

Abstract
GABAergic modulation of retinal bipolar cells plays a crucial role in early visual processing. It helps to form centre-surround receptive fields which filter the visual signal spatially at the bipolar cell dendrites in the outer retina, and it produces temporal filtering at the bipolar cell synaptic terminals in the inner retina. The observed chloride transporter distribution in ON bipolar cells has been predicted to produce an intracellular chloride concentration, [Cl−]i, that is significantly higher in the dendrites than in the synaptic terminals. This would allow dendritic GABA-gated Cl− channels to generate the depolarization needed for forming the lateral inhibitory surround of the cell's receptive field, while synaptic terminal GABA-gated Cl− channels generate the hyperpolarization needed for temporal shaping of the light response. In contrast to this idea, we show here that in ON bipolar cells [Cl−]i is only slightly higher in the dendrites than in the synaptic terminals, and that GABA-gated channels in the dendrites may generate a hyperpolarization rather than a depolarization. We also show that [Cl−]i is controlled by movement of Cl− through ion channels in addition to transporters, that changes of [K+]o alter [Cl−]i and that voltage-dependent equilibration of [Cl−]i in bipolar cells will produce a time-dependent adaptation of GABAergic modulation with a time constant of 8 s after illumination-evoked changes of membrane potential. Time-dependent adaptation of [Cl−]i to voltage changes in retinal bipolar cells may add a previously unsuspected layer of temporal processing to signals as they pass through the retina.
Two classes of retinal bipolar cell, the ON and OFF cells, are depolarized and hyperpolarized respectively when photoreceptors in the centre of their receptive field absorb light. Bipolar cells also receive lateral inhibition at their dendrites; light falling in the bipolar receptive field surround hyperpolarizes horizontal cells, which decreases release of their transmitter GABA (see Fig. 1C) and thus hyperpolarizes ON bipolar cells and depolarizes OFF bipolar cells. GABAergic lateral inhibition plays an important role in early visual processing by removing low spatial frequencies from the visual signal, thereby enhancing the detection of edges and spots (Kuffler, 1953; Marr & Hildreth, 1980; Roska et al. 2000). It is mediated in part by a feedback synapse from horizontal cells to photoreceptors, and in part by a feedforward synapse from horizontal cells to bipolar cells (Kondo & Toyoda, 1983; Yang & Wu, 1991; Vardi & Sterling, 1994). In addition, both ON and OFF bipolars are inhibited at their synaptic terminals by GABA released from amacrine cells (see Fig. 1C). This shapes the bipolar cell light response, making depolarizing responses more transient, with temporal tuning characteristics that depend on the mix of GABAA and GABAC receptors expressed, and broadens the range of light intensity over which the cells respond (Kondo & Toyoda, 1983; Tachibana & Kaneko, 1987; Dong & Werblin, 1998; Euler & Wässle, 1998; Lukasiewicz & Shields, 1998; Hartveit, 1999; Euler & Masland, 2000; Roska et al. 2000; Shields et al. 2000).
Figure 1. Morphology, GABAergic inputs and putative Cl− transporters of ON bipolar cells.
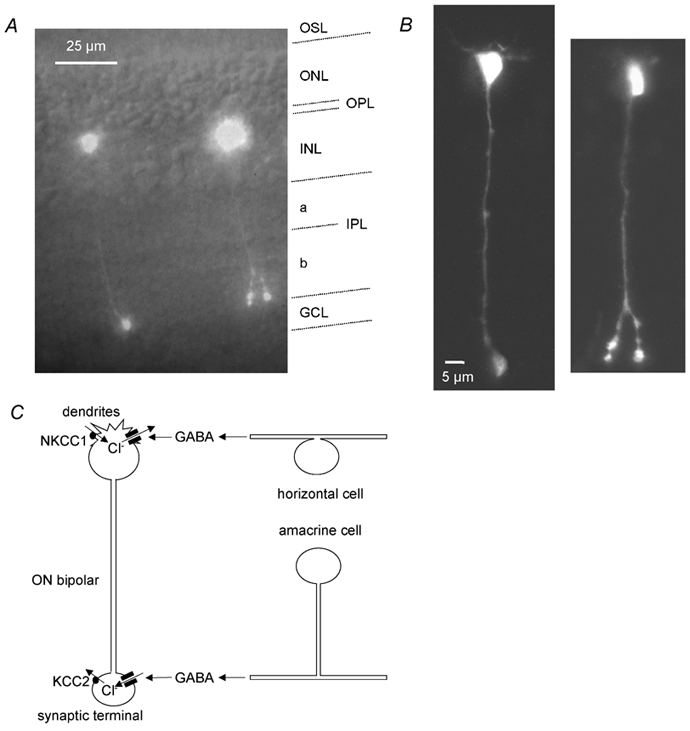
A, fluorescence image of ON bipolars in a retinal slice filled with Lucifer yellow via the whole-cell patch pipette (taken with a CCD camera), with a transmitted light image superimposed to show the retinal layers (labelled at the right-hand side: OSL, photoreceptor outer segment layer; ONL, photoreceptor outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer, IPL, inner plexiform layer divided into the OFF and ON sublaminae a and b, respectively; GCL, ganglion cell layer). Rod ON bipolar cells have their soma in the outermost part of the INL, their dendrites branch in the OPL, and their axon spans the entire IPL before terminating as a large, bulbous axon terminal in the inner part of the IPL (sublamina b), while type 8 and 9 cone ON bipolars have multiple axon terminals less close to the ganglion cell bodies. The cell on the left is a rod ON bipolar cell, whereas the cell on the right could be either a rod bipolar cell or a type 8 cone ON bipolar cell, based on the length of its axon and terminals below the axonal branch point. It is less likely to be a type 9 cone ON bipolar cell because of its restricted dendritic arborization. B, the same cells imaged with a confocal microscope. C, schematic diagram of the GABAergic inputs to ON bipolar cells and the distribution of Cl− transporters in these cells (see Introduction). The dendrites in the OPL receive GABAergic input from horizontal cells, and contain NKCC1 transporters in their membranes, which normally accumulate Cl− and shift ECl positive to the membrane potential, so GABA evokes a Cl− efflux and an inward current. The synaptic terminals in the IPL receive GABAergic input from amacrine cells, and express KCC2 transporters, which normally extrude Cl− and shift ECl negative to the membrane potential, so GABA evokes a Cl− influx and an outward current.
For a feedforward signal at the bipolar cell dendrites to generate an antagonistic surround when GABA release from horizontal cells is decreased, the reversal potential of GABA-gated Cl− channels in the dendrites must be more positive than the resting potential in ON cells and more negative in OFF cells. However, for GABA released from amacrine cells to hyperpolarize bipolar cells, the reversal potential of GABA-gated Cl− channels in the bipolar cell synaptic terminals must be more negative than the resting potential. Thus, for ON bipolar cells, there should be a gradient of intracellular chloride concentration, [Cl−]i, along the cell, being higher in the dendrites and lower in the synaptic terminals. Two transporters contributing to the regulation of [Cl−]i in neurons are a Na+-K+-2Cl− co-transporter (NKCC1) which normally accumulates chloride, and a K+-Cl− co-transporter (KCC2) that normally extrudes Cl− (Delpire, 2000). NKCC1 is expressed in the dendrites of both rod and cone ON bipolar cells, where [Cl−]i is expected to be high, while KCC2 is expressed in OFF bipolar dendrites and in ON and OFF synaptic terminals, where [Cl−]i is expected to be low (Vardi et al. 2000; Vu et al. 2000).
To test the functional significance of this apparent congruence between transporter location and the [Cl−]i needed for GABAergic modulation of bipolar cells, we examined how [Cl−]i varies along the ON bipolar cell, using the response to local application of GABA to measure the value of [Cl−]i. The aim was to determine whether [Cl−]i is indeed significantly higher in the dendrites than in the synaptic terminals of ON bipolar cells. In addition we investigated how [Cl−]i is affected by the extracellular potassium concentration which is known to change during illumination (Steinberg et al. 1980), and by the membrane potential of the bipolar cell, in order to gain a more complete understanding of the mechanisms regulating [Cl−]i in ON bipolar cells.
Methods
Retinal slices
Slices of retina from adult (P35) Sprague-Dawley rats (bred at UCL) were prepared as described by Werblin (1978) for salamander retina, after killing the rat by cervical dislocation according to UK animal use legislation (UK Animals (Scientific Procedures) Act 1986, Schedule 1). Retinal pieces 2 mm square were laid on Millipore filter paper, ganglion cell side down, the sclera was removed and the tissue was covered with normal extracellular solution. The retina was then cut into 200 μm thick slices, each still attached to a strip of Millipore filter paper, with a hand-operated razor blade. The slices were rotated through 90 deg and the attached Millipore filter paper was embedded in lines of Vaseline to hold the slice so that all cell types were visible for perforated patch-clamping using an upright fixed-stage microscope. Experiments were carried out in the light.
Cell identification
Cells were selected that had a soma in the inner nuclear layer closely apposed to the outer plexiform layer (OPL), with dendrites in the OPL and an axon heading down to the inner plexiform layer (IPL). Further identification was provided by checking that the cells responded when GABA was puffed onto the most proximal part of the ON sublamina of the IPL. After studying the response to GABA using perforated patching (see below), approximately 50 % of the bipolar cells from which recordings were made survived long enough to allow further confirmation of their identity by going to whole-cell mode, and allowing Lucifer yellow in the pipette to enter the cell. Cells filled with dye in this way proved to be ON bipolar cells, as described in the Results (Fig. 1A, B). These were rod bipolar cells or the morphologically similar (but less numerous) type 8 or 9 cone bipolars (Euler & Wässle, 1995; Hartveit, 1997), which we are not certain we can reliably distinguish in this kind of experiment (see Results). Experiments were at room temperature (21-25 °C); previous experiments have shown that NKCC1 and KCC2 can raise or lower the chloride reversal potential (ECl) above or below the resting membrane potential even at room temperature (Ehrlich et al. 1999; Kakazu et al. 1999; Ganguly et al. 2001; Jang et al. 2001).
Solutions
Normal extracellular solution contained (mm): NaCl 135, KCl 2.5, CaCl2 1, MgCl2 3, Hepes 10, glucose 10, NaH2PO4 1, pH set to 7.4 with NaOH, bubbled with O2. When recording responses to GABA, synaptic transmission mediated by calcium-dependent exocytosis was blocked by replacing CaCl2 with MgCl2, adding 5 mm sodium EGTA to chelate trace calcium, and adding 20 μm 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo(f)quinoxaline-7-sulphonamide (NBQX) and 50 μmd-aminophosphonovalerate (d-AP5) to block ionotropic glutamate receptors (aminophosphonobutyrate was not used to block the mGluR6 receptors on ON bipolar cells (Neal et al. 1981; Tian & Slaughter, 1994) because, possibly due to glycine contamination, it has been reported to activate a Cl− flux through glycine-gated channels in isolated retinal ganglion cells (Chiba & Saito, 1994) and this would distort our [Cl−]i measurements if it also occurred in bipolar cells). Intracellular solution contained (mm): KCl 135, CaCl2 0.5, Na2 EGTA 5, Hepes 10, MgCl2 2, Mg ATP 2, pH set to 7.2 with KOH, Lucifer yellow (di-potassium salt) 2 g l−1, and gramicidin D (64 μg ml−1 with 0.8 % DMSO final concentration, from an 8 mg ml−1 stock in DMSO). Although the use of Hepes rather than CO2/HCO3− as a buffer can alter the properties of retinal neurons, possibly because intracellular pH is not controlled (Hanitzsch & Küppers, 2001), using Hepes as the buffer in these solutions meant that we could convert measured reversal potentials directly to [Cl−]i values, without the complication of correcting for the permeability of GABA-gated channels to HCO3−, as would be necessary if CO2/HCO3− were used as the buffer (Bormann et al. 1987). In addition, the absence of HCO3− should maximize control of [Cl−]i by NKCC1 and KCC2 by eliminating any changes of [Cl−]i mediated by Cl−-HCO3− exchange, and so provide the best chance of detecting the ability of the non-uniform distribution of NKCC1 and KCC2 to generate a [Cl−]i gradient. The possible effect of a bicarbonate permeability is considered in the Results.
Electrophysiology
Perforated patch-clamping was used, to prevent alteration of the intracellular chloride concentration by the pipette solution. After forming a seal on the cell membrane, we waited ≈30 min for the gramicidin to make sufficient cation selective pores (Ebihara et al. 1995; Akaike, 1996) in the membrane to lower the series resistance to below 100 MΩ (range 30–100 MΩ, generally around 50 MΩ), which will result in series resistance voltage errors of <5 mV for typical currents of <50 pA; for each cell this residual voltage error was calculated as described in the main text, and was subtracted from the apparent reversal potential of the GABA-evoked current to obtain the true reversal potential. The junction potential between the electrode and the external solution before a seal was made on a cell was measured as described by Fenwick et al. (1982) as −2 mV. All membrane potentials were corrected for this, assuming that after cations had equilibrated across the pores formed by gramicidin in the membrane there was no junction potential between the electrode and the inside of the cell (any junction potential present across the perforated patch would alter the absolute values of ECl calculated, but not the difference in ECl values at the two ends of the cell, which is important for our conclusions). A total of around 100 cells were recorded from, but data from three-quarters of these had to be discarded, most often because the perforated patch ruptured during the experiment (allowing the pipette solution to enter the cell and alter [Cl−]i), leaving 24 cells which provided high quality data.
Determination of reversal potential and conductance of GABA-evoked current
GABA was applied by puffing a 100 μm GABA solution (containing blockers of synaptic transmission, as did the superfusion solution, see above) from a pipette (resistance ≈10 MΩ, tip diameter ≈1 μm) resting gently on the surface of the slice (perhaps 10–20 μm above the position of the cell dendrites or synaptic terminals within the slice), using a PicoSpritzer unit (PMI-100, Dagan Corporation, MN, USA), which applied a pressure of 70 kPa (10 p.s.i.) to the top of the pipette. External solution was pumped by a peristaltic pump at a constant rate of about 4 ml min−1 through the bath of 1 ml volume, and the flow was oriented across the retinal slice (parallel to the plexiform layers; Fig. 2A) so that GABA puffed onto the OPL would not be swept over the IPL, and vice versa (dye included in the pipette solution in early experiments confirmed this). The puffer pipette position was adjusted to maximize the response either at the dendrites (selected by placing the pipette over the OPL near the soma) or at the synaptic terminal (selected by placing the pipette over the most proximal part of the IPL next to the ganglion cell layer (GCL), assuming that the axon ran ‘vertically’ through the retina from the soma). The puffer pipette was found to be less than 5 μm from the ‘vertical’ axis of the cell when cells were filled with Lucifer yellow at the end of the experiment. GABA was applied with pressure puffs only 100 ms long, which produced a response with a 10–90 % rise-time of 79 ± 19 ms (n = 5), to restrict the time of ejection and thus minimize the diffusion of GABA laterally (from one plexiform layer to the other). During a time t =100 ms, for a diffusion coefficient of D = 7.6 × 10−10 m2 s−1 (i.e. the value for glutamine; Longsworth, 1953), GABA is expected to diffuse a distance x≈(2Dt)1/2 = 12 μm. Proof of the spatially restricted flow pattern was obtained from the observation that in some cells no response was obtained when GABA was puffed onto the IPL (because the axon and synaptic terminals had been amputated in the slicing procedure), despite the occurrence of a robust response when GABA was puffed onto the OPL; thus GABA does not diffuse laterally sufficiently to activate receptors at one plexiform layer when it is applied at the other plexiform layer. To determine the reversal potential of GABA-evoked currents, voltage ramps (from +8 to −92 mV over 200 ms, from a holding potential of −42 mV) were applied. Because the voltage ramp might activate time-dependent currents, for all measurements a control voltage ramp was first applied, an interval of 40 s was allowed for reversal of any current activation, and then GABA was puffed and at the end of the puff (around the peak of the GABA response) another voltage ramp was applied. Finally, after another 40 s, another control voltage ramp was applied to check for reversibility of the GABA-evoked current; the current response to this ramp normally superimposed on the response to the first control ramp, as shown for the two grey traces in Fig. 3A. The duration of the ramp was not negligible compared to the time course of GABA conductance activation (see Fig. 2), so the magnitude of the GABA conductance was varying (by about 35 %) during the ramp, but as the main parameter of interest was the voltage at which there was no GABA-evoked current (the reversal potential) and we were not interested in the exact shape of the I-V relation of the GABA-evoked current (see below), this was of no consequence to our results. For the speed of voltage ramp used, the current response includes a capacity current, CdV/dt, of <2 pA which will be the same in the presence and absence of GABA and will not affect the reversal potential derived for the GABA-evoked current. The relative conductances of the GABA-evoked currents at the OPL and IPL were calculated from the slope of the I-V relation of the GABA-evoked current (corrected for series resistance voltage errors as described in the main text) at a time in the voltage ramp when the voltage was approximately −40 mV, i.e. close to the reversal potential of the current for a holding potential of −42 mV (Fig. 4B). At this time (100 ms after the end of the GABA puff) the GABA-evoked conductance was about half of its peak value at both the OPL and IPL (see Fig. 2A). Although the conductance gGABA(t) activated by GABA is time dependent, which contributes to the apparent change of current with voltage, this does not affect the derived conductance. If the current is represented as:
where V is membrane potential and Vrev is the reversal potential, then the derived conductance is:
and when V = Vrev the second term is zero, so that dI/dV = gGABA.
Figure 2. Response of ON bipolar cells to GABA application at the dendrites and synaptic terminal.
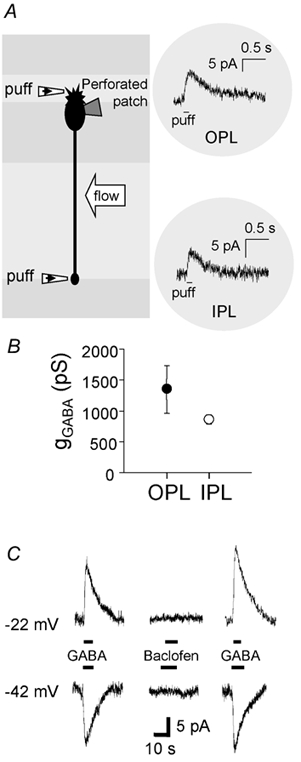
A, schematic diagram of the experiment. GABA (100 μm) was pressure-applied to the dendrites or the axon terminal via a small patch pipette (‘puff’). The flow of the bath solution was oriented parallel to the retinal layers to avoid GABA puffed at one plexiform layer being swept to the other plexiform layer. Insets on the right show specimen current responses at −42 mV to GABA puffs at the OPL or IPL. In this cell the reversal potential of the response was negative to −42 mV at both the OPL and the IPL. B, relative magnitude of the conductance evoked by a GABA puff (gGABA) at the dendrites (OPL) and the axon terminal (IPL) of ON bipolar cells (measurements made at both ends of seven cells). The conductance was obtained as described in the Methods, at a time when it had decreased to approximately half its peak value; peak values would be approximately twice the values plotted here. C, specimen traces of currents evoked by bath application of 30 μm GABA and 100 μm baclofen. The ON bipolar cell was voltage clamped at −22 mV (upper panel) or −42 mV (lower panel), bracketing the reversal potential of the GABA response. Baclofen did not evoke a significant current at either potential.
Figure 3. ECl is more negative in the axon terminal than in the dendrites of ON bipolar cells.
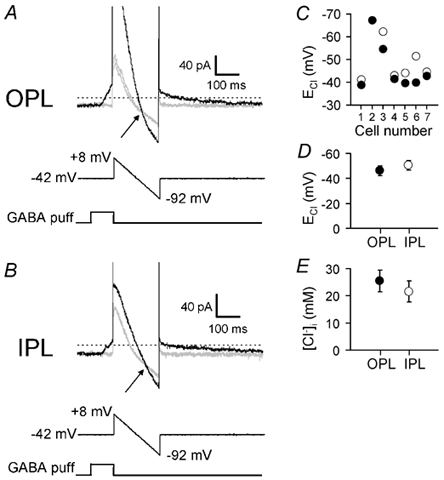
A, top records: specimen current response of an ON bipolar cell to a voltage ramp from +8 to −92 mV (middle trace, 200 ms duration) applied from a holding potential of −42 mV in control solution (two grey traces, obtained 40 s before and 40 s after the GABA puff) or at the end of a 100 ms puff of 100 μm GABA (black trace) to the OPL. Reversal potential of the GABA-evoked current is at the voltage where the current in the control solution and that in GABA cross (arrow). Current traces have been clipped at positive voltages to show data near the reversal potential at a higher gain. B, as for A (same cell), but applying GABA to the IPL. C, values of chloride reversal potential (ECl) at the OPL (•) and IPL (○) obtained from data as in A and B in seven cells. For cell no. 2 the value was the same at the OPL and IPL. D, mean values of ECl from C. E, mean internal chloride concentration ([Cl−]i) at the OPL and IPL calculated from the Nernst equation using ECl measurements as in C.
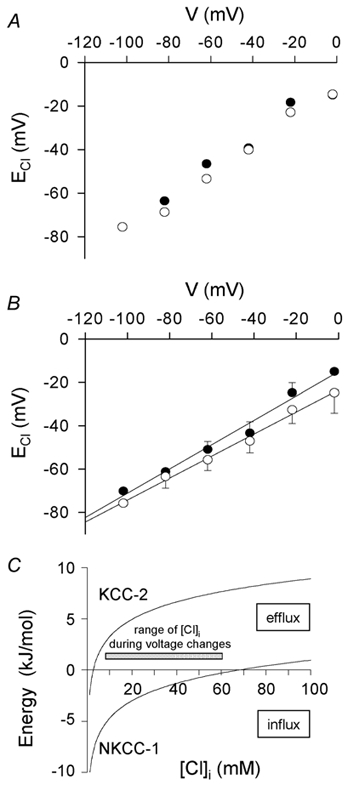
Figure 4. Adaptation of ECl to the membrane potential, and direction of operation of transporters modulating that adaption.
Voltage-clamp quality
Voltage errors caused by the electrode series resistance were compensated for as described above and in the main text. To estimate voltage non-uniformity along the bipolar cell axon, we assumed that the typical input slope resistance of 1 GΩ in the physiological response range was produced by conductance distributed uniformly over an area of 369 μm2 (estimated by approximating the left-hand cell in Fig. 1 to have a soma of mean diameter 7 μm, an axon terminal of mean diameter 3.8 μm, a 70-μm-long axon and 20 μm total length of dendrites, both with a mean diameter 0.6 μm (this last value being the most prone to error)), giving a specific conductance of 2.7 S m2. For an internal resistivity of 1.2 Ωm this predicts an electrical space constant of λ = 215 μm for the axon and dendrites. We then solved the steady-state cable equation d2V/dx2 = V/λ2 to calculate the resulting voltage gradient along the axon when a voltage is imposed at the soma by the patch pipette. The axon was assumed to be terminated by a synaptic terminal of resistance 8.2 GΩ in the absence of GABA (calculated from the fraction of the cell area present in the terminal) and the boundary conditions applied were continuity of voltage and current flow at the junction between the axon and terminal. If the true reversal potential of the GABA-evoked current in the terminal was set to a value different to the cell resting potential, the apparent reversal potential measured at the soma was predicted to overestimate the displacement of the reversal potential from the resting potential by 9 %, e.g. if EGABA were 10 mV negative to the resting potential the apparent reversal potential would be 0.9 mV more negative than the real reversal potential. (A similar calculation for a 10 μm long dendrite with a sealed end predicts negligible errors of less than 0.2 % for measurement of the displacement from the resting potential of EGABA at the dendrites.) Since the reversal potential at the synaptic terminal is expected to be more negative than the resting potential, and indeed was usually observed to be more negative than the apparent cell resting potential (which may, however, be displaced positive by current flow through the seal conductance; Tessier-Lavigne et al. 1988), then the true value of the reversal potential at the synaptic terminal will if anything be more positive than the measured value, reinforcing the conclusion of this paper that there is little difference between ECl at the two ends of the cell.
Statistics
Data are presented as mean ± s.e.m. Statistical P values were obtained from Student's two-tailed t tests.
Results
GABA activates a current at the dendrites and at the synaptic terminals of ON bipolar cells
Filling of recorded bipolar cells with Lucifer yellow (see Methods) showed that we were able to select ON bipolar cells, defined by the position of their somata being close against the outer plexiform layer, into which their dendrites run, and by the fact that their axons terminated in the ON sublamina of the inner plexiform layer close to the ganglion cells (Fig. 1A, B, the left cell of which was typical of most of the cells we filled). Comparison of the morphology of the filled cells with the catalogue of different bipolar cell classes in rat retina established by Euler & Wässle (1995) and Hartveit (1997) revealed that the bipolar cells we recorded from were either rod ON bipolars or class 8 or 9 cone ON bipolars. We were not certain that we could reliably distinguish between these three different types of ON bipolar cell; for example, although the left-hand cell in our Fig. 1A and B looks like a rod ON bipolar and the right-hand cell looks like a class 8 cone ON bipolar (see Fig. 2 of Euler & Wässle, 1995), if the right-hand cell happened to lose part of its synaptic terminal during the retinal slicing procedure it would appear to be a rod bipolar. Consequently, since both rod and cone ON bipolars are predicted to have the same non-uniformity of [Cl−]i based on their transporter distribution (Vardi et al. 2000; Vu et al. 2000), and since all of the cells we recorded gave similar results, we pooled all our data from these types of ON bipolar cells. A rough lower limit to the proportion of rod bipolar cells in our sample can be estimated from the data of Euler & Wässle (1995). They calculated that rod bipolars comprise at least 40 % of all bipolar cells in the rat retina, and 14 out of their sample of 83 filled cone bipolars (17 %) were types 8 or 9. This implies that of the bipolar cells with the morphology we selected (a soma closely apposed to the OPL and synaptic terminals closely apposed to the ganglion cells, i.e. rod plus cone types 8 and 9) at least 80 % (calculated as 40 %/(40 % + 17 % × 60 %)) would be rod bipolars. On average the input resistance of the cells was around 1 GΩ near −40 mV, and the apparent resting potential in the light (which may be made more positive by the seal conductance; Tessier-Lavigne et al. 1988) was −34.6 ± 2.9 mV in 15 cells.
GABA-evoked currents were studied using perforated patch recording, employing the perforant gramicidin, which forms cation channels in the membrane under the electrode but does not alter the intracellular chloride concentration (Ebihara et al. 1995; Akaike, 1996). Puffing 100 μm GABA in solution lacking calcium to block Ca2+-dependent synaptic transmission (see Methods), produced a current that lasted about 0.7 s (Fig. 2A). GABA evoked a response at the dendrites and the synaptic terminals (Fig. 2A), as expected from the presence of GABAA and GABAC receptors at these locations (Karschin & Wässle, 1990; Qian & Dowling, 1995; Enz et al. 1996; Du & Yang, 2000; Shields et al. 2000). The conductance activated (calculated as described in the Methods at a time when the conductance had decayed to approximately half of its peak value) was similar at the dendrites and at the synaptic terminals (Fig. 2B; not significantly different, P = 0.21, by paired t test).
In lower vertebrates there are GABAB receptors on bipolar cell synaptic terminals (Maguire et al. 1989), but these are reported to be absent in the rat (Koulen et al. 1998). Our previous work showed that essentially all of the GABA-evoked current in whole-cell clamped rat bipolar cells was abolished by a combination of GABAA and GABAC blockers (Billups et al. 2000). To confirm that there was no significant contribution of GABAB receptors to the GABA-evoked current recorded in these cells with the less intrusive perforated patch technique, we compared the response to bath-applied GABA and baclofen, a GABAB receptor agonist. In all three cells tested, baclofen (100 μm) generated no current (Fig. 2C), confirming that the GABA response was generated by GABAA and GABAC receptors, with no contribution from GABAB receptors.
The [Cl−]i is higher at the dendrites than at the synaptic terminals of ON bipolar cells
Since the GABA-evoked current is generated by GABAA and GABAC receptors, and there was no bicarbonate in our solutions (which permeates GABA-gated Cl− channels; Bormann et al. 1987), we could use the reversal potential of the GABA-evoked current to determine the value of [Cl−]i in the dendrites and synaptic terminals of the cell. Figure 3A shows the protocol used to determine the reversal potential. From a holding potential of −42 mV (nominally −40 mV but with −2 mV added from the electrode junction potential), a voltage ramp from +8 to −92 mV was applied, initially in the absence of GABA (grey current trace). (It was important that the holding potential be the same for all cells since, as described below, the resting [Cl−]i adapts to the holding potential used.) The voltage ramp was then repeated (40 s later) immediately after a puff of GABA was applied to the outer plexiform layer (black trace). At the point where the current traces, obtained in the presence and absence of GABA, cross in Fig. 3A (arrow) is the apparent reversal potential of the GABA-evoked current, which was −56 mV (including the electrode junction potential) in this cell. Using the net membrane current flowing at this point (-32 pA for Fig. 3A), and the measured residual (uncompensated) series resistance of the perforated patch plus electrode (45 MΩ), this value was then corrected for series resistance voltage error (1.4 mV in this case) to obtain the final reversal potential (Erev) of −54.6 mV. From the Nernst potential for Cl−:
where F is the Faraday constant with [Cl−]o = 145.5 mm and a temperature of T = 24 °C, this gives a value of [Cl−]i = 17.2 mm at the bipolar cell dendrites. F is the Faraday constant. The value of the reversal potential was stable within a cell (e.g. Fig. 6B), but varied quite significantly between different cells, ranging from −35 to −67 mV, corresponding to [Cl−]i ranging from 37 to 11 mm. This variability between cells made it essential to compare the values of [Cl−]i at the dendrites and synaptic terminal of the same cell, rather than comparing between cells.
Figure 6. The effect of elevated [K+]o on ECl.
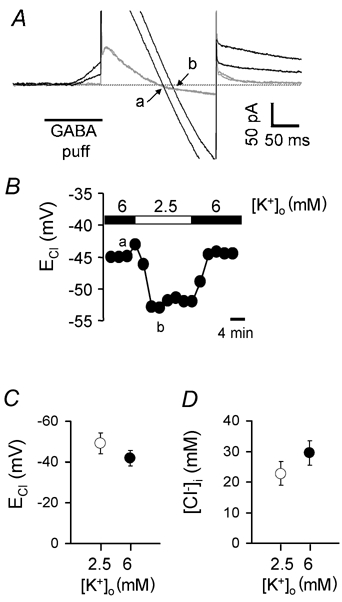
A, specimen current responses to voltage ramps (as in Figs 3 and 5) in the absence (grey traces) and presence (black traces) of a GABA puff in 6 mm[K+]o (smaller outward current in GABA) or in 2.5 mm[K+]o (larger outward current in GABA). The chloride reversal potential is at the voltage where the current in the control solution and that in GABA cross (arrows: a = 6 mm[K+]o, b = 2.5 mm[K+]o). B, the measured ECl as a function of time during the experiment in A. The lower case letters indicate the times of the specimen traces shown in A. C, average chloride reversal potential (ECl) for five cells bathed in 6 mm[K+]o (•) or in 2.5 mm[K+]o (○). D, average internal chloride concentration ([Cl−]i) calculated from the data in C.
The whole procedure was then repeated while puffing GABA at the inner plexiform layer (Fig. 3B), giving a reversal potential of −62 mV, which was 7.4 mV more negative than at the OPL, and corresponded to a value of [Cl−]i = 12.9 mm at the bipolar cell synaptic terminal. Out of seven cells to which GABA was applied at the OPL and the IPL, six had a reversal potential that was slightly more negative at the IPL and in the seventh there was no difference at the two locations (Fig. 3C). The mean difference between the two ends of the cell was 4.2 ± 1.5 mV in the seven cells (significantly different with P = 0.033 by a paired t test, although voltage non-uniformity in the bipolar cell axon could reduce this difference slightly; see Methods); the mean reversal potentials were −46.2 ± 4.0 mV at the OPL and −50.4 ± 3.9 mV at the IPL (Fig. 3D). The corresponding mean values for [Cl−]i were 25.4 ± 3.1 mm at the OPL and 21.5 ± 2.8 mm at the IPL (Fig. 3E).
[Cl−]i is strongly affected by membrane potential
If the ON bipolar cell has a significant membrane permeability to Cl− ions then, when central light depolarizes the cell, entry of Cl− is expected to displace ECl in the positive direction unless membrane Cl− transporters confer a stronger control of [Cl−]i than do membrane channels. To test the effect of membrane potential on [Cl−]i in ON bipolars, we held the cell at a test membrane potential for 1–2 min, and then applied a puff of GABA to either the dendrites or the synaptic terminals of the cell and used voltage ramps to measure the reversal potential of the GABA-evoked current, as described above.
Specimen data for a single cell obtained in this way are shown in Fig. 4A, while Fig. 4B shows averaged data from a total of five cells. Figure 4B shows a remarkable adaptation of the steady-state value of ECl to the prevailing membrane potential. At both the dendrites and the synaptic terminal, the steady-state ECl varied roughly linearly with holding potential. This relationship had a slope of about 0.5, and ECl was approximately equal to the membrane potential at around −40 to −45 mV. Although these data, like those in Fig. 3, showed [Cl−]i to be slightly higher in the dendrites than in the synaptic terminal (see below), at both locations the steady-state ECl was positive to the membrane potential for membrane potentials more negative than about −50 mV, while ECl was negative to the membrane potential for membrane potentials positive to about −40 mV. This remained the case even after correcting the derived values of ECl for a possible contribution of intracellular bicarbonate to the reversal potential of the GABA-evoked current, as described in detail below.
These results imply, first, that there is indeed a significant membrane permeability to chloride and, second, that at positive potentials there is an active extrusion of Cl− from the cell while at negative potentials there is active accumulation of Cl−. Although in principle the extrusion and accumulation could be done by a single mechanism (e.g. KCC2) operating in different directions as [Cl−]i alters, considering the extent to which [Cl−]i alters at positive and negative extremes of potential suggests that two separate mechanisms (presumably KCC2 and NKCC1) must mediate the extrusion and accumulation. Fig. 4C shows the energy needed to extrude one mole of Cl− using either KCC2 (with a stoichiometry such that one K+ ion moves with each Cl− ion; Payne, 1997) or NKCC1 (with a stoichiometry such that one Na+ ion, one K+ ion and two Cl− ions move together; Russell, 2000) as a function of [Cl−]i, which allows prediction of the direction of operation of each transporter. In the steady state at a holding potential of −102 mV the most negative ECl (at the synaptic terminals) was around −76 mV, corresponding to a [Cl−]i of 7.5 mm. From Fig. 4C when [Cl−]i = 7.5 mm, KCC2 still extrudes Cl−, so the Cl− accumulation needed to maintain ECl positive to the membrane potential is assumed to be produced by NKCC1. Conversely, at −22 mV the most positive ECl (at the dendrites) was −24 mV, corresponding to a [Cl−]i of 57 mm. From Fig. 4C when [Cl−]i = 57 mm NKCC1 still accumulates Cl−, so the Cl− extrusion needed to maintain ECl negative to the membrane potential is presumably mediated by KCC2. When the membrane potential is held at −2 mV, ECl at the OPL rises to −15 mV, corresponding to a [Cl−]i of 81 mm. At this value of [Cl−]iFig. 4C predicts that, in addition to KCC2, efflux mediated by NKCC1 may also contribute to ECl being maintained more negative than the membrane potential.
The data in Fig. 4B show the tendency of ECl to be more positive in the dendrites than in the synaptic terminal which was shown in Fig. 3, but the differences between the OPL and IPL data at each voltage are not statistically significant. This is because the prolonged experiment required for these steady-state measurements meant that for most cells the OPL and IPL data could not be obtained for all voltages in the same cell (unlike in Fig. 3), so that cell-to-cell variability contributed a great deal to the error bars.
The kinetics of the adaptation of [Cl−]i to the prevailing membrane potential were investigated by clamping a cell to a potential of −62 mV for different durations (from a holding potential of −42 mV), and then returning to −42 mV, applying a GABA puff to the OPL and using voltage ramps to determine the reversal potential of the GABA-evoked current (Fig. 5A; see legend for details of the ramp protocol). Data from a single cell in Fig. 5B show that the shift in ECl of approximately 11 mV that occurred on stepping the membrane potential from −42 to −62 mV occurred with a time constant of 11 s. On average, the time course of the change in ECl could be approximately fitted with an exponential with a time constant of around 8 s (Fig. 5C).
Figure 5. Kinetics of the voltage-dependent change of ECl.
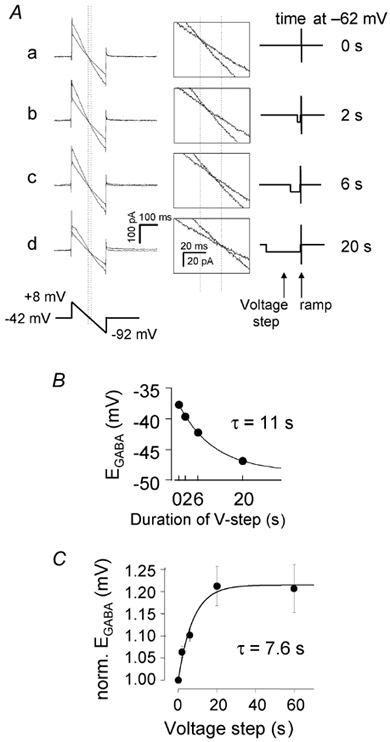
A, specimen current responses to a voltage ramp applied from a holding potential of −42 mV, in the absence (control, less steep traces during the ramps) and presence (steeper traces)of a preceding 100 ms GABA puff to the OPL. The control voltage ramp was applied 300 ms after the end of a voltage step to −62 mV for 0 (a), 2 (b), 6 (c) or 20 (d) s. Then (after a 40 s interval) the voltage pulse to −62 mV was repeated followed (after 200 ms) by a 100 ms GABA puff and the second voltage ramp was applied. Finally the procedure was repeated again without the GABA puff to test that the second control I-V relation superimposed on the first one as in Fig. 3A (data not shown for clarity). The vertical dotted lines indicate the value of ECl when the cell was either not hyperpolarized (left vertical line) or was hyperpolarized for 20 s (right vertical line). Insets in the middle show the data at higher gain. The voltage protocol is shown on the right. B, reversal potential derived from the traces in A, plotted as a function of the duration of the voltage step to −62 mV. Smooth curve is an exponential with a time constant of 11 s and a maximum change of ECl of 10.9 mV. C, average data for the change of ECl, measured as in A (number of cells per point was 5, 5, 3 and 5 for 2, 6, 20 and 60 s pulses respectively; total number of cells studied was six). For each cell the values of ECl were normalized to the control value of ECl (when the membrane was not hyperpolarized before the voltage ramp), to remove variation due to differences in the initial value of ECl. The line shows the fit of the data with a single exponential of time constant 7.6 s.
Both NKCC1 and KCC2 are electroneutral, so their direction (and, for NKCC1 at least, rate) of operation will not be affected by changes of membrane potential (Payne, 1997; Russell, 2000). Consequently the tendency of ECl to track membrane potential changes is most simply explained by there being a significant resting permeability to Cl−. Below we attempt to model the voltage dependence of [Cl−]i based on the assumption that [Cl−]i is determined by a combination of a chloride conductance, NKCC1 and KCC2.
ECl is more positive when [K+]o is higher
From the thermodynamics of their carrier cycle, both the NKCC1 and the KCC2 transporters are expected to produce a higher equilibrium level of [Cl−]i when the extracellular potassium concentration rises (for KCC2 the equilibrium [Cl−]i is given by [K+]o[Cl−]o/[K+]i, and for NKCC1 it is given by {([K+]o[Na+]o[Cl−]o2)/([K+]i[Na+]i)}1/2. Since [K+]o is higher in the outer retina than in the inner retina (by about 0.5 mm) in the dark, and light initially lowers [K+]o in the outer retina and raises it in the inner retina (Steinberg et al. 1980), we examined the effect of changing [K+]o on [Cl−]i.
Raising [K+]o from 2.5 to 6 mm at a holding potential of −42 mV shifted ECl positive (Fig. 6A and B) by a mean value of 7.4 ± 1.6 mV (P < 0.01, Fig. 6C) in five cells to which GABA was applied at the dendrites, corresponding to an increase of [Cl−]i of about 7 mm (from 22.8 ± 3.8 to 29.5 ± 4.0 mm, P = 0.003, Fig. 6D). Similarly, ECl shifted positive by 9 mV when [K+]o was raised from 2.5 to 6 mm in one cell to which GABA was applied at the synaptic terminals. In all five cells studied, the shift of ECl took much longer than that shown for a change of membrane potential in Fig. 5, and was only complete in 6 min or longer (Fig. 6B), consistent with a time constant for an exponential change of more than ≈200 s (the time constant could not be estimated accurately because of the small number of ECl data points that it was possible to measure during the change of ECl). The rate of change of ECl was not limited by the speed of the perfusion, since bath perfusion produced GABA-evoked currents with a time to peak of a few seconds (Fig. 2D). In a non-voltage-clamped cell in vivo, if [K+]o rose uniformly around the cell the resulting depolarization of the membrane potential would produce an extra positive shift of ECl (Fig. 4B).
Modelling the control of [Cl−]i
To gain a more quantitative understanding of how Cl− transporters control [Cl−]i in bipolar cells, we analysed a simplified model in which we ignored the small difference in [Cl−]i between the two ends of the cell, treated the cell as a single well-mixed compartment, and tried to fit the data in Fig. 4B. Chloride fluxes across the cell membrane were assumed to reflect current flow through an ohmic Cl− conductance, GCl (although bipolar cells express hyperpolarization-activated CLC-2 chloride channels, these are only significantly activated at voltages negative to −80 mV; Enz et al. 1999). Extrusion of Cl− by KCC2 was assumed to be at a rate k[Cl−]i, i.e. a non-saturating dependence since KCC2 has a low affinity for Cl− (at least for external Cl−:Km = 101 mm; Payne, 1997). Pumping of Cl− into the cell by NKCC1 was assumed to occur at a constant rate n (inhibition of NKCC1 when [Cl−]i rises was initially ignored for simplicity; see below). Both n and k were assumed to be voltage independent, since both NKCC1 and KCC2 are electroneutral, and NKCC1 has been suggested to be voltage independent (see discussion in Russell, 2000). If F is the Faraday constant and U is the cell volume, this gives the rate of change of [Cl−]i as:
| (1) |
so that in the steady state:
| (2) |
In Fig. 4B, V is approximately equal to the mean of the ECl values at the dendrites and synaptic terminals when V = −40 mV and hence [Cl−]i = 30.5 mm, so (inserting V = ECl into eqn (2)):
| (3) |
and
| (4) |
We then set the parameter Fn/GCl, which sets the ratio of Cl− fluxes mediated by pumping to those mediated by the conductance, so that the relationship between ECl and V predicted by eqn (4) gave as good a fit as possible to the data of Fig. 4B. The dashed line in Fig. 7A, which is superimposed on the data replotted from Fig. 4B, shows a plot of eqn (4) using Fn/GCl = 30 mV, which gives a reasonable fit to the data over the range of potentials between −30 and −80 mV, but predicts values of ECl which are too negative at extremes of potential. Incorporating inhibition of NKCC1 by intracellular [Cl−]i by making the rate of NKCC1 obey the formula n(1 - λ[Cl−]i), with λ a constant, led to an equation which is formally equivalent to eqn (4) and so does not lead to a better fit. We then considered the possible contribution of bicarbonate to the reversal potential of the GABA-evoked currents. Although there is no bicarbonate in our Hepes-buffered superfused solution (see Methods), HCO3− might be present inside the cell at up to a concentration of 15.1 mm (the value predicted for an internal pH of 7.2, as in our pipette solution, and 1.2 mm (40 mmHg) CO2 produced by metabolism). HCO3− passes through GABAA receptor channels with a permeability that is (at most) about 0.3 of that for Cl− (Kaila et al. 1989; Wotring et al. 1999). Thus, from the Goldman-Hodgkin-Katz equation, assuming no extracellular HCO3−, the reversal potential of the GABA-gated current, Erev, could be displaced positive from the value of ECl by the presence of intracellular HCO3− according to:
or using PHCO3/PCl = 0.3 and [HCO3−]i = 15.1 mm:
| (5) |
From eqn (5) we calculated, for the reversal potential data in Fig. 4B, the corrected value of [Cl−]i and thus a corrected value of ECl, and plotted this against the membrane potential. As shown in Fig. 7B, the main effect of correcting for the possible presence of HCO3− is that at more negative membrane potentials the values of ECl derived from Erev become more negative. Fitting eqn (2) to the points in Fig. 7B as described above for Fig. 7A, with ECl equal to V at −60 mV and Fn/GCl = 7 mV, gave the dashed curve shown. As for the uncorrected data in Fig. 7A, the curve fits reasonably well over the voltage range −30 to −80 mV, but predicts ECl values that are too negative at more extreme voltages.
Figure 7. Modelling the voltage dependence of ECl.
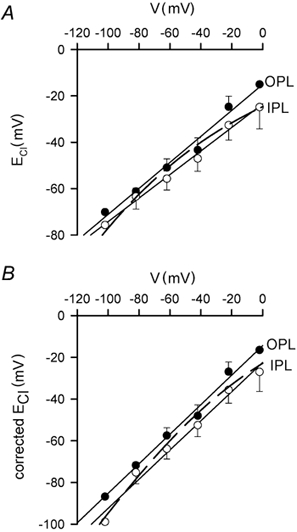
A, data points are the OPL (filled circles) and IPL (open circles) values of ECl from Fig. 4B. The dashed curve is a fit to the data, i.e. eqn (4) with Fn/GCl = 30 mV. B, points show the data in A corrected for the contribution of internal bicarbonate to the reversal potential of the GABA-evoked current. The dashed curve is a fit to the corrected data (derived using eqn (5)), i.e. a modified version of eqn (4) such that ECl and V are equal at −60 mV: ECl = V + (Fn/GCl)(1 - [Cl−]i/14 mm) with Fn/GCl = 7 mV. Lines are linear regressions.
To determine whether this model (not including the possible presence of bicarbonate) could account for the shift of ECl seen when [K+]o was raised from 2.5 to 6 mm, we assumed for simplicity that raising [K+]o stimulates Cl− influx mediated by NKCC1 (i.e. alters the value of n) but has no effect on the Cl− efflux mediated by KCC2 (in fact raising [K+]o shifts ECl positive even in neurons which have had NKCC1 knocked out (Sung et al. 2000), presumably by slowing KCC2 extrusion, so this is an oversimplification, see below). The shift of ECl predicted by eqn (2) at constant voltage (-42 mV), when the rise of [K+]o elevates [Cl−]i from [Cl−]iold to [Cl−]inew, is given by:
or
Using Fn(2.5 mm K+)/GCl = 30 mV from the fit to Fig. 7A, and n(2.5 mm K+) = k[Cl−]iold near −40 mV from eqn (3), this becomes:
Inserting [Cl−]inew/[Cl−]iold = 1.335 to give ΔECl = 7.4 mV as observed experimentally (Fig. 6C and D), this gives:
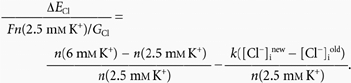 |
i.e. a 2.4-fold increase of [K+]o from 2.5 to 6 mm is predicted to increase the rate of NKCC1 by a factor of 1.58. If the rate of NKCC1 is assumed to depend in a Michaelis-Menten fashion on [K+]o, i.e.:
then to produce a 1.58-fold rise in the rate when [K+]o is raised from 2.5 to 6 mm, the EC50 for activation by [K+]o must be 4.2 mm. This predicted value is in reasonable agreement with the EC50 of 2 mm measured for activation of human and mouse NKCC1 by external Rb+ and K+ (Isenring et al. 1998; Glanville et al. 2001). A value of 2 mm (rather than 4.2 mm) for the EC50 would allow some of the effect of external K+ on ECl to be mediated by a slowing of KCC2.
Despite the reasonable success of the model above in predicting the steady-state dependence of [Cl−]i on membrane potential and [K+]o, the large difference in kinetics of the ECl shift in response to a change of membrane potential (τ ≈8 s) and to a rise of [K+]o (τ > 200 s) is not predicted by the model. Linearizing eqn (1) for small changes of [Cl−]i predicts a time constant for the equilibration of [Cl−]i which is similar when the membrane potential is changed or when n is altered by raising [K+]o. (This can be seen quickly by noting that the initial rate of change of [Cl−]i predicted by eqn (1) for the 20 mV voltage change experiment of Fig. 5 is GCl20 mV/FU, while that for the [K+]o change experiment of Fig. 6 which produces a comparable change of ECl is (n(6 mm K+) - n(2.5 mm K+))/U = 0.58n(2.5 mm K+)/U from the factor of 1.58 calculated above. Thus, the ratio of these rates of change is GCl20 mV/(0.58n(2.5 mm K+)F, or 1.15 using Fn/GCl = 30 mV, so [Cl−]i changes at a similar rate in response to the voltage or [K+]o change). This raises the question of whether the effect of raising [K+]o is not a direct effect on Cl− transporters, as we have assumed, but may instead reflect an inherently slower process, such as stimulation of the Na+-K+ pump by the raised [K+]o leading secondarily to a lowering of [Na+]i (if this is not well controlled by diffusion from the patch pipette) and subsequent stimulation of NKCC1.
Discussion
The aim of this study was to determine whether the presence in the dendrites of the NKCC1 transporter, which imports Cl− into cells, and the presence in the synaptic terminals of the transporter KCC2, which exports Cl− from cells, does indeed lead to [Cl−]i being higher in the dendrites than in the synaptic terminals of ON bipolar cells.
Factors controlling [Cl−]i in ON bipolar cells
As described in the Introduction, the concentration of chloride in the dendrites and synaptic terminals of retinal bipolar cells will determine the sign of the voltage change produced by the GABAergic input mediating lateral inhibition at the dendrites and mediating temporal shaping at the synaptic terminal. The data reported here show that the intracellular chloride concentration in ON bipolar cells is neither constant, nor in equilibrium with the membrane potential, but is controlled by the membrane potential, by the extracellular potassium concentration and by two separate Cl− transporters, probably KCC2 which extrudes K+ and Cl−, and NKCC1 which pumps Na+, K+ and 2Cl− into the cell. Around a membrane potential of −40 mV, in the steady state ECl is approximately equal to the membrane potential. Altering the membrane potential leads to a time-dependent alteration of the chloride reversal potential in the same direction, as expected if there is a passive membrane conductance to Cl−, but in the steady state the shift of ECl is only about half that of the membrane potential change (Fig. 4B). Consequently, at more positive potentials ECl is more negative than the membrane potential probably because of Cl− extrusion by KCC2, and at more negative potentials ECl is more positive than the membrane potential probably because NKCC1 raises [Cl−]i. Raising the extracellular [K+] leads to a positive shift of ECl, presumably because the raised [K+]o stimulates Cl− entry by NKCC1 and inhibits Cl− efflux on KCC2. A simple mathematical model, incorporating a membrane chloride conductance and KCC2 and NKCC1 transporters could account approximately for the steady-state voltage and [K+]o dependence of ECl, but was not successful at accounting for the different kinetics of the change of ECl in response to a change of membrane potential and a rise of [K+]o (see Fig. 7 and associated text). In other CNS neurons, control of [Cl−]i by membrane potential has been reported by Ehrlich et al. (1999), and control by [K+]o has been reported by Thompson et al. (1988), Kakazu et al. (1999), DeFazio et al. (2000) and Sung et al. (2000).
These data are consistent with demonstrations of a role for NKCC1 and KCC2 in controlling [Cl−]i in other neurons (superior olive neurons: Kakazu et al. 1999; hippocampal pyramidal cells: Rivera et al. 1999; neocortical pyramidal cells: DeFazio et al. 2000; dorsal root ganglion cells: Sung et al. 2000; spinal motoneurons: Hübner et al. 2001; amygdala and neocortical cells: Martina et al. 2001). Indeed, these transporters have the power to change the sign of GABA-evoked signalling, since an increase in the expression of KCC2 during development can lead to a shift of ECl from being more positive to being more negative than the resting potential of central neurons, so that GABA or glycine changes from being initially excitatory during development to being inhibitory in the adult animal (Ben-Ari et al. 1994; Ehrlich et al. 1999; Rivera et al. 1999; Ganguly et al. 2001).
Non-uniformity of [Cl−]i along the ON bipolar cell
The observed distribution of Cl− transporters in ON bipolar cells, with NKCC1 at the dendrites and KCC2 at the synaptic terminals, suggested that the intracellular chloride concentration should be higher at the dendrites than at the synaptic terminals (Vardi et al. 2000; Vu et al. 2000). If this difference were large, so that ECl were significantly more depolarized than the resting potential at the dendrites, and significantly more hyperpolarized than the resting potential at the synaptic terminals, then it would allow GABA released by horizontal cells to produce a depolarization at the dendrites and GABA released by amacrine cells to produce a hyperpolarization at the synaptic terminals. As discussed below, this would fit in with current ideas about the sign of inhibition at the two plexiform layers.
Experimentally, although ECl was consistently more positive at the dendrites than at the synaptic terminal, i.e. in the direction predicted by the asymmetrical transporter distribution in ON bipolar cells, the difference was only 4 mV, corresponding to [Cl−]i being approximately 4 mm higher (25 versus 21 mm). Although our experiments were done at room temperature (21-25 °C), slowed transporter operation at this temperature is unlikely to be the cause of the small size of the [Cl−]i gradient along the cell, because previous experiments have shown that NKCC1 and KCC2 can shift ECl to be more positive or more negative than the resting membrane potential even at room temperature (Ehrlich et al. 1999; Kakazu et al. 1999; Ganguly et al. 2001; Jang et al. 2001), and because in our experiments, at negative and positive potentials respectively, NKCC1 and KCC2 did indeed shift ECl to be more positive and more negative than the holding potential, as shown in Fig. 4. Our experiments were carried out in Hepes-buffered solution, with the aim of eliminating any contribution of Cl−-HCO3− and Na+-dependent Cl−-HCO3− exchange to the control of [Cl−]i (which may in fact be relatively unimportant; Ballanyi & Grafe, 1985), and thus maximizing any difference of [Cl−]i which could be produced between the dendrites and synaptic terminal of the cell. Even in this optimal situation, however, the difference in reversal potential for GABA-evoked currents at the two ends of the cell does not seem large enough to produce a significant functional difference between the effects of GABA at the two ends of the cell, for the reasons described below.
In the intact retina, in the dark, the extracellular [K+] is approximately 0.5 mm higher around the bipolar cell dendrites than around the synaptic terminals (Steinberg et al. 1980) because of K+ efflux from the photoreceptors (which are depolarized in the dark). Steinberg et al. (1980, Fig. 15) showed that during prolonged illumination, as was present in our experiments, this difference is essentially abolished. The data in Fig. 6 (where a 3.5 mm increase of [K+]o produces a 7 mV positive shift of ECl) suggest that the 0.5 mm higher [K+]o at the dendrites in the dark might increase the difference in ECl between the two ends of the cell by 1 mV, from the 4 mV that we measured in the light to approximately 5 mV in the dark. If the dark resting potential of the bipolar cell (around −45 mV; Euler et al. 1996) was exactly half-way between the values of ECl at the two ends of the cell in the dark, it would result in only 2.5 mV of driving force at each end of the cell for GABA-evoked chloride currents to produce a depolarization at the dendrites and a hyperpolarization at the synaptic terminals, and the driving force for depolarization at the dendrites would disappear as soon as the bipolar cell was depolarized by central illumination. Furthermore, despite being consistently slightly more positive than the value of ECl in the synaptic terminal, there was great variability in the absolute value of ECl in the dendrites of ON bipolar cells, ranging from −35 to −67 mV. Although we did not measure the dark resting potential of the cells (our experiments were done in the light), the more negative ECl values we measured would require an extremely negative dark resting potential for ECl to be more positive than the dark resting potential. We conclude that the average ECl is not sufficiently positive for GABA to reliably produce a depolarization at the dendrites of ON bipolar cells.
Our data differ from those of Satoh et al. (2001) who measured the reversal potential of GABA-evoked currents in mouse retinal bipolar cells. Satoh et al. (2001) reported no difference between the reversal potential at the dendrites and at the synaptic terminals of ON bipolar cells. The reason for this is unclear, although their long (5 s) pressure applications of GABA, which may allow more GABA to diffuse between the dendrites and synaptic terminal than is the case for our brief GABA puffs (which lasted only 100 ms), may contribute to their not observing a more positive ECl at the dendrites. Satoh et al. (2001) also reported that ECl was more positive in rod bipolar cells than in cone ON or OFF bipolars (which had a similar ECl). We were not certain that we could distinguish dye-filled rod and cone ON bipolar cells but, although our grouping of them together may account for some of the variability in the values of ECl that we measured, Satoh et al. reported a similar variability of −23 to −61 mV even in cells they defined to be rod ON bipolars.
Implications for visual processing: lateral inhibition in ON bipolar cells
Early visual processing depends on changes in the activation of GABAA and GABAC receptors at the dendrites and synaptic terminals of retinal bipolar cells. The effect of these receptors is determined by the value of the chloride reversal potential relative to the membrane potential. We had expected that there would be a gradient of intracellular chloride concentration, [Cl−]i, along ON bipolar cells, being higher in the dendrites and lower in the synaptic terminals. This distribution is predicted, not only from the non-uniform expression of NKCC1 and KCC2 transporters (Vardi et al. 2000; Vu et al. 2000), but also from current ideas on how bipolar cells integrate lateral inhibitory signals arriving from horizontal and amacrine cells with the direct synaptic input they receive from photoreceptors (see Introduction and references therein). ON bipolar cells are depolarized when light falling on the centre of their receptive field suppresses glutamate release from photoreceptors. Thus, to produce an antagonistic surround to the receptive field, the suppression of GABA release from horizontal cells which is produced by peripheral light must lead to a hyperpolarization. This can only occur if ECl in the dendrites is more positive than the resting potential (around −45 mV in the dark; Euler et al. 1996). At the same time, release of GABA from amacrine cells onto the synaptic terminal needs to produce a hyperpolarization to induce transience into the depolarizing voltage response of the bipolar cell to central light. Our data show that ECl in the dendrites is unlikely to be more than a few millivolts more positive than the resting potential in the dark and as soon as central light has depolarized the cell (by up to 20 mV; Euler & Masland, 2000) the membrane potential will be positive to ECl so that suppression of GABA release from horizontal cells by surround light will lead to further depolarization. By contrast, inhibition at the synaptic terminal will be able to function, since depolarization of ON bipolar cells by central light will move the membrane potential more positive than ECl.
The generation of a centre-surround receptive field for bipolar cells by lateral inhibitory signals from horizontal cells is well established for amphibian retinae (Werblin & Dowling, 1969; Kaneko, 1970; Stone & Schutte, 1991; Hare & Owen, 1996), but less so for mammalian retinae particularly for rod bipolar cells. Anatomically defined synapses from horizontal cells to bipolar cells are reported to be rare in mammals (Kolb, 1979), but they have been reported for rod (ON) bipolar cells in human retina (Linberg & Fisher, 1988), and GABA receptors are located on bipolar cell dendrites (Haverkamp et al. 2000), while vesicular GABA transporters are present in horizontal cell axon terminals (Cueva et al. 2002); it is also possible that GABA is released at this synapse by reversed uptake (Schwartz, 1987) rather than by exocytosis. Cone bipolar cells (ON and OFF) show an antagonistic surround to their receptive field (Dacey et al. 2000), but rod (ON) bipolar cells have been reported to show no antagonistic surround (Bloomfield & Xin, 2000) although earlier work did suggest antagonistic horizontal cell input (Dacheux & Raviola, 1986). Our observation that ECl in the dendrites of ON bipolar cells is similar to the dark potential of the cells could explain why rod bipolar cells show no antagonistic surround mediated by suppression of GABA release from horizontal cells.
Implications for visual processing: time-dependent adaptation of [Cl−]i and inhibitory signals
A striking result of our study is that [Cl−]i in ON bipolar cells, and thus the driving force for inhibitory signals generated by GABAA and GABAC receptors, adapts on a time scale of ≈8 s following a change of the bipolar cell membrane potential. This has important implications for the time course of inhibition following membrane potential changes produced either by central illumination or by GABAergic input. When central light depolarizes the cell, initially the membrane potential will be substantially more positive than the value of ECl at the synaptic terminal, and GABA released from amacrine cells will have a large hyperpolarizing effect on the bipolar cell. However, during a maintained depolarization induced by central light, ECl will move positive (with a time constant of 8 s, by about half the depolarization produced by the central light (Fig. 4), resulting in a halving of the driving force for the inhibitory input. Similarly, if GABA receptors in the bipolar cell membrane are activated by a change of input from horizontal or amacrine cells (e.g. due to a change of illumination in the receptive field surround) then after an initial voltage change the resulting adaptation of ECl towards the new membrane potential will decrease the strength of the GABAergic signal, inducing an inherent transience to this signal. Thus, time-dependent adaptation of [Cl−]i in ON bipolar cells may add a previously unsuspected layer of temporal processing to signals as they pass through the retina.
Acknowledgments
We thank Martine Hamann, Païkan Marcaggi, Peter Mobbs and David Rossi for comments on the paper. Supported by the Wellcome Trust and a Wolfson-Royal Society Award.
References
- Akaike N. Gramicidin perforated patch recording and intracellular chloride activity in excitable cells. Progress in Biophysics and Molecular Biology. 1996;65:251–264. doi: 10.1016/s0079-6107(96)00013-2. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P. An intracellular analysis of gamma-aminobutyric-acid-associated ion movements in rat sympathetic neurones. Journal of Physiology. 1985;365:41–58. doi: 10.1113/jphysiol.1985.sp015758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Tseeb V, Raggozzino D, Khazipov R, Gaiarsa JL. γ-Aminobutyric acid (GABA), a fast excitatory transmitter which may regulate the development of hippocampal neurones in early postnatal life. Progress in Brain Research. 1994;102:261–273. doi: 10.1016/S0079-6123(08)60545-2. [DOI] [PubMed] [Google Scholar]
- Billups D, Hanley JG, Orme M, Attwell D, Moss SJ. GABAC receptor sensitivity is modulated by interaction with MAP1B. Journal of Neuroscience. 2000;20:8643–8650. doi: 10.1523/JNEUROSCI.20-23-08643.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield SA, Xin D. Surround inhibition of mammalian AII amacrine cells is generated in the proximal retina. Journal of Physiology. 2000;523:771–783. doi: 10.1111/j.1469-7793.2000.t01-1-00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and γ-aminobutyric acid in mouse cultured spinal neurones. Journal of Physiology. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba C, Saito T. APB (2-amino-4-phosphonobutyric acid) activates a chloride conductance in ganglion cells isolated from newt retina. Neuroreport. 1994;5:489–492. doi: 10.1097/00001756-199401120-00030. [DOI] [PubMed] [Google Scholar]
- Cueva JG, Haverkamp S, Reimer RJ, Edwards R, WäSSLE H. Vesicular γ-aminobutyric acid transporter expression in amacrine and horizontal cells. Journal of Comparative Neurology. 2002;445:227–237. doi: 10.1002/cne.10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacey D, Packer OS, Diller L, Brainard D, Peterson B, Lee B. Center surround receptive field structure of cone bipolar cells in primate retina. Vision Research. 2000;40:1801–1811. doi: 10.1016/s0042-6989(00)00039-0. [DOI] [PubMed] [Google Scholar]
- Dacheux RF, Raviola E. The rod pathway in the rabbit retina: a depolarizing bipolar and amacrine cell. Journal of Neuroscience. 1986;6:331–345. doi: 10.1523/JNEUROSCI.06-02-00331.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defazio RA, Keros S, Quick MW, Hablitz JJ. Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. Journal of Neuroscience. 2000;20:8069–8076. doi: 10.1523/JNEUROSCI.20-21-08069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E. Cation-chloride cotransporters in neuronal communication. News in Physiological Science. 2000;15:309–312. doi: 10.1152/physiologyonline.2000.15.6.309. [DOI] [PubMed] [Google Scholar]
- Dong CJ, Werblin FS. Temporal contrast enhancement via GABAC feedback at bipolar terminals in the tiger salamander retina. Journal of Neurophysiology. 1998;79:2171–2180. doi: 10.1152/jn.1998.79.4.2171. [DOI] [PubMed] [Google Scholar]
- Du J, Yang X. Subcellular localization and complements of GABAA and GABAC receptors on bullfrog retinal bipolar cells. Journal of Neurophysiology. 2000;84:666–676. doi: 10.1152/jn.2000.84.2.666. [DOI] [PubMed] [Google Scholar]
- Ebihara S, Shirato K, Harata N, Akaike N. Gramicidin-perforated patch recording: GABA response in mammalian neurones with intact intracellular chloride. Journal of Physiology. 1995;484:77–86. doi: 10.1113/jphysiol.1995.sp020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Lohrke S, Friauf E. Shift from depolarizing to hyperpolarizing glycine action in rat auditory neurones is due to age-dependent Cl− regulation. Journal of Physiology. 1999;520:121–137. doi: 10.1111/j.1469-7793.1999.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enz R, Brandstatter JH, WäSSLE H, Bormann J. Immunocytochemical localization of the GABAC receptor ρ subunits in the mammalian retina. Journal of Neuroscience. 1996;16:4479–4490. doi: 10.1523/JNEUROSCI.16-14-04479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enz R, Ross BJ, Cutting GR. Expression of the voltage-gated Cl− channel CLC-2 in rod bipolar cells of the rat retina. Journal of Neuroscience. 1999;19:9841–9847. doi: 10.1523/JNEUROSCI.19-22-09841.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler T, Masland RH. Light-evoked responses of bipolar cells in a mammalian retina. Journal of Neurophysiology. 2000;83:1817–1829. doi: 10.1152/jn.2000.83.4.1817. [DOI] [PubMed] [Google Scholar]
- Euler T, Schneider H, WäSSLE H. Glutamate responses of bipolar cells in a slice preparation of the rat retina. Journal of Neuroscience. 1996;16:2934–2944. doi: 10.1523/JNEUROSCI.16-09-02934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler T, WäSSLE H. Immunocytochemical identification of cone bipolar cells in the rat retina. Journal of Comparative Neurology. 1995;361:461–478. doi: 10.1002/cne.903610310. [DOI] [PubMed] [Google Scholar]
- Euler T, WäSSLE H. Different contributions of GABAA and GABAC receptors to rod and cone bipolar cells in a rat retinal slice preparation. Journal of Neurophysiology. 1998;79:1384–1395. doi: 10.1152/jn.1998.79.3.1384. [DOI] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E. A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. Journal of Physiology. 1982;331:577–597. doi: 10.1113/jphysiol.1982.sp014393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–532. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- Glanville M, Kingscote S, Thwaites DT, Simmons NL. Expression and role of sodium, potassium, chloride cotransport (NKCC1) in mouse inner medullary collecting duct (mIMCD-K2) epithelial cells. Pflügers Archiv. 2001;443:123–131. doi: 10.1007/s004240100629. [DOI] [PubMed] [Google Scholar]
- Hanitzsch R, KüPPERS L. The influence of Hepes on light responses of rabbit horizontal cells. Vision Research. 2001;41:2165–2172. doi: 10.1016/s0042-6989(01)00116-x. [DOI] [PubMed] [Google Scholar]
- Hare WA, Owen WG. Receptive field of the retinal bipolar cell: a pharmacological study in the tiger salamander. Journal of Neurophysiology. 1996;76:2005–2019. doi: 10.1152/jn.1996.76.3.2005. [DOI] [PubMed] [Google Scholar]
- Hartveit E. Functional organization of cone bipolar cells in the rat retina. Journal of Neurophysiology. 1997;77:1716–1730. doi: 10.1152/jn.1997.77.4.1716. [DOI] [PubMed] [Google Scholar]
- Hartveit E. Reciprocal synaptic interactions between rod bipolar cells and amacrine cells in the rat retina. Journal of Neurophysiology. 1999;81:2923–2936. doi: 10.1152/jn.1999.81.6.2923. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Grunert U, WäSSLE H. The cone pedicle, a complex system in the retina. Neuron. 2000;27:85–95. doi: 10.1016/s0896-6273(00)00011-8. [DOI] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Isenring P, Jacoby SC, Forbush B III. The role of transmembrane domain 2 in cation transport by the Na-K-Cl cotransporter. Proceedings of the National Academy of Sciences of the USA. 1998;95:7179–7184. doi: 10.1073/pnas.95.12.7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang I, Jeong H, Akaike N. Contribution of the Na-K-Cl cotransporter on GABAA receptor-mediated presynaptic depolarization in excitatory nerve terminals. Journal of Neuroscience. 2001;21:5962–5972. doi: 10.1523/JNEUROSCI.21-16-05962.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Pasternack M, Saarikoski J, Voipio J. Influence of GABA-gated bicarbonate conductance on potential, current and intracellular chloride in crayfish muscle fibres. Journal of Physiology. 1989;416:161–181. doi: 10.1113/jphysiol.1989.sp017755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. Journal of Neuroscience. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko A. Physiological and morphological identification of horizontal, bipolar and amacrine cells in goldfish retina. Journal of Physiology. 1970;207:623–633. doi: 10.1113/jphysiol.1970.sp009084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschin A, WäSSLE H. Voltage- and transmitter-gated currents in isolated rod bipolar cells of rat retina. Journal of Neurophysiology. 1990;63:860–876. doi: 10.1152/jn.1990.63.4.860. [DOI] [PubMed] [Google Scholar]
- Kolb H. The inner plexiform layer in the retina of the cat: electron microscopic observations. Journal of Neurocytology. 1979;8:295–329. doi: 10.1007/BF01236124. [DOI] [PubMed] [Google Scholar]
- Kondo H, Toyoda J. GABA and glycine effects on the bipolar cells of the carp retina. Vision Research. 1983;23:1259–1264. doi: 10.1016/0042-6989(83)90101-3. [DOI] [PubMed] [Google Scholar]
- Koulen P, Malitschek B, Kuhn R, Bettler B, WäSSLE H, Brandstatter JH. Presynaptic and postsynaptic localization of GABAB receptors in neurons of the rat retina. Pflügers Archiv. 1998;10:1446–1456. doi: 10.1046/j.1460-9568.1998.00156.x. [DOI] [PubMed] [Google Scholar]
- Kuffler SW. Discharge patterns and functional organization of mammalian retina. Journal of Neurophysiology. 1953;16:37–68. doi: 10.1152/jn.1953.16.1.37. [DOI] [PubMed] [Google Scholar]
- Linberg KA, Fisher SK. Ultrastructural evidence that horizontal cell axon terminals are presynaptic in the human retina. Journal of Comparative Neurology. 1988;268:281–297. doi: 10.1002/cne.902680211. [DOI] [PubMed] [Google Scholar]
- Longsworth LG. Diffusion measurements at 25 °C of aqueous solutions of amino acids, peptides and sugars. Journal of the American Chemical Society. 1953;75:5705–5709. [Google Scholar]
- Lukasiewicz PD, Shields CR. Different combinations of GABAA and GABAC receptors confer distinct temporal properties to retinal synaptic responses. Journal of Neurophysiology. 1998;79:3157–3167. doi: 10.1152/jn.1998.79.6.3157. [DOI] [PubMed] [Google Scholar]
- Maguire G, Maple B, Lukasiewicz P, Werblin F. γ-Aminobutyrate type B receptor modulation of L-type calcium channel current at bipolar cells in the retina of the tiger salamander. Proceedings of the National Academy of Sciences of the USA. 1989;86:10144–10147. doi: 10.1073/pnas.86.24.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr D, Hildreth E. Theory of edge detection. Proceedings of the Royal Society B. 1980;207:187–217. doi: 10.1098/rspb.1980.0020. [DOI] [PubMed] [Google Scholar]
- Martina M, Royer S, Pare D. Cell-type-specific GABA responses and chloride homeostasis in the cortex and amygdala. Journal of Neurophysiology. 2001;86:2887–2895. doi: 10.1152/jn.2001.86.6.2887. [DOI] [PubMed] [Google Scholar]
- Neal MJ, Cunningham JR, James TA, Joseph M, Collins JF. The effect of 2-amino-4-phosphonobutyrate (APB) on acetylcholine release from the rabbit retina: evidence for on-channel input to cholinergic amacrine cells. Neuroscience Letters. 1981;26:301–305. doi: 10.1016/0304-3940(81)90149-x. [DOI] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. American Journal of Physiology. 1997;273:C1516–1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Qian H, Dowling JE. GABAA and GABAC receptors on hybrid bass retinal bipolar cells. Journal of Neurophysiology. 1995;74:1920–1928. doi: 10.1152/jn.1995.74.5.1920. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Roska B, Nemeth E, Orzo L, Werblin FS. Three levels of lateral inhibition: a space-time study of the retina of the tiger salamander. Journal of Neuroscience. 2000;20:1941–1951. doi: 10.1523/JNEUROSCI.20-05-01941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiological Reviews. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Satoh H, Kaneda M, Kaneko A. Intracellular chloride concentration is higher in rod bipolar cells than in cone bipolar cells of the mouse retina. Neuroscience Letters. 2001;310:161–164. doi: 10.1016/s0304-3940(01)02120-6. [DOI] [PubMed] [Google Scholar]
- Schwartz EA. Depolarization without calcium can release gamma-aminobutyric acid from a retinal neuron. Science. 1987;238:350–355. doi: 10.1126/science.2443977. [DOI] [PubMed] [Google Scholar]
- Shields CR, Tran MN, Wong RO, Lukasiewicz PD. Distinct ionotropic GABA receptors mediate presynaptic and postsynaptic inhibition in retinal bipolar cells. Journal of Neuroscience. 2000;20:2673–2682. doi: 10.1523/JNEUROSCI.20-07-02673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg RH, Oakley B, II, Niemeyer G. Light-evoked changes in [K+]o in retina of intact cat eye. Journal of Neurophysiology. 1980;44:897–921. doi: 10.1152/jn.1980.44.5.897. [DOI] [PubMed] [Google Scholar]
- Stone S, Schutte M. Physiological and morphological properties of off- and on-center bipolar cells in the Xenopus retina: effects of glycine and GABA. Visual Neuroscience. 1991;7:363–376. doi: 10.1017/s0952523800004855. [DOI] [PubMed] [Google Scholar]
- Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. Journal of Neuroscience. 2000;20:7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Kaneko A. γ-Aminobutyric acid exerts a local inhibitory action on the axon terminal of bipolar cells: evidence for negative feedback from amacrine cells. Proceedings of the National Academy of Sciences of the USA. 1987;84:3501–3505. doi: 10.1073/pnas.84.10.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Attwell D, Mobbs P, Wilson M. Membrane currents in retinal bipolar cells of the axolotl. Journal of General Physiology. 1988;91:49–72. doi: 10.1085/jgp.91.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Relative contributions of passive equilibrium and active transport to the distribution of chloride in mammalian cortical neurons. Journal of Neurophysiology. 1988;60:105–124. doi: 10.1152/jn.1988.60.1.105. [DOI] [PubMed] [Google Scholar]
- Tian N, Slaughter MM. Pharmacological similarity between the retinal APB receptor and the family of metabotropic glutamate receptors. Journal of Neurophysiology. 1994;71:2258–2268. doi: 10.1152/jn.1994.71.6.2258. [DOI] [PubMed] [Google Scholar]
- Vardi N, Sterling P. Subcellular localization of GABAA receptor on bipolar cells in macaque and human retina. Vision Research. 1994;34:1235–1246. doi: 10.1016/0042-6989(94)90198-8. [DOI] [PubMed] [Google Scholar]
- Vardi N, Zhang LL, Payne JA, Sterling P. Evidence that different cation chloride cotransporters in retinal neurons allow opposite responses to GABA. Journal of Neuroscience. 2000;20:7657–7663. doi: 10.1523/JNEUROSCI.20-20-07657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TQ, Payne JA, Copenhagen DR. Localization and developmental expression patterns of the neuronal K-Cl cotransporter (KCC2) in the rat retina. Journal of Neuroscience. 2000;20:1414–1423. doi: 10.1523/JNEUROSCI.20-04-01414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werblin FS. Transmission along and between rods of the tiger salamander retina. Journal of Physiology. 1978;280:449–470. doi: 10.1113/jphysiol.1978.sp012394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werblin FS, Dowling JE. Organization of the retina of the mudpuppy, Necturus maculosus. II. Intracellular recording. Journal of Neurophysiology. 1969;32:339–355. doi: 10.1152/jn.1969.32.3.339. [DOI] [PubMed] [Google Scholar]
- Wotring VE, Chang Y, Weiss DS. Permeability and single channel conductance of human homomeric ρ1 GABAC receptors. Journal of Physiology. 1999;521:327–336. doi: 10.1111/j.1469-7793.1999.00327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XL, Wu SM. Feedforward lateral inhibition in retinal bipolar cells: input-output relation of the horizontal cell-depolarizing bipolar cell synapse. Proceedings of the National Academy of Sciences of the USA. 1991;88:3310–3313. doi: 10.1073/pnas.88.8.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]