

Abstract
Information processing in the nervous system is achieved primarily at chemical synapses between neurons. Recent evidence suggests that glia-neuron interactions contribute in multiple ways to the synaptic process. In the present study we used the frequency of spontaneous postsynaptic currents (sPSC) in Purkinje neurons in acute cerebellar brain slices from juvenile rats (13-19 days old) as a measure of synaptic activity. Following 50 depolarizing pulses to an adjacent Bergmann glial cell (50 mV; duration 0.5 s; 1 Hz) the sPSC frequency of the Purkinje neuron was reduced to 65 ± 7 % of control values within 10 min after glial stimulation and remained depressed for at least 40 min. Depolarizing pulses to 0 mV had a comparable effect (70 ± 5 % of control). The frequency of miniature PSCs, as recorded in 300 nm TTX, was not modulated after glial stimulation. Blockade of ionotropic glutamate receptors (iGluRs) with kynurenic acid (1 mm) or 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 5 μm) suppressed the reduction of neuronal activity induced by glial depolarization, whereas the glial modulation of synaptic activity was not inhibited by a block of N-methyl-d-aspartate iGluRs, metabotropic glutamate receptors, cannabinoid receptors or GABAB receptors. Fluorometric measurements of the intraglial Ca2+ concentration revealed no glial Ca2+ transients during the depolarization series, and glial cell stimulation reduced the neuronal sPSC frequency even after loading the glial cell with 20 mm of the Ca2+ chelator BAPTA. Our results indicate a glia-induced long-lasting depression of neuronal communication mediated by iGluRs.
In the last 20 years, the image of glial cells has changed from ‘deaf servants’ of neurons to ‘listening coworkers’ that are endowed with means to perceive the communication of surrounding neurons. They express receptors for most neuronal signalling transmitters and communicate with each other by Ca2+ waves (for review see Deitmer et al. 1998; Verkhratsky et al. 1998). Recently, experiments with co-cultures of neurons and glial cells generated the idea of glia as a neuronal ‘trialog partner’ (Araque et al. 1999). It was shown that astroglial cells not only recognize neuronal activity (Clark & Barbour, 1997; Kulik et al. 1999) but also modulate neuronal properties, such as intracellular Ca2+ transients or electrical excitability (Pasti et al. 1997; Parpura & Haydon, 2000; Haydon, 2001). In addition to the slow modulatory influence on neuronal development via growth factors and other neuromodulators released by glial cells, astrocytes have several faster ways for signalling neuronal neighbours and, thus, for influencing neuronal cross-talk. Possible mechanisms are substrate transfer through gap junctions between neurons and glia (Alvarez-Maubecin et al. 2000), or glial release of ATP (Cotrina et al. 2000) or glutamate, which may be released in a Ca2+-dependent manner, presumably by use of vesicular exocytosis (Calegari et al. 1999; Maienschein et al. 1999; Parpura & Haydon, 2000). Another mode of glial modification of neuronal transmission could be a modulation of transmitter clearance by astrocytes (Bordey & Sontheimer, 2000), since a block of glial glutamate uptake can prolong postsynaptic currents in adjacent neurons (Mennerick & Zorumski, 1994; Bergles & Jahr, 1998). In pyramidal cells of hippocampal brain slices, astrocytes were found to modulate the frequency of miniature inhibitory postsynaptic currents by a pathway that depended on glutamate receptor activity (Kang et al. 1998).
In the cerebellum, Bergmann glial cells are positioned in the direct neighbourhood of the Purkinje cell somata and their processes pass through the molecular layer with very close contact to dendrites and synapses of the Purkinje cells (Grosche et al. 1999). This intimate morphological relation of both cell types makes these cells a preferred model for studying glia-to-neuron signalling. The functional significance of Bergmann glia for motor coordination was shown by astrocyte ablation in adult mice (Cui et al. 2001).
In the present study, we used cerebellar brain slices from juvenile rats to investigate the influence of glial activity on the neuronal communication in double patch-clamp recordings. The spontaneous postsynaptic currents (sPSC) recorded in a Purkinje neuron were used as a measure of neuronal activity. Most of these sPSCs are inhibitory (Konnerth et al. 1990; Farrant & Cull-Candy, 1991) and originate from local interneurons (Llinas & Walton, 1990) in the molecular layer. Our results indicate that a series of depolarizing voltage steps to an adjacent voltage-clamped Bergmann glial cell reduced the sPSC frequency in the Purkinje cell. This glial modulation of neuronal activity was suppressed by blocking AMPA/kainate receptors, indicating a key role of glutamate and iGluRs. Preliminary reports have been given in abstract form (Brockhaus & Deitmer, 2001a,b).
Methods
Cerebellar brain slices were obtained from juvenile rats (13-19 days old) following a standard procedure (Edwards et al. 1989). The rats were bred and housed in our facility in accordance with the current German animal protection laws. The killing protocol was approved by the regional animal care and use committee (Landesuntersuchungsamt Rheinland-Pfalz, Germany). In short, after decapitation the cerebellum was isolated in ice-cold bath solution (see below for composition) with a reduced concentration of CaCl2 (0.5 mm; MgCl2 increased to 2.5 mm). Sagittal slices of the vermis (300 μm thick) were obtained by use of a vibratome (Campden, Loughborough, UK) and stored in the same solution, gassed with carbogen (95 % O2 −5 % CO2) for 1 h at 30 °C and later at room temperature (21-24 °C).
For electrophysiological recordings of a Purkinje neuron and an adjacent Bergmann glial cell, slices were fixed in a recording chamber with a U-shaped platinum wire and nylon grid on the stage of an upright microscope (Axioscope, Zeiss, Oberkochen, Germany). The chamber (0.5 ml) was continuously perfused (3-4 ml min−1) with carbogen-gassed bath solution containing (mm): NaCl 125, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, MgCl2 1, CaCl2 2, glucose 25 and d-lactate 0.5. Recordings were controlled with pCLAMP6 software (Axon Instruments, Foster City, CA, USA) with a digidata 1200 interface and an Axopatch-1D amplifier (Axon Instruments) for the neuron and an EPC-7 amplifier (HEKA, Lambrecht, Germany) for the glial cell. Pipettes were pulled with a horizontal puller (P-87, Brown & Sutter, Novato, CA, USA) and heat-polished to a tip resistance of 2-3.5 MΩ for the neuron, filled with an intracellular solution containing (mm): CsCl 120, tetraethylammonium chloride 20, MgCl2 2, Na-ATP 2, EGTA 10 and Hepes 10; pH adjusted to 7.3 with CsOH. This solution strongly reduced the background K+ conductance and allowed a better observation of sPSCs (Konnerth et al. 1990). The patch pipettes for glia recordings had a tip resistance of 3-6 MΩ when filled with (mm): potassium gluconate 140, MgCl2 1, CaCl2 1, EGTA 11, Na-ATP 2 and Hepes 10; pH adjusted to 7.3 with KOH. In a few experiments, glial intracellular Ca2+ transients were suppressed by use of a pipette solution containing 20 mm BAPTA instead of EGTA. Series resistance was 4-12 MΩ for the Purkinje neuron recordings (compensation 80-90 %) and 6-16 MΩ for the Bergmann glia recording (compensation 55-70 %). In some experiments, the spontaneous firing rate of interneurons was recorded extracellularly with a bath solution-filled patch-clamp electrode in the loosely attached patch configuration.
All drugs, applied via bath solution, were obtained from Sigma-Aldrich (Taufkirchen, Germany) except that 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM-251), (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP55845) and kynurenic acid (KynA) were from Tocris (Bristol, UK) and tetrodotoxin (TTX) was purchased from Alomone Labs (Jerusalem, Israel). CNQX, AM-251, CGP55845, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxalline-7-sulphonamide (NBQX), dl-2-amino-5-phosphonovaleric acid (APV) and (+)-α-methyl-4-carboxyphenylglycine ((+)-MCPG) were added from stock solutions in dimethyl sulphoxide (DMSO); the final concentration of DMSO did not exceed 0.1 %.
Electrical stimulation of Bergmann glia consisted of a train of depolarizing stimuli. The standard stimulation protocol was a series of 50 voltage steps from the holding potential (-70 mV) to +50 mV. We normally used this strong depolarization because of the expected voltage decline along the thin, long processes and especially at the stalks of microdomains (Grosche et al. 1999), to achieve significant depolarizations even in more distal parts of the cell (see also Kang et al. 1998). However, the very complex morphology of Bergmann glial processes with main dendrites going up to the glial endfeet at the pia mater and microdomains, which were connected to the principal processes via very fine stalks, does not allow a precise estimation of the voltage decay. A recent study (Grosche et al. 2002) even suggested that the microdomains are virtually uncoupled electrically from the main dendrites. The voltage steps in our standard protocol lasted 500 ms and were applied at a frequency of 1 Hz. Alternatively, the cells were depolarized to 0 or −20 mV instead of +50 mV or the voltage steps of a train were divided into triplets consisting of three shorter steps of 100 ms duration with 100 ms intervals. Consistent with the standard protocol, 50 triplets were applied at 1 Hz.
The sPSC frequency in the Purkinje neurons was calculated from continuous recordings (sampling rate 2-5 kHz; filter 1-2 kHz; duration 50 s) with pCLAMP6 software with a threshold in the first derivative of the current. Threshold values were set at twice the noise maximum, normally 16-25 pA ms−1. The first derivative was used to minimize problems with slow changes of the baseline level and allowed the counting of postsynaptic events that occurred during the decay phase of a previous sPSC. To test the reliability of this method, the sPSC frequency of five experiments was evaluated from the original recordings with software for analysis of synaptic currents (MiniAnalysis, Synaptosoft Inc., Decatur, GA, USA); this led to very similar results. Data are given as arithmetic means ± s.e.m. Significance was calculated with PlotIT (Scientific Programming Enterprises, Haslett, MI, USA) and was determined at P < 0.05 (*) or ‘highly significant’ at P < 0.01 (**).
In some experiments, fluorescent dyes were added to the pipette solutions for observation of the spatial relationship between a Purkinje neuron (Alexa 568, 200 μm; Molecular Probes, Eugene, OR, USA) and an adjacent Bergmann glial cell (Alexa 488, 200 μm). After the experiment, slices were fixed overnight in 4 % paraformaldehyde in PBS, in part after pretreatment with 1-ethyl-3-(3-methylaminopropyl)carbodiimide (50 mg ml−1 bath solution; modified from Tymianski et al. 1997) for 40 min to increase dye binding to the stained cells. The morphological relation was then studied with a confocal laser-scanning microscope (LSM 510, Zeiss, Oberkochen, Germany). Images were obtained using a ×40 water immersion lens (Zeiss). For each cell pair, a stack of optical sections completely covering the stained Purkinje neuron and Bergmann glial cell was acquired with an axial interval of 1-2 μm. The image stack was deconvoluted using Huygens software (Bitplane, Zürich, Switzerland) and reconstructed three-dimensionally using Imaris software (Bitplane).
For microfluorometric measurements of intracellular Ca2+ concentration changes the Ca2+ indicator fura-2 (100 μm; Molecular Probes) was added to a patch pipette solution containing (mm): KCl 145, MgCl2 1, EGTA 0.5, Na-ATP 2 and Hepes 10; pH adjusted to 7.3 with KOH. Experiments were performed with an imaging system (T.I.L.L. Photonics, Martinsried, Germany) equipped with a scanning monochromator. The fura-2 was excited alternately at 340 and 380 nm for 5-10 ms and the fluorescence intensity (>510 nm) images were recorded at 0.5 or 1 Hz. The intensity ratio (340 : 380 nm), which indicates the intracellular Ca2+ concentration, was calculated from different regions of interest (as indicated in Fig. 6B).
Figure 6. Effects of glial stimulation on the glial cell itself.
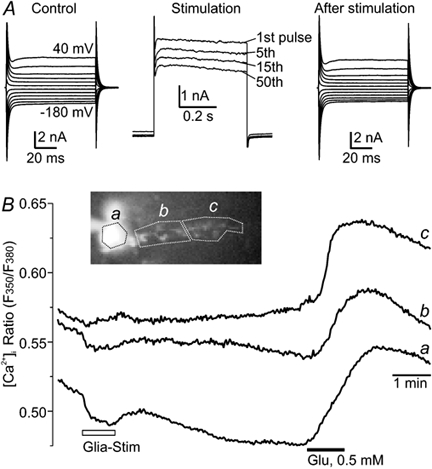
A, membrane currents of the Bergmann glia before, during and 3 min after the stimulation, which did not change membrane currents of the glial cell itself. Glial membrane currents that were induced by the 0.5 s voltage steps to +50 mV decline moderately within a stimulation series consisting of 50 of these depolarizations (example traces given in the middle). B, fluorometric measurement of intracellular Ca2+ concentration changes in a fura-2-loaded Bergmann glial cell during a patch-clamp experiment showed that the depolarization series did not induce an increase in intracellular Ca2+ in the soma (c) or proximal processes (b) and only minute changes in distal parts (a). Bath-applied glutamate, in contrast, evoked the expected Ca2+ response in all regions of interest chosen (a-c).
Results
In acute cerebellar brain slices from 13- to 19-day-old rats, a Purkinje neuron and an adjacent Bergmann glial cell were voltage clamped in the whole cell mode of the patch-clamp technique to investigate glial influence on neuronal communication (Fig. 1A). Confocal reconstruction of six cell pairs showed a strong overlay of dendritic areas of the Purkinje neuron and the glial processes (Fig. 1B), but the processes of a single Bergmann glial cell covered only part of the Purkinje neuron dendritic tree. Intensive dye coupling of Bergmann glial cells was not observed in our experiments, since the loading of one Bergmann glial cell induced only a weak loading of a maximum of three neighbouring Bergmann glial cells. In experiments where only one cell was dye loaded, either a Purkinje neuron (n = 7) or a Bergmann glial cell (n = 9), we never observed dye coupling between neuron and glial cell.
Figure 1. Morphological relation of Bergmann glia and Purkinje neurons.
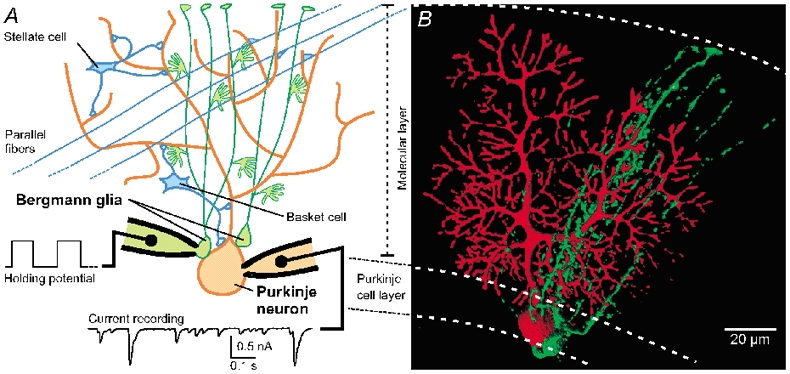
A, schematic drawing of a Purkinje neuron (red) and Bergmann glial cells (green) with close somatic contact. The somata of both cell types are positioned in the Purkinje cell layer and their processes project throughout the molecular layer up to the pia mater. Paired recordings of a Purkinje neuron and a Bergmann glial cell allowed the study of the influence of glial cells on the spontaneous synaptic input to the neighbouring neuron. This input arises predominantly from the local inhibitory interneurons (basket and stellate cells) and not from the excitatory parallel fibre synapses. B, three-dimensional reconstruction of a double labelling of a Bergmann glial cell (Alexa 488, green) and a Purkinje neuron (Alexa 568, red) showing that the processes of a Bergmann glial cell overlap with part of the dendritic tree of a Purkinje neuron.
Spontaneous postsynaptic currents in Purkinje neurons
Purkinje neurons, voltage clamped at −70 mV, show a high rate of spontaneous postsynaptic currents (sPSC), originating mainly from local interneurons (stellate and basket cells, Fig. 1; Llinas & Watson, 1990). Due to the high internal Cl− concentration, inhibitory and excitatory inputs both induced inward currents at this holding potential. The frequency of the sPSCs was recorded as a measure of presynaptic neuronal activity. The mean frequency of the sPSCs was 7.7 ± 0.6 Hz (n = 102), as counted by using the first derivative of the original trace (see Methods). This minimized problems caused by small changes of the baseline holding current during prolonged recordings and avoided the possibility of missing sPSCs during the decay phase of an immediately preceding sPSC (Fig. 2A). The amplitude distribution of the sPSCs had a wide range, with maximal values above 5 nA and a mean amplitude of 189 ± 32 pA (n = 10 cells, with >200 single sPSCs for each cell.
Figure 2. Spontaneous synaptic input to Purkinje neurons.
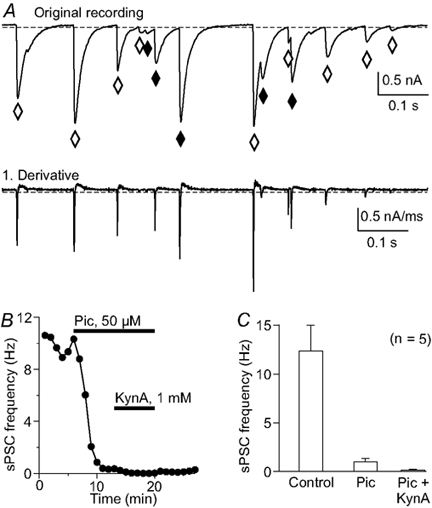
A, the original recording shows several spontaneous postsynaptic currents (sPSC) that would be missed by counting with a standard threshold method (♦) due to their initialization during the recovery phase of the previous PSC. Counting events using the first derivative of the original trace (lower panel in A) overcomes this problem. ⋄, events that are detected by both methods. B, the frequency of sPSCs after blocking inhibitory and excitatory receptors. Most events are identified as GABAergic using a block with picrotoxin (Pic; 50 μm); the remaining sPSCs were blocked by the iGluR antagonist kynurenic acid (KynA; 1 mm). Each point indicates the mean sPSC frequency of a 50 s continuous recording, counted with the method described in A. C, histogram summarizing the sPSC frequency reduction from 5 experiments like that in B with the same antagonists.
In order to identify inhibitory and excitatory synaptic input we used pharmacological tools to block GABAA and ionotropic glutamate receptors (Fig. 2). Most (93 ± 3 %) of the sPSCs were inhibitory, as indicated by a block with 50 μm picrotoxin; the remaining sPSCs were inhibited by 1 mm kynurenic acid (n = 6; Fig. 2B and C).
A block of AMPA/kainate-type glutamate receptors with 5 μm CNQX induced a transient increase of the sPSC frequency to 348 ± 39 % and a plateau phase of 189 ± 15 % (n = 14, not shown) of the control value after 9 min. This increase was not abolished by a combined application of CNQX and the NMDA receptor antagonist AP5 (50 μm; n = 4). In contrast, in the presence of NBQX (n = 7) or the less specific glutamate receptor antagonist kynurenic acid (KynA, 1 mm; n = 12), the sPSC frequency did not increase as in CNQX. The CNQX-specific effect on the sPSC frequency of Purkinje neurons resembles that previously described for IPSCs in CA3 pyramidal cells (McBain et al. 1992), but has not been studied any further here.
Glial cell stimulation modulates neuronal communication
During the recording of sPSCs in a Purkinje neuron, an adjacent Bergmann glial cell was stimulated with trains of 50 depolarizing voltage steps that repetitively clamped the glial cell to +50 mV for 0.5 s at 1 Hz. This large, presumably unphysiological, depolarization was chosen to ensure a potential change of at least +50 to +80 mV even in the fine distal cell processes of the Bergmann glial cells (see Methods). After this glial cell stimulation, the sPSC frequency of the Purkinje cell began to decrease in 17 out of 22 paired recordings (Fig. 3). Ten minutes after the stimulation period, the sPSC frequency was reduced to 65 ± 5 % of control values (n = 17; P < 0.01) and remained at this level (67 ± 7 % after 20 min, n = 8; 67 ± 3 % after 30 min, n = 7; Fig. 3B). This indicates that the glial cell depolarization activated a long-lasting reduction of the sPSC frequency of the neighbouring Purkinje neuron. The frequency reduction did not recover during the observation period, which lasted up to 40 min, and was not accompanied by a change in the holding current in the neuron. A second glia stimulation 12-20 min after the first stimulation did not induce a further decrease in the sPSC frequency (n = 6, not shown).
Figure 3. Modulation of synaptic neuronal activity by stimulation of an adjacent Bergmann glial cell.
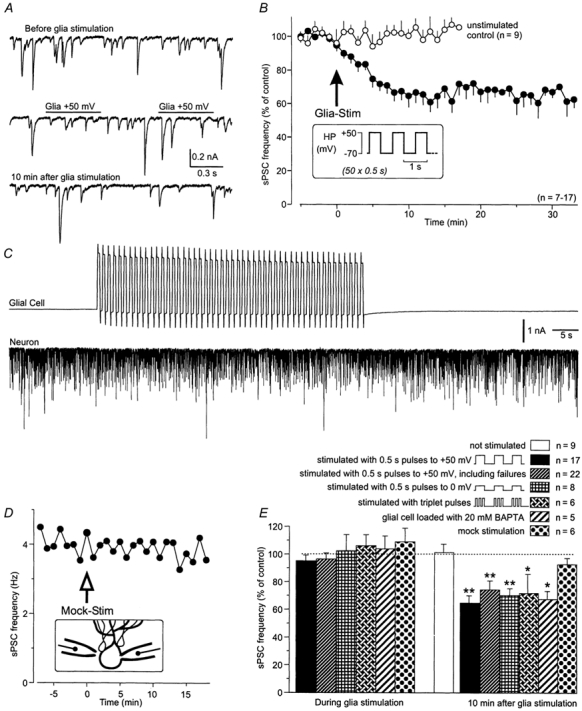
A, continuous recordings of sPSCs in a Purkinje neuron before, during and after stimulation of a neighbouring Bergmann glia. B, modulation of the sPSC frequency of Purkinje neurons (•) following a series of depolarizations of adjacent Bergmann glia (50 voltage steps from −70 to +50 mV for 0.5 s, at 1 Hz; see inset). The sPSC frequency (the mean frequency of 5 min control period = 100 %) decreased within 10 min after glial stimulation and remained at the reduced level for more than 30 min. In control experiments without glial cell stimulation the sPSC frequency remained unchanged (n = 9; ○). C, continuous recording of glial and neuronal membrane currents during a glial stimulation period. D, using the same protocol with the patch pipette close to a Purkinje cell soma (‘mock stimulation’; see inset) the sPSC frequency was not affected. E, histogram showing the changes in sPSC frequency under different experimental conditions: no glia stimulation; glia stimulated with 0.5 s pulses to +50 mV (standard) and to 0 mV; and including the 5 paired recordings in which no effect was observed after glia stimulation (‘failures’); glia stimulated with triplet pulses to +50 mV; glial Ca2+ transients suppressed by loading with 20 mm BAPTA; and mock stimulation. The mean sPSC frequency over a period of 5 min before glial stimulation was used as the control value for each condition. Asterisks indicate significant difference from the unstimulated control.
In a series of experiments, the glial cell was depolarized to 0 or −20 mV instead of +50 mV. Glial depolarization to 0 mV reduced the sPSC frequency of the Purkinje neuron by 30 ± 5 % after 10 min (P < 0.01; n = 9; Fig. 3E). However, a depolarization series consisting of 50 pulses to −20 mV failed to decrease the sPSC frequency significantly (92 ± 8 % of control values; n = 6). Glial cell stimulation with triplet pulses (each 0.5 s pulse substituted by a triplet of three 0.1 s pulses; see Methods) had a similar effect on the spontaneous synaptic activity compared to the standard protocol; 10 min after glial stimulation with 50 triplet pulses to +50 mV, the sPSC frequency was reduced by 28 ± 13 % (n = 6; P < 0.05; Fig. 3E). In contrast, neither a ‘mock stimulation’ (n = 6; standard stimulation protocol with a patch pipette next to a Bergmann glial cell without voltage clamping it; see Fig. 3D inset and Fig. 3E), nor hyperpolarizing the glial cell from −70 to −190 mV (n = 6, not shown) decreased the sPSC frequency significantly. Stimulation of a more distant Bergmann glial cell (≈100 μm distance) also had no significant effect on the sPSC frequency (91 ± 8 % of control; P > 0.05; n = 5; not shown).
To evaluate the importance of intracellular Ca2+ transients in the depolarized glial cell for the glia-to-neuron signalling, we clamped the Bergmann glial cell with a pipette solution containing 20 mm of the Ca2+ chelator BAPTA, to suppress an increase of the intracellular Ca2+ concentration. In paired recordings with blocked glial Ca2+ transients, the reduction of neuronal activity 10 min after glial stimulation averaged 32 ± 5 % (n = 5; Fig. 3E; 1 additional cell pair without reduction of sPSC frequency) and was not significantly different from the values obtained in experiments using BAPTA-free pipette solution. This indicates that an increase in intraglial Ca2+ is not likely to be involved in the reduction of sPSC frequency.
Within the 50 s period of glial cell stimulation, the frequency of the sPSCs was not significantly modified (95 ± 4 % of control, n = 17). No shift in the Purkinje cell holding current at −70 mV was observed, either during glial cell depolarization or throughout the depolarization series (Fig. 3A and C).
As stated above, in 5 out of 22 paired recordings we observed no effect of glial depolarization on the sPSC frequency of the Purkinje neuron. This could have been due to the morphological relation of the chosen cell pair, since in one out of six paired recordings with fluorescently labelled pipette solutions, we found no overlap of the dendritic areas of the Purkinje neuron and the processes of the Bergmann glial cell chosen for recording. In this paired recording no sPSC frequency decrease was observed after glial stimulation, although the somata were positioned next to each other. When these recordings showing no glia-induced reduction of the neuronal sPSC frequency were included in the evaluation, the sPSC frequency was still significantly reduced to 74 ± 7 % after glial stimulation (n = 22; P < 0.01; Fig. 3E).
A postsynaptic locus of a synaptic modulation or modified transmitter uptake is normally indicated by changes in the kinetics or amplitude of PSCs. Analysis of the rise time and the amplitude of more than 250 sPSCs before and 10 min after the glial cell stimulation (n = 3), however, showed no differences in the rise time, the decay time or the amplitude distribution as indicated by an overlay of the traces in cumulative plots (Fig. 4A-C).
Figure 4. The sPSC kinetics and miniature PSCs are not affected by glial stimulation.

Comparing the rise time (A), amplitude distribution (B) and decay time (C) of >250 consecutive single sPSCs in cumulative plots before and 10 min after glial cell stimulation shows that these parameters were not changed. D, the frequency of miniature PSCs, as isolated by blocking action potential firing with TTX (300 nm), was not reduced after glial cell stimulation.
The effect of glial stimulation on miniature PSCs (mPSCs) was measured during a block of sodium currents with TTX (300 nm). TTX blocked 83 ± 8 % of the sPSCs of the Purkinje neurons, but stimulation of an adjacent Bergmann glial cell did not significantly change the frequency of the remaining mPSCs (97 ± 17 % of control; n = 7; Fig. 4D). Thus, while glial stimulation affected sPSCs dependent on neuronal activity, it had no effect on spontaneous transmitter release from the nerve terminals forming synapses with Purkinje neurons.
Single depolarizing pulses to a Purkinje cell (1-5 s) transiently reduce the sPSC frequency, an effect that has been described as ‘depolarization-induced suppression of inhibition’ (DSI; Llano et al. 1991; Alger & Pitler, 1995). Recent reports identified cannabinoids as the retrograde messenger released by Purkinje neurons, showing that DSI can be inhibited by the cannabinoid CB1 receptor antagonist AM251 (Kreitzer & Regehr, 2001; Diana et al. 2002). To investigate a possible relation of DSI with the glia-to-neuron signalling described here, the effect of a DSI-inducing depolarization of the Purkinje neuron (0 mV, 2 s) on the sPSC frequency was compared before and 12-15 min after glial cell stimulation. The sPSC frequency in the first 10 s after the pulse was reduced by 41 ± 5 % under control conditions. After glial cell stimulation the same DSI-inducing depolarization of the Purkinje neuron suppressed the sPSC frequency by 38 ± 6 % (n = 5; Fig. 5A and B), which was not significantly different from control values. However, DSI was suppressed by the CB1 antagonist AM251, as also shown by Kreitzer & Regehr (2001) and by Diana et al. (2002). In contrast, the reduction of the sPSC frequency induced by glial stimulation was not significantly different after induction of DSI or in the presence of the CB1 antagonist AM251 (23 ± 3 %; n = 7; Fig. 5C and D; P > 0.05, compared to the glial effect on the sPSC frequency without the antagonist). Thus, DSI and the long-lasting glia-to-neuron signalling described here do not appear to use the same pathways.
Figure 5. Depolarization-induced suppression of inhibition (DSI).
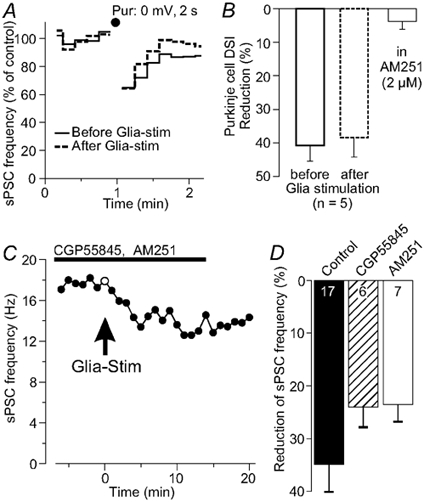
A, depolarizing a Purkinje neuron for 2 s to 0 mV (•) transiently reduced the sPSC frequency by a mechanism known as DSI. This reduction occurred before and after a glial cell stimulation with similar strength. B, histogram showing that the reduction in the sPSC frequency in the first 10 s after depolarizing the Purkinje neuron is not modified after stimulation of an adjacent Bergmann glial cell, whereas the CB1 receptor antagonist AM251 eliminated DSI. C, during a combined block of GABAB and CB1 receptors with CGP55845 and AM251, respectively, glial stimulation still reduced the sPSC frequency of Purkinje neurons. D, reduction of the sPSC frequency following application of either CGP or AM251, indicating that GABAB and CB1 receptors do not mediate the glial modulation of the synaptic activity; values for n include two experiments using a cocktail of both receptor antagonists as shown in C.
The glial influence on synaptic force from GABAergic interneurons to pyramidal cells in the hippocampus included activity of GABAB receptors (Kang et al. 1998). Glial stimulation still reduced the sPSC frequency of the Purkinje neuron in the presence of the GABAB antagonist CGP55845 (2 μm) by 24 ± 4 % (n = 6; Fig. 5C and D). This reduction showed no significant difference from the reduction of the sPSC frequency under control conditions (P > 0.05).
Modulation of interneuron activity
The finding that miniature PSCs were not affected by the glial cell stimulation suggests an influence on the spontaneous activity of presynaptic interneurons. We therefore recorded the firing rate of interneurons in the molecular layer near to the voltage-clamped Bergmann glial cell extracellularly. The mean action potential frequency of the interneurons in the molecular layer was 5.3 ± 1.1 Hz under control conditions, and it was significantly reduced 10 min after glial stimulation with the standard series of depolarizing pulses to +50 mV (4.0 ± 1.0 Hz; P < 0.05; n = 6). Thus, Bergmann glial stimulation reduced the spontaneous interneuron firing by 29 ± 8 % (n = 6). However, no direct effect on the firing frequency was observed during glial depolarization (98 ± 3 % of control).
Effect of depolarization series on the Bergmann glia itself
The Bergmann glial cells showed outward currents in response to the depolarizations that declined by 17 ± 4 % (n = 18) during the stimulation series lasting 50 s, while the baseline current was shifted to more negative values (Fig. 3C and Fig. 6A). Similar results were obtained with triplet pulses (not shown). Glial membrane currents, however, were not permanently modified by the depolarization series (compare left and right part of Fig. 6A), indicating that the strong depolarizing pulses themselves did not modulate the neuronal cross-talk by damaging the glial cells.
Fluorometric measurements of the intraglial Ca2+ concentration with the Ca2+ indicator fura-2 (100 μm) showed only minor or no increases of the intracellular Ca2+ concentration as a result of glial stimulation (n = 6, Fig. 6B) compared to the transients induced by glutamate (0.5 mm; n = 4) or ATP (0.1 mm; n = 3; not shown). This result, which was obtained with a pipette solution with reduced Ca2+ buffering capacity, further indicates that intraglial Ca2+ transients were not part of the glia-to-neuron signalling cascade. A minor Ca2+ increase in the most distal part of the Bergmann glial cell, as seen in the recording in Fig. 6Bc, was observed in only two of six cells and may be a secondary effect of an increased extracellular concentration of signalling molecules, as would be expected, for example, with glutamate (Müller et al. 1992).
Glutamate receptors as mediators of glia-to-neuron signalling
A possible candidate for glia-to-neuron signalling is glutamate, which was found to be essential for the glial modulation of synaptic transmission in cell culture (Araque et al. 1998; Haydon, 2001) and in a hippocampal slice preparation (Kang et al. 1998). If glutamate is involved in the sPSC frequency reduction induced by glial depolarization, a blockade of ionotropic or metabotropic glutamate receptors (iGluR, mGluR) should affect the frequency decrease as observed in our experiments. Glial stimulation reduced the neuronal sPSC frequency during a block of mGluRs with 0.5 mm (+)-MCPG by 33 ± 6 % (10 min after glial stimulation; n = 5; Fig. 7D and F) and during a block of NMDA-iGluRs with 50 μm APV by 30 ± 5 % (n = 5; Fig. 7E), which is not significantly different from the control experiments. However, when iGluRs were blocked with 5 μm CNQX (n = 5; Fig. 7A) or 5 μm NBQX (n = 6; Fig. 7B), both specific for AMPA/kainate type receptors, or 1 mm KynA (n = 5; Fig. 7C), the sPSC frequency reduction following glial stimulation was suppressed (Fig. 7F). This suggested an involvement of iGluRs of the fast AMPA/kainate type in the mechanism leading to the reduction of the neuronal sPSC frequency induced by glial stimulation.
Figure 7. Ionotropic glutamate receptors (iGluR) mediating glial modulation of sPSC frequency.
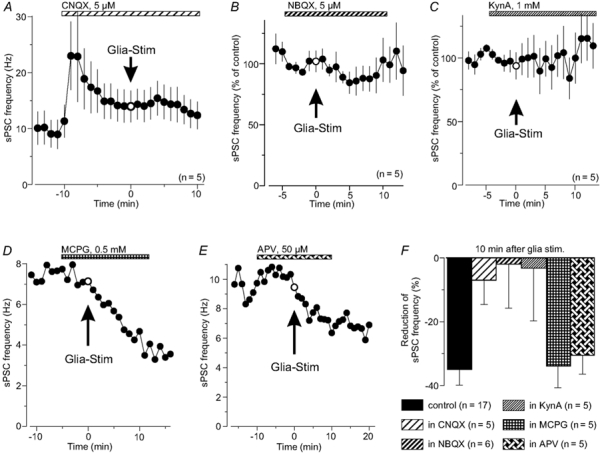
A, the sPSC frequency of a Purkinje neuron increased in response to CNQX (5 μm). In the presence of CNQX, stimulation of an adjacent glial cell has no influence on the sPSC frequency. B and C, similar effect of NBQX (5 μm) and KynA (1 mm), which also suppressed the modulation of sPSC frequency following glial stimulation. D, block of metabotropic glutamate receptors with (+)-MCPG (0.5 mm), which did not suppress the glial modulation of synaptic activity. E, a block of NMDA iGluRs with APV (50 μm) also failed to inhibit the glia-induced reduction of neuronal activity. F, histogram summarizing the changes of the sPSC frequency as induced by glial stimulation during a block of AMPA/kainate GluR with CNQX, NBQX or KynA, respectively, a block of NMDA receptors with APV or during a block of mGluRs with (+)-MCPG, indicating the role of iGluRs of the AMPA/kainate type for the synaptic modulation.
Bath application of glutamate (20-50 μm; n = 7; Fig. 8A and D) for 50 s or increasing extracellular K+ concentration from 2.5 to 27.5 mm (n = 8; Fig. 8B and D), both known to induce long-term depression (LTD) in Purkinje neurons (Konnerth et al. 1992; Daniel et al. 1998), did not mimic the long-lasting effect of glial cell stimulation on the sPSC frequency. Instead, both applications induced a more than threefold increase of the sPSC frequency followed by a transient reduction that lasted less than 3 min before frequency returned to control values. Thus, stimuli that are described as inducing LTD of evoked parallel fibre EPSCs do not induce a long-lasting modulation of the sPSC frequency as it is seen here following Bergmann glial stimulation. Since glial depolarization may result in a reduction of the electrogenic glutamate uptake and might thus reduce the sPSC frequency, we mimicked a condition of reduced glutamate uptake. In the presence of the uptake blocker dl-threo-β-hydroxyaspartic acid (THA, 200 μm, 1 min), the sPSC frequency transiently increased but was not significantly reduced 10 min later (n = 6; Fig. 8C and D). Thus, the effect of THA on the sPSC frequency was similar to that of glutamate but was different from the long-lasting glia-induced reduction of neuronal communication.
Figure 8. Mimicking possible effects of glial stimulation.
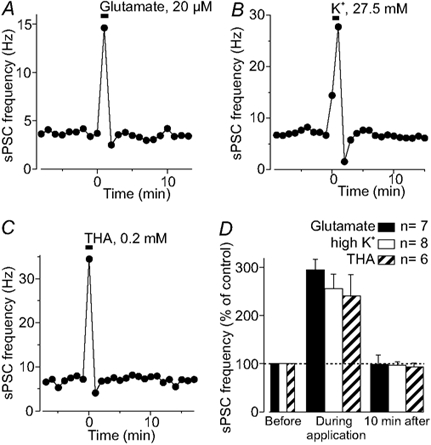
Bath application of glutamate (A) to mimic glial glutamate release, increasing the extracellular K+ concentration (B) or reducing glial glutamate uptake with THA (C) induced a transient increase of the sPSC frequency, but had no long-lasting effect, which is in contrast to the glial stimulation-induced long-lasting reduction of the sPSC frequency. D, histogram summarizing the changes in the sPSC frequency before, during and 10 min after application of the agents that mimic potential effects of glial stimulation.
Discussion
In paired patch-clamp recordings of Purkinje neurons and Bergmann glial cells, a focused electrical treatment of the glial cell was found to induce a decrease of the frequency of spontaneous synaptic input to the Purkinje neuron by one-third within 10 min. The sPSC frequency remained reduced over the duration of the experiment, which was up to 40 min. Activation of iGluRs was found to mediate this glia-induced modulation of neuronal activity.
Possible locus of glial influence on synaptic activity
The frequency of sPSCs depends on the spontaneous activity of presynaptic neurons, and accordingly an effect on presynaptic elements is assumed for modulation of the sPSC frequency. This implies that glial depolarization might affect the neuronal communication primarily at presynaptic sites. The observation of a reduced spontaneous firing of interneurons in the cerebellar molecular layer after glial stimulation, and the lack of glial modulation of miniature PSCs, as obtained in the presence of TTX, supports this view. The intimate co-localization of Purkinje neuron dendrites and processes of the Bergmann glial cell, however, suggests an involvement of presynaptic boutons next to the postsynaptic terminals of the Purkinje neuron as a target of the glial signalling.
Since the reduction of the sPSC frequency is suppressed during a block of iGluRs, one may speculate that glial stimulation specifically affected glutamatergic sPSCs. However, more than 90 % of the observed sPSCs were GABAergic, as indicated by a block with picrotoxin, and as described previously (Konnerth et al. 1990; Farrant & Cull-Candy, 1991). Since glial stimulation reduced the frequency by more than 30 %, most of these synaptic inputs affected by glial stimulation are likely to be inhibitory.
Implications for the role of iGluRs
Activation of iGluRs appeared essential for the glial modulation of neuronal communication as described here; thus, release of glutamate appears to be involved in this process. The simplest explanation is that the depolarized Bergmann glial cells release glutamate, which activates iGluRs on presynaptic terminals. Glutamate may be released from the glial cells by reversed glutamate transport (Szatkowski et al. 1990), following cell swelling (Rutledge & Kimelberg, 1996), or via Ca2+-dependent exocytosis (Pasti et al. 1997; Bezzi et al. 1998). The latter mechanism requires a rise in intraglial Ca2+, which was, however, neither observed during glial depolarization nor necessary for the glial modulation of the sPSC frequency. This is in line with the observation that Bergmann glial cells lack voltage-sensitive Ca2+ channels (Verkhratsky & Steinhäuser, 2000). In addition, the glial stimulation-induced reduction of sPSC frequency was not affected by suppression of intraglial Ca2+ transients by BAPTA.
Alternatively, depolarizing the Bergmann glial cell may induce a release of l-homocysteate (HCA), a glutamate-like excitatory amino acid that preferentially acts on NMDA receptors (Do et al. 1986, 1997), but that was shown also to modulate Purkinje cell currents via AMPA-type GluRs (Yuzaki & Connor, 1999). Thus, a modulation of neuronal communication in the cerebellum that is antagonized by iGluR blockers, as shown for the glia-to-neuron signalling in this study, could be mediated by HCA as well as by glutamate itself.
Another possible glia-derived neuromodulator is d-serine, which acts on the glycine site of NMDA receptors (Wolosker et al. 1999). However, bath application of NMDA increases the IPSC frequency in Purkinje cells (Farrant & Cull-Candy, 1991; Glitsch & Marty, 1999), which is in contrast to the decreased sPSC frequency following glial depolarization. In addition, the NMDA receptor antagonist APV had no effect on the glial sPSC frequency modulation.
Impaired glial uptake
The glial depolarization may also induce an increase in the extracellular glutamate concentration by impairing glial clearance of neuronally released glutamate. A suppression of glial glutamate uptake increased the duration of EPSCs by prolonging the presence of the transmitter in synaptic domains (Barbour et al. 1994; Mennerick & Zorumski, 1994; Bergles & Jahr, 1998; Iino et al. 2001). A depolarization of Bergmann glial cells has been reported to enhance both the amplitude and frequency of isolated spontaneous EPSCs in Purkinje cells (Bordey & Sontheimer, 2000), which was explained by glial K+ release and inhibition of glial glutamate uptake. The glutamate receptor-dependent glial influence on neuronal activity, described here, may increase extracellular glutamate in this way, although bath-applied glutamate was not able to mimic the effect of a glial cell stimulation. In contrast, glutamate induced a transient sPSC frequency increase of relatively short duration. Likewise, a block of glutamate uptake with THA induced a transient increase of the sPSC frequency but no long-lasting reduction as the glial depolarization did.
Comparison with hippocampal glia-to-neuron signalling
In hippocampal slices, the inhibitory synaptic input in CA1 pyramidal neurons was increased following a series of depolarizing stimuli to adjacent astrocytes (Kang et al. 1998). This potentiation of IPSCs depended both on activation of iGluRs and on intraglial Ca2+ transients. Potentiation of IPSCs was strongest in the first 5 min after glial stimulation and then declined with a time course of 15-20 min. The increase of inhibitory input in the pyramidal neurons, however, reversed to a decrease in the IPSC frequency with a comparable time course, when the intraglial Ca2+ transients were suppressed with thapsigargin or by intracellular BAPTA (20 mm). In our study in cerebellar slices, the synaptic input of Purkinje neurons decreased following glial cell depolarization. Since Bergmann glial cells did not show major Ca2+ transients in response to the depolarization in our experiments, the reduction of IPSCs described by Kang et al. (1998) when intraglial Ca2+ transients were suppressed may reflect the same mechanism that was found in this study. Hence, glial modulation of synaptic activity of the type reported here may be a more widespread glia-neuron interaction, and not limited to the Bergmann glia-to-Purkinje neuron interaction in the cerebellum.
Comparison with other modulations of the synaptic activity in the cerebellum
Depolarizing a Purkinje cell for a few seconds reduces its sPSC frequency for up to 2 min, an effect that has been described as ‘depolarization-induced suppression of inhibition’ (DSI; Llano et al. 1991; Alger & Pitler, 1995). This modulation involves a depolarization-induced calcium increase in the postsynaptic cell and activation of presynaptic metabotropic glutamate receptors (Glitsch et al. 1996) and cannabinoid receptors (Kreitzer & Regehr, 2001; Diana et al. 2002). If the glia-to-neuron signalling presented in this study employs the same mechanism as DSI, then DSI should be suppressed after glial stimulation. However, DSI was not impaired after glial cell depolarization, and the glia-to-neuron signalling was modulated neither by AM251 nor by MCPG; hence both forms of sPSC frequency reduction seem to employ independent signalling pathways. In addition, the sPSC frequency reduction brought about by glial cell stimulation lasted much longer than DSI and depended on activation of ionotropic glutamate receptors, in contrast to DSI.
Long-lasting, activity-dependent depression of synaptic transmission is referred to as LTD (for a recent review see Daniel et al. 1998). Here, we describe a long-lasting decrease of synaptic activity input of Purkinje neurons that was induced by glial cell depolarization. However, several of our findings are in contrast to classical LTD. The induction of the depression is not directly induced in the pre- or postsynaptic neuron. High potassium concentration or glutamate application, which are known to induce LTD at the parallel fibre-Purkinje cell synapse (Crepel et al. 1994; Daniel et al. 1998), had no long-lasting effect on the frequency of sPSCs, which mainly originate from the activity of local inhibitory interneurons. Classical LTD refers to a decreased efficacy of a given synaptic connection, whereas the lowered frequency described in this study may rather be due to reduced activity of several presynaptic neurons and may not be a feature of a single synaptic connection.
Physiological relevance of glia-to-neuron signalling
We can only speculate about the physiological relevance of the decrease in neuronal activity following a glial depolarization to at least 0 mV at this point. However, substantial glial depolarizations may occur, e.g. when glutamate, as released from neurons during neuronal activity, depolarizes the glial membrane potential by activating ionotropic receptors and electrogenic transporters. The possibility of a modulation of neuronal communication via a focused manipulation of glial cells may become relevant in the light of several recent findings. If astrocytes release glutamate, either in a Ca2+-dependent manner as shown for cultured cells (Pasti et al. 1997; Bezzi et al. 1998), or by other mechanisms, this would offer a new type of glia-to-neuron communication. If we assume that the glial depolarization in our experiments induced an increase of extracellular glutamate (or another iGluR agonist, e.g. homocysteate) and that this ‘glio-transmitter’ elicited the long-lasting reduction of neuronal synaptic activity, then a physiological stimulus that activates glial glutamate release, perhaps via intraglial Ca2+ transients, may use the same signalling cascade to modulate neuronal communication.
Acknowledgments
We thank Dr C. Lohr and R. Zur Nieden for help with the confocal imaging and for comments on the manuscript. S. Bergstein, D. Dressel and C. Hecker gave excellent technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (BR-1831.2-1 and DE-231/11-2).
References
- Alger BE, Pitler TA. Retrograde signaling at GABAA-receptor synapses in the mammalian CNS. Trends in Neurosciences. 1995;18:333–340. doi: 10.1016/0166-2236(95)93923-l. [DOI] [PubMed] [Google Scholar]
- Alvarez-Maubecin V, Garcia-Hernandez F, Williams JT, Van Bockstaele EJ. Functional coupling between neurons and glia. Journal of Neuroscience. 2000;20:4091–4098. doi: 10.1523/JNEUROSCI.20-11-04091.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. European Journal of Neuroscience. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends in Neurosciences. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. Journal of Neuroscience. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzino BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bordey AF, Sontheimer HW. Glial modulation of glutamatergic neurotransmission demonstrated by paired recordings in cerebellar slices. Society for Neuroscience Abstracts. 2000;26:707.15. [Google Scholar]
- Brockhaus J, Deitmer JW. Modulation of neuronal communication by stimulation of Bergmann glial cells in acute cerebellar slices from juvenile rats. In: Elsner N, Kreutzberg GW, editors. Göttingen Neurobiology Report 2001. Stuttgart: Thieme; 2001a. p. 768. [Google Scholar]
- Brockhaus J, Deitmer JW. Reduction of neuronal activity after glial stimulation. Society for Neuroscience Abstracts. 2001b;27:715.1. [Google Scholar]
- Calegari F, Coco S, Taverna E, Bassetti M, Verderio C, Corradi N, Matteoli M, Rosa P. A regulated secretory pathway in cultured hippocampal astrocytes. Journal of Biological Chemistry. 1999;274:22 539–22 547. doi: 10.1074/jbc.274.32.22539. [DOI] [PubMed] [Google Scholar]
- Clark BA, Barbour B. Currents evoked in Bergmann glial cells by parallel fibre stimulation in rat cerebellar slices. Journal of Physiology. 1997;502:335–350. doi: 10.1111/j.1469-7793.1997.335bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JH-C, López-García JC, Naus CCG, Nedergaard M. ATP-mediated glia signaling. Journal of Neuroscience. 2000;20:2835–2844. doi: 10.1523/JNEUROSCI.20-08-02835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel F, Audinat E, Daniel H, Hemart N, Jaillard D, Rossier J, Lambolez B. Cellular locus of the nitric oxide-synthase involved in cerebellar long-term depression induced by high external potassium concentration. Neuropharmacology. 1994;33:1399–1405. doi: 10.1016/0028-3908(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Cui W, Allen ND, Skynner M, Gusterson B, Clark AJ. Inducible ablation of astrocytes shows that these cells are required for neuronal survival in the adult brain. Glia. 2001;34:272–282. doi: 10.1002/glia.1061. [DOI] [PubMed] [Google Scholar]
- Daniel H, Levenes C, Crépel F. Cellular mechanisms of cerebellar LTD. Trends in Neurosciences. 1998;21:401–407. doi: 10.1016/s0166-2236(98)01304-6. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Verkhratsky AJ, Lohr C. Calcium signalling in glial cells. Cell Calcium. 1998;24:405–416. doi: 10.1016/s0143-4160(98)90063-x. [DOI] [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A. Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. Journal of Neuroscience. 2002;22:200–208. doi: 10.1523/JNEUROSCI.22-01-00200.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do KQ, Benz B, Sorg O, Pellerin L, Magistretti PJ. β-Adrenergic stimulation promotes homocysteic acid release from astrocyte cultures: evidence for a role of astrocytes in the modulation of synaptic transmission. Journal of Neurochemistry. 1997;68:2386–2394. doi: 10.1046/j.1471-4159.1997.68062386.x. [DOI] [PubMed] [Google Scholar]
- Do KQ, Herrling PL, Streit P, Turski WA, Cuenod M. In vitro release and electrophysiological effects in situ of homocysteic acid, an endogenous N-methyl-(d)-aspartic acid agonist, in the mammalian striatum. Journal of Neuroscience. 1986;6:2226–2234. doi: 10.1523/JNEUROSCI.06-08-02226.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Archiv. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Farrant M, Cull-Candy SG. Excitatory amino acid receptor-channels in Purkinje cells and thin cerebellar slices. Proceedings of the Royal Society B. 1991;244:179–184. doi: 10.1098/rspb.1991.0067. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Llano I, Marty A. Glutamate as a candidate retrograde messenger at interneurone-Purkinje cell synapses of rat cerebellum. Journal of Physiology. 1996;497:531–537. doi: 10.1113/jphysiol.1996.sp021786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. Journal of Neuroscience. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosche J, Kettenmann H, Reichenbach A. Bergmann glial cells form distinct morphological structures to interact with cerebellar neurons. Journal of Neuroscience Research. 2002;68:138–149. doi: 10.1002/jnr.10197. [DOI] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Möller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nature Neuroscience. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- Haydon PG. Glia: listening and talking to the synapse. Nature Reviews Neuroscience. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, Ozawa S. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nature Neuroscience. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Dreessen J, Augustine GJ. Brief dendritic calcium signals initiate long-lasting synaptic depression in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the USA. 1992;89:7051–7055. doi: 10.1073/pnas.89.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. Journal of Neuroscience. 2001;21:RC174. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik A, Haentzsch A, Luckermann M, Reichelt W, Ballanyi K. Journal of Neuroscience. 1999;19:8401–8408. doi: 10.1523/JNEUROSCI.19-19-08401.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synatic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Llinas RR, Walton KD. Cerebellum. In: Shepherd GM, editor. The Synaptic Organization of the Brain. Oxford: Oxford University Press; 1990. pp. 214–245. [Google Scholar]
- McBain CJ, Eaton JV, Brown T, Dingledine R. CNQX increases spontaneous inhibitory input to CA3 pyramidal neurones in neonatal rat hippocampal slices. Brain Research. 1992;592:255–260. doi: 10.1016/0006-8993(92)91683-6. [DOI] [PubMed] [Google Scholar]
- Maienschein V, Marxen M, Volknandt W, Zimmermann H. A plethora of presynaptic proteins associated with ATP-storing organelles in cultured astrocytes. Glia. 1999;26:233–244. doi: 10.1002/(sici)1098-1136(199905)26:3<233::aid-glia5>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Glial contributions to excitatory neurotransmission in cultured hippocampal cells. Nature. 1994;368:59–62. doi: 10.1038/368059a0. [DOI] [PubMed] [Google Scholar]
- Müller T, Möller T, Berger T, Schnitzer J, Kettenmann H. Calcium entry through kainate receptors and resulting potassium-channel blockade in Bergmann glial cells. Science. 1992;256:1563–1566. doi: 10.1126/science.1317969. [DOI] [PubMed] [Google Scholar]
- Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proceedings of the National Academy of Sciences of the USA. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. Journal of Neuroscience. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge EM, Kimelberg HK. Release of [3H]-d-aspartate from primary astrocyte cultures in response to raised external potassium. Journal of Neuroscience. 1996;16:7803–7811. doi: 10.1523/JNEUROSCI.16-24-07803.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–446. doi: 10.1038/348443a0. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Bernstein GM, Abdel-Hamid HM, Sattler R, Velumian A, Carlen PL, Razavi H, Jones OT. A novel use for a carbodiimide compound for the fixation of fluorescent and non-fluorescent calcium indicators in situ following physiological experiments. Cell Calcium. 1997;21:175–183. doi: 10.1016/s0143-4160(97)90042-7. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiological Reviews. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Steinhäuser C. Ion channels in glial cells. Brain Research Reviews. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing d-serine to regulate glutamate-N-methyl-d-aspartate neurotransmission. Proceedings of the National Academy of Sciences of the USA. 1999;96:13 409–13 414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M, Connor JA. Characterization of l-homocysteate-induced currents in Purkinje cells from wild-type and NMDA receptor knockout mice. Journal of Neurophysiology. 1999;82:2820–2826. doi: 10.1152/jn.1999.82.5.2820. [DOI] [PubMed] [Google Scholar]