonicity response was evoked by use of solutions of very high tonicity (500 mm sucrose was added to normal saline). On the other hand, at rat neuromuscular junctions, Losavio & Muchnik (1997) compared the hypertonicity response induced by a solution with relatively low hypertonicity (200 mosmol above normal saline) before and after application of EGTA-AM or BAPTA-AM, and found that although the hypertonicity response remained, the amplitude of the response was reduced. Similar results were obtained at the frog neuromuscular junction with solutions of 10–20 mosmol above normal saline (Kashani et al. 2001). These results are in accord with our finding that Ca2+ enhances the hypertonicity response induced by a solution with relatively low hypertonicity (Fig. 3).
The hypertonicity-induced enhancement of spontaneous release and the enhancement of nerve-evoked synaptic transmission by mechanical stretch have many properties in common. Both responses remain in the absence of external Ca2+ and even after the internal Ca2+ is strongly buffered to a low level (Chen & Grinnell, 1997; Rosenmund & Stevens, 1996; Mochida et al. 1998), and both are reduced by treatment with RGD peptides (Chen & Grinnell, 1995, 1997; Kashani et al. 2001; Suzuki et al. 2002). Responses to hypertonicity and stretch co-varied in magnitude among different muscle fibres in the frog (Kashani et al. 2001). Thus it seems likely that these responses, at least to some degree, share a common underlying mechanism. Because of the close temporal relationship between stretch and the enhancement of synaptic transmission, the stretch effect is considered to be purely mechanical (Chen & Grinnell, 1997). On the other hand, the elimination of external Ca2+ reduced by half the stretch effect on synaptic transmission, and buffering external Ca2+ with BAPTA further reduced it to one-fifth of that recorded in normal saline (Chen & Grinnell, 1997). These results are in accord with our finding that the enhancing hypertonicity response is augmented by Ca2+.
We have proposed the following hypothesis for the enhancing effect of hypertonicity on release: independent of Ca2+, hypertonicity facilitates vesicle fusion as well as the recruitment of vesicles for release. The magnitude of the hypertonicity response may be determined mainly by factors such as the intensity and time course of the enhanced rate of vesicle fusion induced by hypertonicity and the recruitment rate of vesicles for release (Suzuki et al. 2002). Our observation that external Ca2+ augments the enhancing hypertonicity response (Fig. 3) is in accord with the idea that the recruitment of vesicles to the release sites is facilitated by Ca2+ (Von Ruden & Neher, 1993; Von Gersdorff & Matthews, 1997; Wang & Kaczmarek, 1998).
The hypertonicity response appears to be complex. The enhancing effect of release has at least two components, integrin-mediated and integrin-independent. The integrin-mediated response is coupled to the cAMP/PKA cascade (Suzuki et al. 2002). The depressing hypertonicity response is independent of the enhancing effect and is probably caused by the inhibitory effect of hypertonicity on presynaptic voltage-gated Ca2+ channels. Knowing these properties of the hypertonicity response, we can now use it as a tool to dissect presynaptic events in mutants.
Acknowledgments
We thank Professor Alan D. Grinnell for invaluable comments on the manuscript and for improvement of English. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan to Y.K.
References
- Aravamudan B, Fergestad T, Davis WS, Roedesch CK, Broadie K. Drosophila Unc-13 is essential for synaptic transmission. Nature Neuroscience. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- Barton SB, Cohen IS, Van Der Kloot W. The calcium dependence of spontaneous and evoked quantal release at the frog neuromuscular junction. Journal of Physiology. 1983;337:735–751. doi: 10.1113/jphysiol.1983.sp014652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Body J, Nishino H, Zhou Y, Branton WD, Kimura T, Sakakibara S. Synthesis of an O-palmitoylated 44-residue peptide amide (PLTX II) blocking presynaptic calcium channels in Drosophila. Peptide Research. 1995;8:228–235. [PubMed] [Google Scholar]
- Branton WD, Kolton L, Jan YN, Jan LY. Neurotoxins from Plectreurys spider venom are potent presynaptic blockers in Drosophila. Journal of Neuroscience. 1987;7:4195–4200. doi: 10.1523/JNEUROSCI.07-12-04195.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K, Bate M. Innervation directs receptor synthesis and localization in Drosophila embryo synaptogenesis. Nature. 1993a;361:350–353. doi: 10.1038/361350a0. [DOI] [PubMed] [Google Scholar]
- Broadie K, Bate M. Activity-dependent development of the neuromuscular synapse during Drosophila embryogenesis. Neuron. 1993b;11:607–619. doi: 10.1016/0896-6273(93)90073-z. [DOI] [PubMed] [Google Scholar]
- Broadie K, Bellen H, Diantonio A, Littleton JT, Schwarz TL. Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proceedings of National Academy of Sciences of the USA. 1994;91:10727–10731. doi: 10.1073/pnas.91.22.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, McKinney RA, O'Connor V, GÄhwiler BH, Thompson SM. Ca2+ or Sr2+ partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. Journal of Neuroscience. 1997;17:7190–7202. doi: 10.1523/JNEUROSCI.17-19-07190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B-M, Grinnell AD. Integrins and modulation of transmitter release from motor nerve terminals by stretch. Science. 1995;269:1578–1580. doi: 10.1126/science.7667637. [DOI] [PubMed] [Google Scholar]
- Chen B-M, Grinnell AD. Kinetics, Ca2+ dependence, and biophysical properties of integrin-mediated mechanical modulation of transmitter release from frog motor nerve terminals. Journal of Neuroscience. 1997;17:904–916. doi: 10.1523/JNEUROSCI.17-03-00904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. Journal of Neuroscience. 1998;15:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailit J, Ruoslahti E. Regulation of the fibronectin receptor affinity by divalent cations. Journal of Biological Chemistry. 1988;263:12927–12932. [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hubbard JI, Jones SF, Landau EM. An examination of the effects of osmotic pressure changes upon transmitter release from mammalian motor nerve terminals. Journal of Physiology. 1968;197:639–657. doi: 10.1113/jphysiol.1968.sp008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashani A, Chen B-M, Grinnell AD. Hypertonic enhancement of transmitter release from frog motor nerve terminals: Ca++ independence and role of integrins. Journal of Physiology. 2001;530:243–252. doi: 10.1111/j.1469-7793.2001.0243l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidokoro Y, Nishikawa K. Miniature endplate currents at the newly formed neuromuscular junction in Drosophila embryos and larvae. Neuroscience Research. 1994;19:143–154. doi: 10.1016/0168-0102(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Kirchhofer D, Grzesiak J, Pierschbacher MD. Calcium as a potential physiological regulator of integrin-mediated cell adhesion. Journal of Biological Chemistry. 1991;266:4471–4477. [PubMed] [Google Scholar]
- Kita H, Van Der Kloot W. Time course and magnitude of effects of changes in tonicity on acetylcholine release at frog neuromuscular junction. Journal of Neurophysiology. 1977;40:212–224. doi: 10.1152/jn.1977.40.2.212. [DOI] [PubMed] [Google Scholar]
- Leung H-T, Branton WD, Phillips HS, Jan L, Byerly L. Spider toxins selectively block calcium currents in Drosophila. Neuron. 1989;3:767–772. doi: 10.1016/0896-6273(89)90245-6. [DOI] [PubMed] [Google Scholar]
- Losavio A, Muchnik S. Spontaneous acetylcholine release in mammalian neuromuscular junctions. American Journal of Physiology. 1997;273:C1835–1841. doi: 10.1152/ajpcell.1997.273.6.C1835. [DOI] [PubMed] [Google Scholar]
- Matzner O, Bentabou S, Nussinomitch I. Hyperosmotic regulation of voltage-gated calcium currents in rat anterior pituitary cells. Journal of Neurophysiology. 1996;75:1894–1900. doi: 10.1152/jn.1996.75.5.1894. [DOI] [PubMed] [Google Scholar]
- Mochida S, Yokoyama CT, Kim DK, Itoh K, Catterall WA. Evidence for a voltage-dependent enhancement of neurotransmitter release mediated via the synaptic protein interaction site of N-type Ca2+ channels. Proceedings of the National Academy of Sciences of the USA. 1998;95:14523–14528. doi: 10.1073/pnas.95.24.14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami K, O'Donnell PT, Bernstein SI, Wright TR, Emerson D P., Jr Mutations of the Drosophila myosin heavy-chain gene: Effects on transcription, myosin accumulation, and muscle function. Proceedings of the National Academy of Sciences of the USA. 1986;83:1393–1397. doi: 10.1073/pnas.83.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Kidokoro Y. Junctional and extrajunctional glutamate receptor channels in Drosophila embryos and larvae. Journal of Neuroscience. 1995;15:7905–7915. doi: 10.1523/JNEUROSCI.15-12-07905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierschbacher MD, Ruoslahti E. Influence of stereochemistry of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. Journal of Cell Biology. 1987;262:17294–17298. [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Saito M, Wu C-F. Expression of ion channels and mutational effects in giant Drosophila neurons differentiated from cell division-arrested embryonic neuroblasts. Journal of Neuroscience. 1991;11:2135–2150. doi: 10.1523/JNEUROSCI.11-07-02135.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proceedings of the National Academy of Sciences of the USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu C-F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. Journal of Comparative Physiology. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Grinnell AD, Kidokoro Y. Hypertonicity-induced transmitter release at Drosophila neuromuscular junctions is partly mediated by integrins and cAMP/protein kinase A. Journal of Physiology. 2002;538:103–119. doi: 10.1113/jphysiol.2001.012901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe N, Kijima H. Transmitter release at frog endplate loaded with a Ca2+ chelator, BAPTA: hypertonicity and erythrosin B augment the release independently of internal Ca2+ Neuroscience Letters. 1988;92:52–57. doi: 10.1016/0304-3940(88)90741-0. [DOI] [PubMed] [Google Scholar]
- Thesleff S. Motor end-plate ‘desensitization’ by repetitive nerve stimuli. Journal of Physiology. 1959;148:659–664. doi: 10.1113/jphysiol.1959.sp006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. Journal of Neuroscience. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Ruden L, Neher E. A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science. 1993;262:1061–1065. doi: 10.1126/science.8235626. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Ueda A, Zhang D, Deitcher DL, Schwarz TL, Kidokoro Y. Selective effects of neuronal-synaptobrevin mutations on transmitter release evoked by sustained versus transient Ca2+ increases and by cAMP. Journal of Neuroscience. 1999;19:2432–2441. doi: 10.1523/JNEUROSCI.19-07-02432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Suzuki K, Kidokoro Y. Two independent pathways mediated by cAMP and protein kinase A enhance spontaneous transmitter release at Drosophila neuromuscular junctions. Journal of Neuroscience. 2000;20:8315–8322. doi: 10.1523/JNEUROSCI.20-22-08315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-Y, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wu C-F, Sakai K, Saito M, Hotta Y. Giant Drosophila neurons differentiated from cytokinesis-arrested embryonic neuroblasts. Journal of Neurobiology. 1990;21:499–507. doi: 10.1002/neu.480210310. [DOI] [PubMed] [Google Scholar]
- Zhang D, Kuromi H, Kidokoro Y. Activation of metabotropic glutamate receptors enhances synaptic transmission at the Drosophila neuromuscular junction. Neuropharmacology. 1999;38:645–657. doi: 10.1016/s0028-3908(98)00232-9. [DOI] [PubMed] [Google Scholar]