

Abstract
The objective of this study was to examine the effects of isoproterenol (isoprenaline) and carbachol upon voltage-dependent inactivation of L-type Ca2+ current (ICa,L). ICa,L was recorded in guinea-pig isolated ventricular myocytes in the presence and absence of extracellular Ca2+ to separate total inactivation and voltage-dependent inactivation. In the presence of Ca2+, isoproterenol and carbachol had ‘competitive’ effects upon the relationships between membrane voltage and ICa,L amplitude and inactivation. Neither agonist had a marked effect upon the decay of inward ICa,L carried by Ca2+. In the absence of Ca2+, isoproterenol severely reduced and slowed ICa,L inactivation; this effect was reversed by carbachol. Under control conditions decay was dominated by fast inactivation. Isoproterenol reduced fast-inactivating and increased time-independent currents in a dose-dependent manner. These effects were counteracted by carbachol. There was a reciprocal relationship between the amplitude of fast-inactivating and time-independent currents with agonist stimulation. It is concluded that agonist modulation of rapid voltage-dependent inactivation of L-type Ca2+ channels involves an ‘on-off’ switch.
The heart is subject to sympathetic and parasympathetic regulation, which is reflected in the responses of L-type Ca2+ channel currents (ICa,L) in isolated cardiac myocytes. ICa,L is increased by β-adrenergic stimulation and subsequently reduced upon the application of muscarinic agonists (Trautwein & Hescheler, 1990). Inactivation is an important characteristic of ICa,L. It limits Ca2+ entry and contributes to the effective translation of electrical into mechanical activity. ICa,L inactivates according to voltage- and intracellular Ca2+-dependent mechanisms (Pelzer et al. 1990). The effect of β-adrenergic stimulation upon inactivation of ICa,L in cardiac muscle varies between acceleration, no effect and slowing down, according to the report (Pelzer et al. 1990).
The mechanism of sympathetic and parasympathetic regulation of the cardiac muscle L-type Ca2+ channel involves the phosphorylation of one or more serine-threonine residues upon one or more of the channel subunits by protein kinase A (PKA) (Kamp & Hell, 2000). Interaction between the β-adrenergic and the muscarinic agonists occurs at different levels in the intracellular pathways leading to the activation of PKA, not at the level of the calcium channel (Kamp & Hell, 2000). Thus, if we are to understand the contribution of ICa,L to the physiology of cardiac muscle it is necessary to understand the effects of phosphorylation by PKA upon the ion channel. Consideration that the L-type Ca2+ channel molecule or molecular complex might show distinct behaviour under basal conditions and following β-adrenergic stimulation is a surprisingly recent concept (Mitarai et al. 2000; Findlay, 2002b).
The consensus of opinion has been that Ca2+-induced inactivation dominated the decay of cardiac muscle ICa,L (Linz & Meyer, 1998), since voltage-dependent inactivation was exceedingly slow (Matsuda, 1986) and could not contribute significantly to the decay of ICa,L under physiological conditions. In a pioneering study, Bean et al. (1984) showed that, whereas β-adrenergic stimulation accelerated the decay of inward currents carried by Ca2+, in the same cells in the continued presence of Ca2+, it slowed the decay of outward Ca2+ currents carried by Cs+. Here, a clear dichotomy concerning the effects of β-adrenergic stimulation upon the inactivation of ICa,L was revealed. In the first situation, the Ca2+ current would have been subject to total inactivation arising from Ca2+ influx and Ca2+-induced Ca2+ release (CICR) from the sarcoplasmic reticulum as well as membrane voltage. In the second situation, the Ca2+ current would have been subject to only voltage-dependent inactivation since, positive to the reversal potential, Ca2+ current would be carried by the efflux of intracellular monovalent cations (Tsien et al. 1987) and neither Ca2+ influx nor CICR would contribute to the decay. The importance of this observation was not appreciated, though several groups reported β-adrenergic slowing of the decay of ICa,L carried by Ba2+ (Tsien et al. 1986; Tiaho et al. 1991). Unfortunately, a large number of studies of either the influence of Ca2+ and CICR upon the inactivation of ICa,L (Sham, 1997) or the voltage-dependent decay of ICa,L (Matsuda, 1986) involved the use of particular experimental conditions to either impede ‘run-down’ or maximise currents, and/or to maximise interactions between the channel and sarcoplasmic reticulum, without taking into account that such treatments could alter the behaviour of the ion channel and thus bias the conclusions towards the importance of Ca2+-induced inactivation.
In 1987, Hadley and Hume took advantage of the asymmetric permeation of the L-type Ca2+ channel to monitor the inactivation of ICa,L in the absence of ion flux through the channels. When extracellular Ca2+ is removed, extracellular Mg2+ blocks inward but allows outward current through Ca2+ channels (Fukushima & Hagiwara, 1985; Hess et al. 1986). This outward current can be used to describe the inactivation of ICa,L in the absence of Ca2+ and in the absence of inward ion flux where inactivation is a single and voltage-dependent process (Hadley & Hume, 1987; Hadley & Lederer, 1991; Findlay, 2002b). In this manner, Findlay (2002b) confirmed the observation of Bean et al. (1984) by showing that the decay of ICa,L flowing outward through Ca2+ channels was slowed by β-adrenergic stimulation. However, in agreement with Mitarai et al. (2000), Findlay (2002b) showed that this was due to alteration of the kinetic make-up of the channel population rather than to a change in the process of inactivation. The principal aim of that previous study was to evaluate the relative contributions of voltage and Ca2+ to the decay of ICa,L in isolated cardiac myocytes. It was shown that under basal conditions inactivation of ICa,L depended largely upon membrane voltage while with β-adrenergic stimulation the reduction of the voltage-dependent kinetics of inactivation enabled the intervention of Ca2+-induced inactivation. It was therefore clear that the relative contributions of these two processes to the overall decay of ICa,L depended upon the kinetics of voltage-dependent decay and that this was the target of agonist stimulation of the channel. The objective of this present report was to further understanding of the effects of agonist stimulation upon voltage-dependent inactivation of the Ca2+ ion channel, by examining in some detail the influence of different doses of the β-adrenergic agonist isoproterenol (isoprenaline) and by testing, for the first time, the effects of the muscarinic agonist carbachol. The results revealed a surprising simplicity. Under basal conditions, the majority of Ca2+ channels show rapid voltage-dependent inactivation. In a dose-dependent manner, β-adrenergic agonists converted the channels to an inactivation-resistant form, whereas carbachol provoked a return to rapid voltage-dependent inactivation. It is therefore proposed that PKA phosphorylation of the L-type Ca2+ channel serves as an on-off switch for voltage-dependent inactivation.
Methods
Cell preparation
All animal experiments were conducted according to the ethical standards of the Ministère Français de l'Agriculture (Licence number B37-261-4). Male guinea-pigs (250-400 g) were killed by cervical dislocation and the hearts were removed. Single ventricular myocytes were isolated using collagenase and protease digestion, as described elsewhere (Le Guennec et al. 1993). Myocytes isolated from the left ventricle were aliquoted into 35 mm diameter plastic Petri dishes that served as experimental chambers. The storage solution consisted of the standard extracellular solution described below. Dishes that contained myocytes were kept on the laboratory bench and used within 6-8 h after isolation.
Experimental procedures
Whole-cell current voltage-clamp experiments were conducted with an Axon Instruments 202A patch-clamp amplifier in resistive feedback mode (Axon Instruments, CA, USA). Pipettes were fabricated from thin-walled borosilicate glass capillary tubes (Clark Electromedical Instruments, Pangbourne, UK) with a Narishige PB7 double-stage puller (Narishige Instruments, Tokyo, Japan). Pipettes were coated with Sylgard (Dow Corning, MI, USA) and then heat polished. Finished pipettes had a resistance of < 2 MΩ when filled with standard intracellular solution. Plastic Petri dishes containing isolated myocytes were placed upon the stage of an Olympus CK2 inverted microscope. Isolated myocytes were superfused with experimental solutions via a parallel pipes system lowered into the vicinity of the cells. Experimental voltage-clamp protocols and data acquisition were controlled with Acquis1 software (Dipsi Industrie, Chatillon, France) installed upon a 386-20 PC computer. Data were filtered at either 1 or 2 kHz and acquired at 2 or 5 kHz, respectively. Cell capacitance and series resistance were compensated (≈80 %) with the Axon Instruments amplifier. Cell currents are expressed as current density, pA pF−1. Data analysis was performed with Acquis1 and Origin 4.1 (Microcal Software, MA, USA). Results are shown as mean ± s.e.m. values obtained from n different ventricular myocytes. Isolated myocytes were voltage clamped at −80 mV using the whole-cell configuration of the patch-clamp cell current recording technique (Hamill et al. 1981). Voltage-clamp protocols were delivered to the cells from this holding potential. Each voltage-clamp protocol was preceded by a voltage step to −50 mV for a period of 1000 ms to inactivate Na+ current remaining after the application of 10 μm tetrodotoxin (TTX) and to inactivate any T-type Ca2+ current (Balke et al. 1992). The standard method of evoking cell currents consisted of a double-pulse voltage-clamp protocol with 1000 ms pre-pulse voltage steps to between −50 and +80 mV in 10 mV increments, and a 1000 ms duration test pulse voltage-clamp step to +80 mV. A 10 ms interval at −50 mV separated pre- and test pulse voltage steps. Current-voltage (I-V) relationships were established from the peak amplitude of currents evoked during pre-pulse voltage steps. Availability-voltage (A-V) relationships were established by normalising the amplitude of the current evoked by the test pulse to that evoked following a pre-pulse voltage step to −50 mV. The test pulse voltage step to +80 mV would evoke an outward ICa,L carried by K+ in experiments in which cells were bathed in normal extracellular solution containing 2 mm Ca2+ and also in which cells were bathed in zero calcium extracellular solution (see Fig. 1). The amplitude of this outward ICa,L served as a measure of the extent of inactivation occurring during the pre-pulse voltage steps. In this way, (1) the effects of evaluating inactivation of ICa,L at different voltages would be avoided (Hadley & Hume, 1987; and see Gera & Byerly, 1999), (2) the test pulse current would not be subject to Ca2+-induced inactivation whether recorded in the presence or absence of extracellular Ca2+, and (3) direct comparisons could be made between curves recorded in the presence and absence of extracellular Ca2+ because extracellular Mg2+ in the zero calcium solution assured the maintenance of membrane surface charge (Findlay, 2002b). Voltage-gated currents through L-type Ca2+ channels were extracted from the ensemble whole-cell current with 200 μm CdCl2. The limitations of this method are discussed by Linz & Meyer (1998). All experiments were conducted at room temperature (≈23 °C).
Figure 1. Effects of β-adrenergic and muscarinic stimulation upon ICa,L.
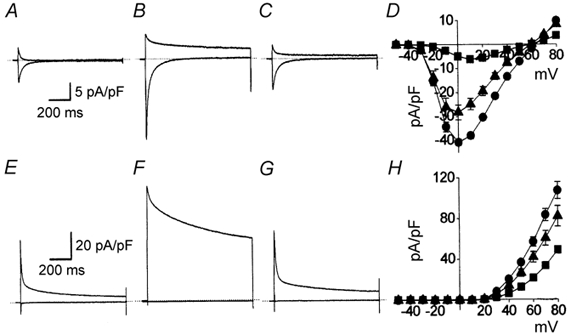
Cell current records (A-C and E-G) represent superimposed cell current traces recorded during 1000 ms duration voltage steps to +10 mV (lower traces) and +80 mV (upper traces). The dotted lines indicate the 0 pA current level and the time and current scale bars in A and E, respectively, apply also to the records shown in B and C, and F and G. In one series of experiments (A-D), ventricular myocytes were bathed in extracellular solution containing 2 mm Ca2+. A, cell currents recorded from a ventricular myocyte under basal conditions. B, cell currents recorded from a ventricular myocyte in the presence of 100 nm isoproterenol. C, cell currents recorded from the same myocyte following the addition of 10 μm carbachol. D, I-V relationships for ICa,L recorded in the presence of 2 mm Ca2+ under basal conditions (▪, n = 14), in the presence of 100 nm isoproterenol (•, n = 7) and following the addition of 10 μm carbachol (▴). In a separate series of experiments (E-H), ventricular myocytes were bathed in extracellular solution that did not contain Ca2+ (see Methods). E, cell currents recorded from a ventricular myocyte under basal conditions. F, cell currents recorded from a ventricular myocyte in the presence of 100 nm isoproterenol. G, cell currents recorded from the same myocyte following the addition of 10 μm carbachol. H, I-V relationships for ICa,L recorded in the absence of extracellular Ca2+ under basal conditions (▪, n = 8), in the presence of 100 nm isoproterenol (•, n = 8) and following the addition of 10 μm carbachol (▴).
Experimental solutions
The standard extracellular solution used to fill the Petri dishes and store myocytes prior to experiments contained (mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 Hepes, pH 7.4 with NaOH. TTX (citrate salt, 10 μm; from Alomone Labs, Jerusalem, Israel) and ryanodine (5 μm) were added to extracellular solutions used to superfuse cells during experiments. Calcium-free (zero calcium) extracellular solution contained 250 μm EGTA-NaOH, 3 mm MgCl2 and no added Ca2+. The standard intracellular solution used to fill the patch pipettes contained (mm): 140 KCl, 5 EGTA-KOH, 3.4 MgCl2, 0.1 CaCl2, 2 ATP-2Na+, 0.1 GTP, 10 glucose, 10 Hepes, pH 7.3 with KOH. The estimated free concentrations of Mg2+ and Ca2+ in this solution were 1 mm and 1 nm, respectively. Isoproterenol and carbachol were prepared daily as 100 μm and 1 mm stock solutions, respectively, in distilled water. Ryanodine was dissolved as a 1 mm stock solution in distilled water.
Results
Effects of agonists upon ICa,L
Figure 1 illustrates the effects of 100 nm isoproterenol and the subsequent addition of 10 μm carbachol upon ICa,L recorded in the presence (Fig. 1A-D) and absence (Fig. 1E-H) of extracellular Ca2+. Isoproterenol increased the amplitude of ICa,L irrespective of whether this was an inward current carried by Ca2+ (Fig. 1A-D) or an outward current carried by K+ in the presence (Fig. 1A-D) or absence (Fig. 1E-H) of extracellular Ca2+. The addition of carbachol reduced the stimulation of ICa,L induced by isoproterenol. Neither agonist altered the reversal potential of ≈+55 mV between inwardly and outwardly directed currents through L-type Ca2+ channels (Fig. 1D; and see Bean et al. 1984).
The whole-cell current recordings shown in Fig. 1A-C and E-G illustrate the principal objectives of this study. The time-dependent decay of currents flowing thorough L-type Ca2+ channels was altered by agonist stimulation. In comparison with basal conditions (Fig. 1A and E), β-adrenergic stimulation provoked an apparent acceleration of the inactivation of ICa,L carried by Ca2+ (Fig. 1B, inward current) while it apparently slowed down the inactivation of ICa,L carried by K+ (Fig. 1B, outward current, and F). These results are similar to previous observations (Bean et al. 1984; Tiaho et al. 1991; Mitarai et al. 2000; Findlay, 2002b). Here it is shown for the first time that the muscarinic agonist carbachol counteracted these effects of β-adrenergic stimulation. The inactivation of ICa,L carried by Ca2+ (Fig. 1C, inward current) was apparently slowed by the application of carbachol in the continued presence of isoproterenol while the inactivation of ICa,L carried by K+ (Fig. 1C, outward current, and G) was accelerated.
The obvious difference between these effects of the agonists upon the time course of inactivation of ICa,L was investigated in two stages. First, the effects of the agonists upon the total inactivation of ICa,L were examined. These experiments were conducted upon ventricular myocytes which were bathed in extracellular solution containing 2 mm Ca2+. Under these experimental conditions, inactivation of inwardly directed ICa,L carried by Ca2+ would be due to both voltage- and Ca2+-dependent processes (McDonald et al. 1994). Normally, Ca2+-induced inactivation would result from both Ca2+ influx via cell surface channels and Ca2+ released from the sarcoplasmic reticulum. This last and variable component of the inactivation of ICa,L (Adachi-Akahane et al. 1996; Hussain & Orchard, 1997; Sham, 1997; Findlay, 2002b) was excluded from this study by the inclusion of 5 μm ryanodine in all of the experimental solutions. Total inactivation of ICa,L was therefore represented by voltage-dependent inactivation and Ca2+-induced inactivation due to Ca2+ influx through L-type Ca2+ channels. Second, the effects of the agonists upon voltage-dependent inactivation was examined. These experiments were conducted upon ventricular myocytes bathed in extracellular solutions that did not contain Ca2+ but where Ca2+ had been replaced by Mg2+. This method has been shown to adequately isolate the voltage-dependent process of inactivation of ICa,L without disturbing the membrane voltage field (Findlay, 2002b; and see Hadley & Hume, 1987), which is a drawback of recording inward ICa,L carried by Na+ (Fukushima & Hagiwara, 1985; Mitarai et al. 2000; Findlay, 2002a).
The effects of agonists upon total inactivation of ICa,L
Figure 2 illustrates the effects of agonists upon the total inactivation of ICa,L, where this was represented by the quasi-steady-state availability curve recorded in normal extracellular solution containing 2 mm Ca2+. Under basal conditions in the absence of agonists, the A-V relationship of ICa,L was U-shaped with a half-inactivation voltage (V0.5) of −20 mV (□, Fig. 2A). The application of isoproterenol enhanced inactivation of ICa,L in a dose-dependent manner, such that V0.5 was shifted to more negative values: −25 mV in 2 nm isoproterenol (▪, Fig. 2A), −30 mV in 20 nm isoproterenol (▪, Fig. 2B) and −29 mV in 100 nm isoproterenol (▪, Fig. 2C). In each case recovery of ICa,L, which followed pre-pulse voltage steps to membrane potentials more positive than +20 mV, was enhanced compared with basal conditions. The addition of 10 μm carbachol reversed the negative shift of the A-V relationship caused by 2 nm isoproterenol (V0.5, −19 mV; •, Fig. 2A), had a slight effect upon that recorded in the presence of 20 nm isoproterenol (V0.5, −26 mV; •, Fig. 2B), but had no effect upon that recorded in the presence of 100 nm isoproterenol (V0.5, −30 mV; •, Fig. 2C).
Figure 2. Effects of agonists upon the availability of ICa,L.
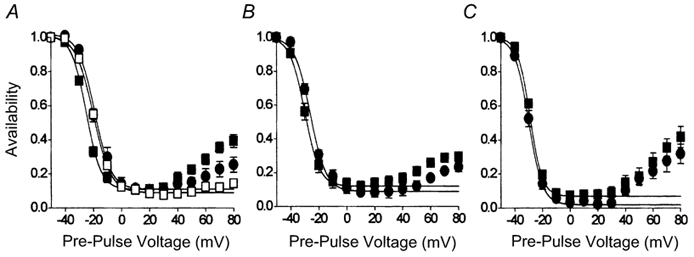
These experiments were conducted in extracellular solution containing 2 mm Ca2+. A-V relationships were recorded with the double-pulse voltage-clamp protocol described in Methods. A, effects of 2 nm isoproterenol (▪, n = 9) and 10 μm carbachol (•). For comparison, data obtained from cells under basal conditions (□, n = 14) are also shown. B, effects of 20 nm isoproterenol (▪, n = 6) and 10 μm carbachol (•). C, effects of 100 nm isoproterenol (▪, n = 7) and 10 μm carbachol (•). The lines represent fits of the Boltzmann equation (Origin 4.1) between the maximum and minimum data points, and which were then extrapolated to +80 mV. See text for further details.
A-V relationships of ICa,L evaluate inactivation at an approximation of steady-state conditions where it is assumed that the processes of voltage- and Ca2+-dependent inactivation will have run their course. The influence of one or other of the processes may then be difficult to distinguish from the sum of their effects. In order to examine the relative influence of these processes, it is necessary to either examine the availability of the current at different and shorter intervals in time after activation of the current (Findlay, 2002b) or to follow the development of inactivation by recording the time course of decay of the current.
The effects of isoproterenol and carbachol upon the time course of decay of ICa,L evoked by a voltage step to +10 mV were examined (Fig. 3). This voltage was chosen since it evoked nearly maximum inward Ca2+ current (Fig. 1D), was close to maximal to induce inactivation (Fig. 2), and was the highest voltage that in the absence of extracellular Ca2+ did not evoke current flow through L-type Ca2+ channels (Fig. 1H). This final point will be important for comparisons with subsequent experiments which were conducted in zero calcium extracellular solution (Fig. 5). Figure 3A illustrates ICa,L recorded from four representative myocytes. Each recording consists of two cell current traces which were normalised to their respective peak amplitude and superimposed to allow a direct visual comparison of the time course of their inactivation. In the presence of extracellular Ca2+, a voltage step to +10 mV evoked inward Ca2+ currents and carbachol was without discernable effect upon the time course of inactivation of ICa,L recorded in the presence of three concentrations of isoproterenol. Carbachol induced a slight acceleration of the inactivation of ICa,L recorded under basal conditions. The inactivation of ICa,L evoked at +10 mV could be described by a double-exponential function (Origin 4.1), which under these circumstances declined to completion in a time-dependent manner. Isoproterenol increased the initial rate of inactivation of ICa,L in a dose-dependent manner; τf + 25 ± 2, 17 ± 2, 17 ± 1 and 11 ± 1 ms under basal conditions and in the presence of 2, 20 and 100 nm isoproterenol, respectively. Neither isoproterenol nor carbachol had any significant effect upon the relative proportions of the fast and slow components of biphasic decay of inward ICa,L (Fig. 3B). It should be emphasised that these experiments were conducted in the presence of ryanodine in order to exclude any influence of Ca2+ release from the sarcoplasmic reticulum upon the inactivation of ICa,L. These results therefore show that neither agonist had a particularly marked effect upon the time course of inactivation of ICa,L when carried by Ca2+ and when it was subject to both inactivation associated with membrane voltage and that caused by the influx of extracellular calcium. A different picture was to emerge from experiments conducted in the absence of extracellular Ca2+.
Figure 3. Effects of agonists upon the time-dependent decay of ICa,L.
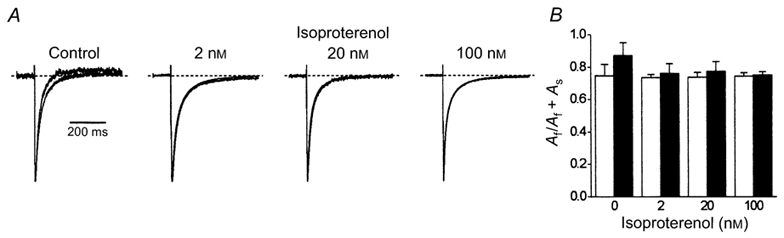
A, ICa,L was evoked by voltage steps to +10 mV in cells bathed in extracellular solution containing 2 mm Ca2+. These normalised records were obtained from four representative cells. Each record consists of two superimposed traces which were recorded first under the indicated experimental condition and then following the addition of 10 μm carbachol. The dotted lines indicate the zero level. The time scale to the left applies to all four sets of traces. B, the biphasic decay of ICa,L evoked by a voltage step to +10 mV was analysed and the amplitudes of the fast and slow components of the double-exponential function (Origin 4.1) were extracted. Here, the amplitude of the fast component of decay is expressed as a proportion of the total current amplitude (Af/Af + As). Cell currents were recorded first either under basal conditions (n = 8) or in the presence of 2 (n = 9), 20 (n = 6) or 100 nm (n = 5) isoproterenol (□) and then following the addition of 10 μm carbachol (▪).
Figure 5. Effects of agonists upon the availability of ICa,L in the absence of Ca2+.
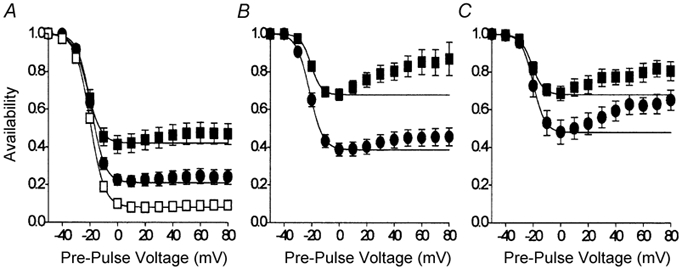
A-V relationships were recorded with the double-pulse voltage-clamp protocol described in Methods. These experiments were conducted with cells bathed in zero calcium extracellular solution. A, effects of 2 nm isoproterenol (▪, n = 7) and 10 μm carbachol (•). For comparison, data obtained from cells under basal conditions (□, n = 8) are also shown. B, effects of 20 nm isoproterenol (▪, n = 6) and 10 μm carbachol (•). C, effects of 100 nm isoproterenol (▪, n = 8) and 10 μm carbachol (•). The lines represent fits of the Boltzmann equation (Origin 4.1) between the maximum and minimum data points, and then extrapolated to +80 mV. See text for further details.
The effects of agonists upon the voltage-dependent inactivation of ICa,L
The cell current records shown in Fig. 4 illustrate the effects of agonists upon the inactivation of ICa,L occurring in the absence of current flow through the channels. Under basal conditions, the test pulse voltage step to +80 mV delivered from a pre-pulse voltage of −50 mV evoked a large outward and rapidly decaying current through L-type Ca2+ channels (Fig. 4A). When this voltage step was delivered after a 1000 ms pre-pulse voltage step to +10 mV, the outward current was severely reduced (arrow in Fig. 4A). No current had flowed through L-type Ca2+ channels during the pre-pulse voltage step to +10 mV. It is concluded that ICa,L had been activated and then inactivated during the voltage step to +10 mV in the absence of ion flux and that in consequence few L-type Ca2+ channels remained available for activation by the test pulse voltage step. The full range of the A-V relationship for ICa,L recorded in the absence of extracellular Ca2+ under basal conditions is shown in Fig. 5A (□). This relationship was sigmoid and could be described by the Boltzmann equation over the entire voltage range between −50 and +80 mV tested here, and half inactivation (V0.5) was assessed to have occurred at −20 mV. These results strongly suggest that, under these experimental conditions, with cells bathed in zero calcium extracellular solution, ICa,L inactivated via a single and voltage-dependent process (Hadley & Hume, 1987; Findlay, 2002b).
Figure 4. Effects of agonists upon the inactivation of ICa,L in the absence of extracellular Ca2+.
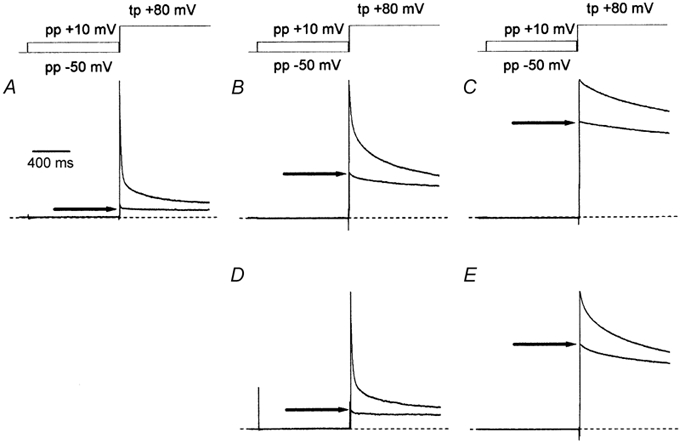
Representative cell currents were recorded in the absence of extracellular Ca2+ using the double-pulse voltage-clamp protocol described in Methods. Each of the normalised traces shown are composed of two superimposed current records which were obtained with pre-pulse (pp) voltage steps to either −50 or +10 mV. The voltage-clamp protocol is indicated schematically above the current records. The amplitude of the current evoked by the test pulse (tp) following a pre-pulse to +10 mV is indicated by an arrow in each recording. The dashed lines indicate the zero level. The time scale in A applies to all traces. A, cell currents recorded under basal conditions. B, cell currents recorded in the presence of 2 nm isoproterenol. C, cell currents recorded in the presence of 100 nm isoproterenol. D, cell currents recorded in the presence of 2 nm isoproterenol and 10 μm carbachol. E, cell currents recorded in the presence of 100 nm isoproterenol and 10 μm carbachol. Cell current records shown in D and E were recorded from the same myocytes as those in B and C, respectively.
The effect of different concentrations of isoproterenol upon the voltage-dependent inactivation of ICa,L recorded in the absence of extracellular Ca2+ is shown in Fig. 4B and C. These recordings differed from the results obtained under basal conditions (Fig. 4A) in two ways. First, ICa,L evoked by the test pulse voltage step showed less inactivation in isoproterenol in a dose-dependent manner (Fig. 4B and C). Second, the amount of ICa,L remaining available for activation following a pre-pulse voltage step to +10 mV was increased by isoproterenol in a dose-dependent manner (arrows in Fig. 4B and C). These two observations strongly suggest a slowing and/or an inhibition of the process of voltage-dependent inactivation of ICa,L by β-adrenergic stimulation. Figure 5 illustrates the A-V relationships for ICa,L recorded in the absence of extracellular Ca2+ following stimulation by 2 nm (▪, Fig. 5A), 20 nm (▪, Fig. 5B) and 100 nm (▪, Fig. 5C) isoproterenol. Isoproterenol had little effect upon the voltage dependence of the A-V relationships recorded in the absence of Ca2+, withV0.5 values of −22 mV in 2 nm isoproterenol, −20 mV in 20 nm isoproterenol and −21 mV in 100 nm isoproterenol, compared with −20 mV for V0.5 recorded under basal conditions. However, isoproterenol clearly reduced the amount of inactivation recorded over 1000 ms in a dose-dependent manner. Thus ≈40 % of the total cell current in 2 nm isoproterenol and ≈70 % of the total cell current in 20 and 100 nm isoproterenol remained available for activation at the minima of the A-V relationships compared with only ≈6 % of the total cell current under basal conditions. Isoproterenol was also associated with the recovery of channel availability following positive pre-pulse voltage steps, such that the A-V relationship that was sigmoid in form under basal conditions was U-shaped.
The effect of carbachol upon the voltage-dependent inactivation of ICa,L is shown in the cell current records illustrated in Fig. 4D and E. In the presence of 2 nm isoproterenol (Fig. 4D), carbachol clearly enhanced the inactivation of ICa,L evoked by the test pulse voltage step and increased the inactivation of ICa,L during the pre-pulse voltage step to +10 mV (arrow in Fig. 4D). The addition of 10 μm carbachol to a cell bathed in 100 nm isoproterenol was less effective (Fig. 4E). The effects of carbachol upon the A-V relationships of ICa,L stimulated by isoproterenol are shown in Fig. 5. Whether in the presence of 2 nm isoproterenol (•, Fig. 5A), 20 nm isoproterenol (•, Fig. 5B) or 100 nm isoproterenol (•, Fig. 5C), carbachol had no effect upon the voltage dependence of the A-V relationships with V0.5 values of −19, −21 and −20 mV, respectively. However, carbachol had enhanced the amount of inactivation since the proportions of the total current available for activation at the minima of the A-V relationships were reduced to ≈20 %, ≈40 % and ≈50 % in 2, 20 and 100 nm isoproterenol, respectively.
These results strongly suggest that the β-adrenergic and muscarinic agonists influenced the time course of voltage-dependent inactivation of ICa,L in isolated ventricular myocytes of the guinea-pig. There was no evidence that either agonist influenced the dependence of inactivation upon membrane voltage.
In the absence of extracellular Ca2+, a voltage step to +10 mV did not evoke a measurable current through L-type Ca2+ channels (Fig. 1E-H and Fig. 4). Therefore in order to examine the effects of agonists upon the time course of the development of voltage-dependent inactivation of ICa,L in the absence of Ca2+ flux and to compare these data with those for inactivation of ICa,L due to membrane voltage and Ca2+ flux (Fig. 3), it was necessary to measure inactivation indirectly. The time course of the development of voltage-dependent inactivation was therefore assessed by the application of an envelope type of double-pulse voltage-clamp protocol, which measured the availability of channels for activation by a test pulse voltage step to +80 mV, following pre-pulse voltage steps to +10 mV of 10, 20, 50, 100, 200, 500 and 1000 ms duration (Findlay, 2002b; and see inset to Fig. 6). Figure 6A illustrates the application of this protocol to a myocyte under basal conditions in the absence of external agonists. Although pre-pulse voltage steps to +10 mV of 50, 200 and 1000 ms duration evoked no current flow through L-type Ca2+ channels because the cell was bathed in zero calcium extracellular solution, the amplitude of the current evoked by the subsequent voltage step to +80 mV was progressively reduced. This loss of the availability of ICa,L for activation is considered to reflect the progressive development of inactivation in the channel population during the pre-pulse voltage step. Thus, under basal conditions, voltage-dependent inactivation developed rapidly (Fig. 6A) and with a bi-exponential time course (τf + 32 ms, τs + 380 ms; □, Fig. 6D). In the presence of 100 nm isoproterenol, the development of inactivation with time was severely impaired (Fig. 6B) and progressed with a single slow exponential time course (τ + 255 ms; ▪, Fig. 6D). The addition of 10 μm carbachol, in the continued presence of 100 nm isoproterenol, enhanced the development of inactivation (Fig. 6C), which again showed a biphasic time course (τf + 47 ms, τs + 350 ms; •, Fig. 6D).
Figure 6. Effects of agonists upon the time course of voltage-dependent inactivation of ICa,L.
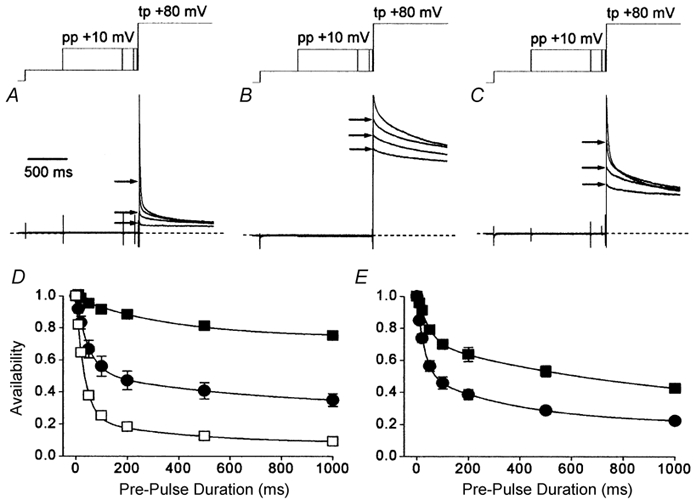
A, current records obtained from a representative myocyte under basal conditions. B, current records obtained from a representative myocyte in the presence of 100 nm isoproterenol. C, current records obtained from the cell shown in B following the addition of 10 μm carbachol. The voltage-clamp protocol is indicated schematically above the current records. These records each consist of four superimposed normalised traces which were recorded with either no pre-pulse, or pre-pulse voltage steps to +10 mV of 50, 200 and 1000 ms duration, as indicated by arrows. The dashed lines indicate the zero level. The time scale in A applies to all three records. D and E, time course of the development of inactivation of ICa,L. The amplitudes of cell currents evoked by test pulse voltage steps were normalised to those recorded in the absence of a pre-pulse. □ (D) represent results obtained from experiments conducted under basal conditions (n = 8). Otherwise, cell currents were recorded first in isoproterenol (▪: D, 100 nm, n = 6; E, 2 nm, n = 6) and then following the addition of 10 μm carbachol (•). The lines represent fits to the data of one or two exponential functions; see text for details.
These results clearly suggest that not only β-adrenergic stimulation but also muscarinic antagonism of the activation of ICa,L affected the time course of the development of voltage-dependent inactivation. Mitarai et al. (2000) and Findlay (2002b) both found that, although the time constants of inactivation of ICa,L were altered by β-adrenergic stimulation, this could be less consequent for ICa,L as a whole than the redistribution of L-type Ca2+ channels between kinetically distinct groups. This was also suggested by analysis of the data shown in Fig. 6E, which illustrates the time course of the development of voltage-dependent inactivation recorded in the presence of 2 nm isoproterenol (▪) and the effect of the addition of 10 μm carbachol (•). Under basal conditions (□, Fig. 6D), inactivation developed with a rapid phase (τf + 32 ms), which had an initial amplitude corresponding to ≈80 % of the current, and a slow phase (τs + 380 ms), which had an initial amplitude of ≈15 % of the current. The data recorded in the presence of 2 nm isoproterenol and 10 μm carbachol declined with a very similar time course (τf + 28 ms, τs + 378 ms; •, Fig. 6E). However, in this case the rapid phase of inactivation corresponded to ≈50 % of the current and the slow phase accounted for ≈30 % of the current. Overall, therefore, less inactivation of the total current was recorded with time in the presence of 2 nm isoproterenol and 10 μm carbachol (Fig. 6E) than under basal conditions (Fig. 6D), while the speed of the process of inactivation was the same under both experimental conditions. It was therefore clear that agonists had more than just an effect upon the process of voltage-dependent inactivation (Mitarai et al. 2000; Findlay, 2002b) and it was decided to examine in further detail the effects of the agonists upon the kinetics of inactivation of ICa,L recorded in the absence of Ca2+.
In this series of experiments, the development of inactivation of ICa,L was measured directly from the time course of decay of ICa,L rather than from the indirect loss of availability of ICa,L. The time course of inactivation evaluated from the loss of availability of ICa,L at +10 mV (τf + 32 ms; amplitude, 0.78; τs + 380 ms; amplitude, 0.15; offset amplitude, 0.08) was quite similar to the results obtained from the fitting of a double-exponential function (Origin 4.1) to the directly recorded inactivation of ICa,L at +80 mV (τf + 15 ± 1 ms; amplitude, 0.74 ± 0.06; τs + 170 ± 3 ms; amplitude, 0.16 ± 0.02; offset amplitude, 0.10 ± 0.02; n = 8). Therefore, although the data were analysed at what could be considered to be an unphysiological membrane voltage, they closely reflected the physiologically relevant kinetics of inactivation. In addition, it was then also possible to perform an equivalent analysis of the decay of ICa,L in the presence of Ca2+, where at the same voltage ICa,L is also outwardly directed (Fig. 1) but not subject to inactivation due to Ca2+ influx (see Fig. 9). In each cell the effect of carbachol was tested either under basal conditions or in the presence of a given concentration of isoproterenol. For clarity these data have been separated into two sections. First the dose-dependent effect of the β-adrenergic agonist will be considered (Fig. 7), then the effect of carbachol will be described (Fig. 8).
Figure 9. Effects of agonists upon voltage-dependent inactivation of ICa,L in the presence of extracellular Ca2+.
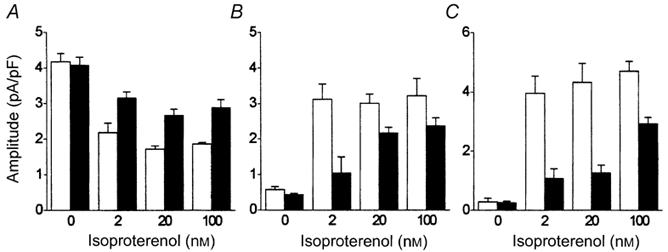
Experiments and analysis were performed as described in Fig. 7 on cells bathed in normal extracellular solution containing 2 mm Ca2+. Cell currents evoked by a voltage step to +80 mV were recorded either under basal conditions (0 nm) or in the presence of isoproterenol (□) and subsequently following the addition of 10 μm carbachol (▪). The initial amplitudes of fast (A) and slow (B) time-dependent and time-independent (C) components of decay are shown. n as for Fig. 3B.
Figure 7. Effect of isoproterenol upon voltage-dependent inactivation of ICa,L.
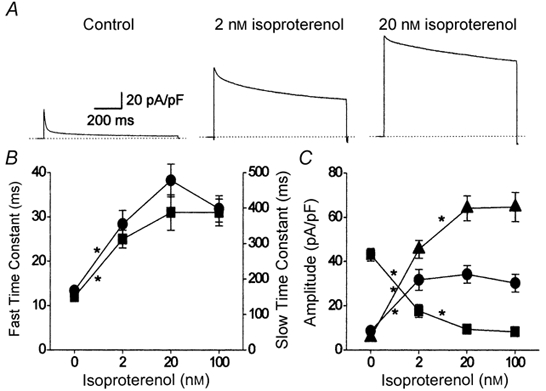
Cells were bathed in zero calcium extracellular solution and currents were evoked by voltage-clamp steps to +80 mV (A). The decay of the evoked cell currents was fitted by a bi-exponential function (Origin 4.1) and the components of that function were extracted (B and C). A, representative cell current records obtained from three different myocytes exposed to either basal conditions in the absence of any external agonist (Control; left), 2 nm isoproterenol (centre) or 20 nm isoproterenol (right). The dotted lines indicate the 0 pA current level and the time and current scales shown on the left apply to all three traces. B, effect of isoproterenol upon the fast (left axis; ▪) and slow (right axis; •) time constants of decay. Note the different y-axis scales for the two time constants. C, effect of isoproterenol upon the initial amplitude of fast (▪) and slow (•) time-dependent, and the time-independent (▴), components of decay. Control (0 nm isoproterenol), n = 8; 2 nm isoproterenol, n = 7; 20 nm isoproterenol, n = 6; 100 nm isoproterenol, n = 7. * P < 0.05, for comparison between adjacent groups with one-way ANOVA (Origin 4.1).
Figure 8. Effect of carbachol upon voltage-dependent inactivation of ICa,L.
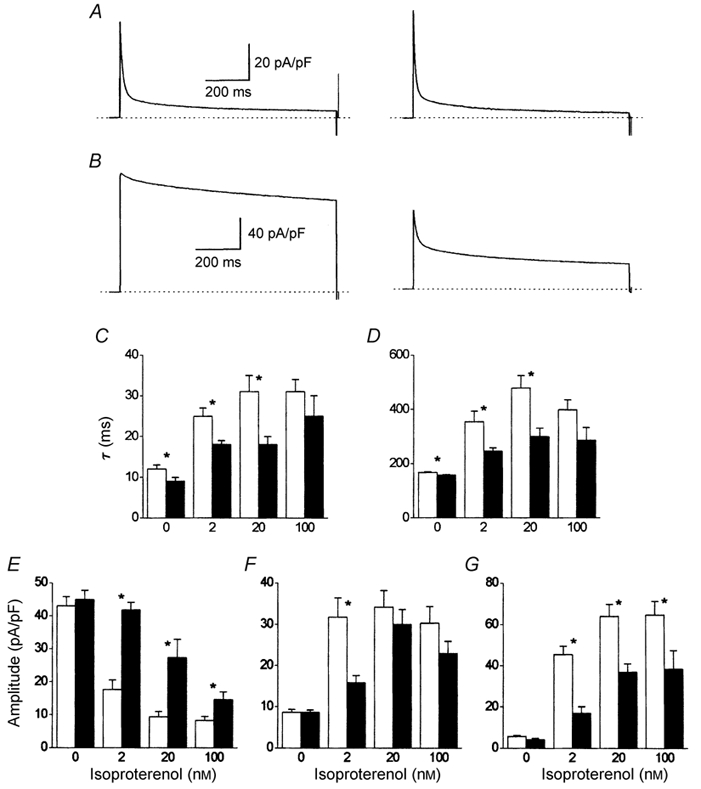
Experiments and analysis were performed as described in Fig. 7. A, effect of 10 μm carbachol (right) upon cell currents recorded in one myocyte under basal conditions (left). B, effect of 10 μm carbachol (right) upon cell currents recorded in one myocyte that was bathed in 20 nm isoproterenol (left). C-G, analysis of the decay of Ca2+ channel currents recorded either under basal conditions or in the presence of isoproterenol (□) and subsequently following the addition of 10 μm carbachol (▪). C, fast time constants of bi-exponential decay. D, slow time constants of bi-exponential decay. E-G, initial amplitudes of fast (E) and slow (F) time-dependent and time-independent (G) components of decay. n as for Fig. 7C. * P < 0.05, for the effect of carbachol determined with one-way ANOVA (Origin 4.1).
In a dose-dependent manner, isoproterenol increased the amplitude and reduced the inactivation of ICa,L recorded in the absence of extracellular Ca2+ (Fig. 7A). The time course of the development of inactivation could be described by a bi-exponential function. From the bi-exponential function, the components of ICa,L could be extracted: a component which inactivated rapidly, a component which inactivated slowly, and a time-independent or offset component. Both the fast and slow time constants of inactivation were significantly increased by 2 nm isoproterenol (Fig. 7B). Higher concentrations of isoproterenol tended to further increase both time constants, but this increase was not statistically significant. The amplitudes of the distinct kinetic components of ICa,L were profoundly altered by β-adrenergic stimulation (Fig. 7C). Under basal conditions, inactivation of ICa,L was dominated by a fast time-dependent component. Isoproterenol depressed the amplitude of the fast component in a dose-dependent manner, which was maximal with 20 nm of the agonist (▪, Fig. 7C). The amplitude of the slow time-dependent component, which was a minor part of the current under basal conditions, was clearly increased by 2 nm isoproterenol, but higher doses of the agonist had no statistically significant further effect (•, Fig. 7C). Under basal conditions, time-dependent inactivation of the current was virtually complete after 1000 ms; there remained only a small time-independent portion of the current. Isoproterenol strongly and significantly increased the amplitude of the time-independent current, which reached a maximum with 20 nm of the agonist (▴, Fig. 7C). It was therefore clear that the main effect of isoproterenol upon voltage-dependent inactivation of ICa,L was to alter the amplitudes of separate time-dependent and time-independent kinetic components of the current (Fig. 7C). β-Adrenergic stimulation had but little effect upon the actual time course of the process of voltage-dependent inactivation (Fig. 7B). It was tempting to speculate that a reciprocal relationship existed between the fast time-dependent (▪, Fig. 7C) and the time-independent (▴, Fig. 7C) kinetic forms of ICa,L.
The effect of carbachol upon the voltage-dependent inactivation of ICa,L is shown in Fig. 8. It was immediately clear that muscarinic stimulation not only reduced the amplitude of ICa,L enhanced by isoproterenol but that it also reversed the effects of β-adrenergic stimulation upon the inactivation of ICa,L (Fig. 8B). Under basal conditions carbachol had little effect upon the total amplitude of ICa,L (Fig. 8A), though it significantly accelerated both fast and slow time constants of inactivation (Fig. 8C and D). Carbachol significantly accelerated the time constants of inactivation of ICa,L in 2 and 20 nm isoproterenol (Fig. 8C and D) but not in 100 nm isoproterenol. Carbachol had little significant effect upon the amplitude of the slow time-dependent component of ICa,L, except to reduce the increase that had been induced by 2 nm isoproterenol (Fig. 8F). The major effects of carbachol were observed upon the amplitudes of the fast time-dependent (Fig. 8E) and the time-independent components of ICa,L (Fig. 8G). Carbachol significantly increased the first and reduced the second at each dose of isoproterenol. The effect of carbachol was ‘competitive’ with that of isoproterenol, being quantitatively less effective with increasing concentrations of the β-adrenergic agonist. The reciprocal relationship between the amplitudes of the fast time-dependent and the time-independent components of ICa,L which was first noted with their response to isoproterenol (Fig. 7) was confirmed therefore with their reaction to carbachol (Fig. 8). A large increase in amplitude of the fast time-dependent component of ICa,L was associated with a large reduction of the time-independent component of ICa,L, and vice versa.
The experiments described in Fig. 7 and Fig. 8 were conducted upon isolated ventricular myocytes which were bathed in zero calcium extracellular solution. Although previous studies have shown that this is propitious for the isolation of the voltage-dependent process of inactivation of ICa,L (Hadley & Hume, 1987; Findlay, 2002b), it could nevertheless be suggested that the removal of extracellular Ca2+ could influence a number of different cellular processes and that its effect would not be confined to the removal of Ca2+-induced inactivation of ICa,L. Figure 9 therefore illustrates analysis of inactivation of ICa,L evoked by voltage steps to +80 mV in isolated myocytes that were bathed in normal extracellular solution containing 2 mm Ca2+. Analysis of the inactivation of these currents through L-type Ca2+ channels with a bi-exponential function extracted fast and slow time-dependent components of ICa,L and a time-independent component of ICa,L. In a manner similar to the results recorded above in the absence of extracellular Ca2+ (Fig. 8), the amplitudes of the fast time-dependent component of ICa,L (Fig. 9A) and the time-independent component of ICa,L (Fig. 9C) were, respectively, decreased and increased by isoproterenol. These effects were counterbalanced by carbachol. These effects of agonist stimulation upon the voltage-dependent inactivation of ICa,L therefore do not reside in a non-specific effect of withdrawal of extracellular Ca2+ upon the physiology of ventricular myocytes.
Discussion
Voltage-dependent inactivation of cardiac muscle ICa,L determines not only the kinetics of decay of Ca2+ currents in the heart but also the extent to which Ca2+-induced inactivation can contribute to this process (Findlay, 2002a,b). In this study, evidence is presented which suggests that modulation of voltage-dependent inactivation of ICa,L by sympathetic and parasympathetic agonists is quite simple. Phosphorylation of the channel by the action of PKA prevents the operation of the mechanism of voltage-dependent inactivation. This can be considered to represent an ‘off’ switch. Under basal conditions, the application of a voltage step provokes the channel molecule to adopt first a configuration that allows the opening of the channel pore and then a configuration where ion passage is occluded. The action of PKA would be to prevent the assumption of the inactivated configuration by the channel. In consequence, during a sustained voltage step, openings and closings of the ion channel would continue. There would no longer be a time-dependent decay of the current.
It was not the objective of this study to determine the intracellular signalling pathways by which β-adrenergic and m2-muscarinic agonists affect their targets. This has been extensively investigated and described in recent reviews by Méry et al. (1997) and Kamp & Hell (2000). It is agreed that neither agonist directly affects ICa,L and that both agonist systems converge upon the modulation of PKA. The effects of carbachol described here therefore correspond to reduction of the activation of PKA and reduction of the phosphorylation of the Ca2+ channel protein(s). The idea therefore that the β-adrenergic dose-dependent redistribution of kinetic behaviour in the ion channel population results from ion channel phosphorylation due to the action of PKA is reinforced by results showing that carbachol reverses the kinetic behaviour of the channels and re-establishes time-dependent decay. The process of voltage-dependent inactivation has been turned on.
There is some evidence that agonist stimulation influenced the time constants of decay of fast and slow components of the channel population (Fig. 7B and Fig. 8C and D). This might suggest a graded response of the L-type Ca2+ channel to agonist stimulation. These effects were significant at low doses of the β-adrenergic agonist, not at high doses. However, Mitarai et al. (2000) and a previous study in this laboratory (Findlay, 2002b) both showed that changes in the time course of decay were of little consequence compared with the very large difference in currents resulting from the redistribution of ion channels between the different kinetic groups. It would be interesting to investigate, using single-channel current recording methods, the kinetic behaviour characterising channels under basal conditions and that seen following agonist stimulation. These experiments would have to be conducted in the absence of divalent cations in order to avoid complications resulting from localised ion-dependent inactivation (Findlay, 2002a).
Voltage-dependent inactivation of Ca2+ channels has been the subject of extensive investigation at the molecular level (see Stotz & Zamponi, 2001, for a recent review). The objectives of that work were to investigate the mechanism and the molecular attributes necessary for the rapid time-dependent decay shown by certain Ca2+ channels. This involved the mix-and-match method of interchanging segments from Ca2+ channel molecules exhibiting different forms of inactivation. Alternatively, the role of accessory subunits of the Ca2+ channel molecular complex has been suggested to determine the behaviour of the central pore-forming α1 protein (Birnbaumer et al. 1998; Bers & Perez-Reyes, 1999). The message from the results described here, and from previous studies (Mitarai et al. 2000; Findlay, 2002b), is that voltage-dependent inactivation of the L-type Ca2+ channel current of cardiac muscle is not a fixed characteristic. Rather that either rapid or no time-dependent decay of the current are characteristics of the same molecular complex, for which an explanation at the molecular level remains to be determined. Some recent studies have suggested that interactions between different subunits of the Ca2+ channel might be subject to modulation by physiological processes (Restituito et al. 2001). It may be that this line of research may lead to the elucidation of the mechanism of the turning on and off of voltage-dependent inactivation of the L-type Ca2+ channel of cardiac muscle.
Two aspects of the experimental approach used in this investigation could be considered unusual in the context of an examination of the behaviour of a native Ca2+ channel current. First, the behaviour of the channel was evaluated from the outward flux of monovalent cations and not from the inward flux of divalent cations. Second, in part the kinetics of inactivation of the current were evaluated at +80 mV, which could be considered not to represent a physiologically relevant membrane potential. Previous studies (Hadley & Hume, 1987; Hadley & Lederer, 1991; Findlay, 2002b) have shown that the replacement of extracellular Ca2+ by Mg2+ and the monitoring of ICa,L carried by the outward flux of intracellular monovalent cations isolated the process of voltage-dependent inactivation of ICa,L (see also Fig. 5). It is admitted that the currents carried by L-type Ca2+ channels under these circumstances (Fig. 1) have the appearance of a transient outward K+-selective ion conductance, and Inoue & Imanaga (1993) described them as an A-type K+ current that was inhibited by the presence of extracellular Ca2+. However, these currents are not K+ selective (Bean et al. 1984), cannot be blocked by blockers of transient outward currents (Inoue & Imanaga, 1993; Findlay, 2002b), but can be blocked by blockers of L-type Ca2+ currents (Hadley & Hume, 1987; Inoue & Imanaga, 1993; Findlay, 2002b). They result from the asymmetric permeation of cations through Ca2+ channels (Tsien et al. 1986). The advantage of this method of recording ICa,L is twofold. First, ion-dependent inactivation of the current is avoided; this would usually result from Ca2+ influx but can also be evoked by Ba2+ and Sr2+ (Ferreira et al. 1997; Findlay, 2002a). Second, disruption of membrane surface charge and the transmembrane voltage field arising from the chelation of extracellular divalent cations to allow monovalent cation influx through L-type Ca2+ channels (Fukushima & Hagiwara, 1985; Mitarai et al. 2000; Findlay, 2002a) is countered by the presence of the impermeant Mg2+ (Hadley & Hume, 1987; Findlay, 2002b). The kinetics of decay of ICa,L were evaluated indirectly (Fig. 6) and directly (Figs 7–9). The former method involved use of the amplitude of the current evoked by a voltage step to +80 mV as a measure of the availability of the ion channel (Hadley & Hume, 1987; Hadley & Lederer, 1991; Findlay, 2002b), in a manner that was no different from the use of double-pulse voltage-clamp protocols involving test pulse voltage steps to 0 or +10 mV, with equivalent results (Findlay, 2002b). The latter method also involved use of the currents evoked by voltage steps to +80 mV as a measure of the process of inactivation. There was little difference between the kinetics of decay obtained using the direct method and those evaluated to occur at +10 mV. Separate experiments (not shown) have shown that the kinetics of voltage-dependent inactivation increase from −30 to ≈+20 mV and that further depolarisation has no further effect. The decay kinetics recorded at +80 mV therefore adequately report the maximal and physiologically relevant time course of inactivation.
The effects of the opposing agonists isoproterenol and carbachol upon ICa,L carried by Ca2+ obtained in this investigation (Fig. 1 and Fig. 2) were very similar to those observed in previous reports involving native cardiac Ca2+ currents. The unusual nature of their effects upon voltage-dependent inactivation therefore does not arise from any particular characteristic of the experiments performed here. Thus β-adrenergic stimulation evoked negative shifts of the peak of the I-V relationship and A-V curves (McDonald et al. 1994). The effects of carbachol were ‘competitive’ with those of isoproterenol (Fischmeister & Shrier, 1989). On the other hand, the observation that neither agonist directly influenced the dependence of inactivation upon membrane voltage recorded in the absence of Ca2+ is new (Fig. 5). This suggests that the shift of the relationship between total inactivation and membrane voltage effected by agonist stimulation (Fig. 2) resulted from enhanced Ca2+ influx and enhanced Ca2+-induced inactivation.
The physiological importance of the ensemble of the results obtained in this investigation lies in the observation that, when Ca2+ carried the Ca2+ current, the effects of the β-adrenergic and muscarinic agonists upon the voltage-dependent decay of the current were not observed (Fig. 3). Their modification of the behaviour of the ion channel had been over-ridden by the influence of Ca2+-induced inactivation (Findlay, 2002b). This therefore identifies quite clearly the manner by which the alteration of the behaviour of the channel molecule could be compensated for by its environment. Any mechanism or pathology interfering with the process of Ca2+-induced inactivation will therefore have consequences that will depend upon the demands placed upon the heart.
Acknowledgments
I thank Joel Derisson for isolating cells and Dr Claire Malecot for her constructive criticism of this text. This work was financed by grants from the Region Centre.
References
- Adachi-Akahane S, Leemann L, Morad M. Cross-signalling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. Journal of General Physiology. 1996;108:435–454. doi: 10.1085/jgp.108.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balke CW, Rose WC, Marban E, Wier WG. Macroscopic and unitary properties of physiological ion flux through T-type Ca2+ channels in guinea-pig heart cells. Journal of Physiology. 1992;456:247–265. doi: 10.1113/jphysiol.1992.sp019335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, Nowycky MC, Tsien RW. β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375. doi: 10.1038/307371a0. [DOI] [PubMed] [Google Scholar]
- Bers DM, Perez-Reyes E. Ca channels in cardiac myocytes: structure and function in Ca influx and intracellular Ca release. Cardiovascular Research. 1999;42:339–360. doi: 10.1016/s0008-6363(99)00038-3. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Quin N, Olcese R, Tareilus E, Platano D, Costantin J, Stefani E. Structures and functions of calcium channel β subunits. Journal of Bioenergetics and Biomembranes. 1998;30:357–375. doi: 10.1023/a:1021989622656. [DOI] [PubMed] [Google Scholar]
- Ferreira G, Yi J, Rios E, Shirokov R. Ion-dependent inactivation of barium current through L-type calcium channels. Journal of General Physiology. 1997;109:449–461. doi: 10.1085/jgp.109.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay I. Voltage- and cation-dependent inactivation of L-type Ca2+ channel currents in guinea-pig ventricular myocytes. Journal of Physiology. 2002a;541:731–740. doi: 10.1113/jphysiol.2002.019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay I. β-Adrenergic stimulation modulates Ca2+- and voltage-dependent inactivation of L-type Ca2+ channel currents in guinea-pig ventricular myocytes. Journal of Physiology. 2002b;541:741–751. doi: 10.1113/jphysiol.2002.019737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmeister R, Shrier A. Interactive effects of isoprenaline, forskolin and acetylcholine on Ca2+ current in frog ventricular myocytes. Journal of Physiology. 1989;417:213–239. doi: 10.1113/jphysiol.1989.sp017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima Y, Hagiwara S. Currents carried by monovalent cations through calcium channels in mouse neoplastic B lymphocytes. Journal of Physiology. 1985;358:255–284. doi: 10.1113/jphysiol.1985.sp015550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gera S, Byerly L. Measurement of calcium channel inactivation is dependent upon the test pulse potential. Biophysical Journal. 1999;76:3076–3088. doi: 10.1016/S0006-3495(99)77460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley RW, Hume JR. An intrinsic potential-dependent inactivation mechanism associated with calcium channels in guinea-pig myocytes. Journal of Physiology. 1987;389:205–222. doi: 10.1113/jphysiol.1987.sp016654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley RW, Lederer WJ. Ca2+ and voltage inactivate Ca2+ channels in guinea-pig ventricular myocytes through independent mechanisms. Journal of Physiology. 1991;444:257–268. doi: 10.1113/jphysiol.1991.sp018876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Calcium channel selectivity for divalent and monovalent cations. Journal of General Physiology. 1986;88:293–318. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M, Orchard CH. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during β-adrenergic stimulation. Journal of Physiology. 1997;505:385–402. doi: 10.1111/j.1469-7793.1997.385bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Imanaga I. Masking of A-type K+ channel in guinea pig cardiac cells by extracellular Ca2+ American Journal of Physiology. 1993;264:C1434–1438. doi: 10.1152/ajpcell.1993.264.6.C1434. [DOI] [PubMed] [Google Scholar]
- Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circulation Research. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- Le Guennec J-Y, Peineau N, Esnard F, Lacampagne A, Gannier F, Argibay J, Gauthier F, Garnier D. A simple method for calibrating collagenase/pronase E ratio to optimise heart cell isolation. Biology of the Cell. 1993;79:161–165. doi: 10.1111/j.1768-322x.1993.tb00906.x. [DOI] [PubMed] [Google Scholar]
- Linz KW, Meyer R. Control of L-type calcium current during the action potential of guinea-pig ventricular myocytes. Journal of Physiology. 1998;513:425–442. doi: 10.1111/j.1469-7793.1998.425bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Matsuda H. Sodium conductance in calcium channels of guinea pig ventricular cells induced by removal of external calcium ions. Pflügers Archiv. 1986;407:465–475. doi: 10.1007/BF00657502. [DOI] [PubMed] [Google Scholar]
- Méry PF, Abi-Gerges N, Vandecasteele G, Jurevicius J, Eschenhagen T, Fischmeister R. Muscarinic regulation of the L-type calcium current in isolated cardiac myocytes. Life Sciences. 1997;60:1113–1120. doi: 10.1016/s0024-3205(97)00055-6. [DOI] [PubMed] [Google Scholar]
- Mitarai S, Kaibara M, Yano K, Taniyama K. Two distinct inactivation processes related to phosphorylation in cardiac L-type Ca2+ channel currents. American Journal of Physiology - Cell Physiology. 2000;279:C603–610. doi: 10.1152/ajpcell.2000.279.3.C603. [DOI] [PubMed] [Google Scholar]
- Pelzer D, Pelzer S, McDonald TF. Properties and regulation of calcium channels in muscle cells. Reviews of Physiology, Biochemistry and Pharmacology. 1990;114:107–207. doi: 10.1007/BFb0031019. [DOI] [PubMed] [Google Scholar]
- Restituito S, Cens T, Rousset M, Charnet P. Ca2+ channel inactivation heterogeneity reveals physiological unbinding of auxiliary β subunits. Biophysical Journal. 2001;81:89–96. doi: 10.1016/S0006-3495(01)75682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham JSK. Ca2+ release-induced inactivation of Ca2+ current in rat ventricular myocytes: evidence for local Ca2+ signalling. Journal of Physiology. 1997;500:285–295. doi: 10.1113/jphysiol.1997.sp022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz SC, Zamponi GW. Structural determinants of fast inactivation of high voltage-activated Ca2+ channels. Trends in Neurosciences. 2001;24:176–181. doi: 10.1016/s0166-2236(00)01738-0. [DOI] [PubMed] [Google Scholar]
- Tiaho F, Nargeot J, Richard S. Voltage-dependent regulation of L-type cardiac Ca channels by isoproterenol. Pflügers Archiv. 1991;419:596–602. doi: 10.1007/BF00370301. [DOI] [PubMed] [Google Scholar]
- Trautwein W, Hescheler J. Regulation of cardiac L-type calcium current by phosphorylation and G proteins. Annual Review of Physiology. 1990;52:257–274. doi: 10.1146/annurev.ph.52.030190.001353. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC. Mechanisms of calcium channel modulation by β-adrenergic agents and dihyropyridine calcium agonists. Journal of Molecular and Cellular Cardiology. 1986;18:691–710. doi: 10.1016/s0022-2828(86)80941-5. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Hess P, McCleskey EW, Rosenberg RL. Calcium channels: mechanisms of selectivity, permeation and block. Annual Review of Biophysics and Biophysical Chemistry. 1987;16:265–290. doi: 10.1146/annurev.bb.16.060187.001405. [DOI] [PubMed] [Google Scholar]