

Abstract
Mutations in the voltage-dependent skeletal muscle chloride channel, ClC-1, result in dominant or recessive myotonia congenita. The Q552R mutation causes a variant of dominant myotonia with a milder phenotype, myotonia levior. To characterise the functional properties of this mutation, homodimeric mutant and heterodimeric wild-type (WT) mutant channels were expressed in tsA201 cells and studied using the whole-cell recording technique. Q552R ClC-1 mutants formed functional channels with normal ion conduction but altered gating properties. Mutant channels were activated by membrane depolarisation, with a voltage dependence of activation that was shifted by more than +90 mV compared to WT channels. Q552R channels were also activated by hyperpolarisation, and this process was dependent upon the intracellular chloride concentration ([Cl−]i). Together, these alterations resulted in a substantial reduction in the open probability at −85 mV at a physiological [Cl−]i. Heterodimeric WT-Q552R channels did not exhibit hyperpolarisation-activated gating transitions. As was the case for WT channels, activation occurred upon depolarisation, but the activation curve was shifted by 28 mV to more positive potentials. The functional properties of heterodimeric channels suggest a weakly dominant effect, a finding that correlates with the inheritance pattern and symptom profile of myotonia levior.
Vertebrate skeletal muscle is distinguished by its large resting chloride conductance (gCl), a property that is maintained by intact, voltage-gated skeletal muscle chloride channels, ClC-1 (Steinmeyer et al. 1991). Mutations in CLCN1, the gene encoding the ClC-1, lead to autosomal dominant or recessive myotonia congenita in humans (Koch et al. 1992; George et al. 1993). This disorder of skeletal myofibre excitability is due to a reduction in the sarcolemmal gCl with a consequent increase in the length constant of the sarcolemma (Bryant, 1962; Bryant & Morales Aguilera, 1971; Rüdel & Lehmann-Horn, 1985). As a result, the elevation of potassium concentration in the t-tubular lumen following electrical activity results in depolarisation of the sarcolemma, triggering a train of spontaneous action potentials (Adrian & Bryant, 1974). This ‘irritability’ of the muscle membrane manifests clinically as myotonia, an impaired relaxation of muscle following sudden, forceful contraction.
Following the linkage of Thomsen (dominant) and Becker (recessive) myotonia to CLCN1, many disease-causing mutations were identified and their properties investigated in heterologous expression systems, providing important insights into channel structure and function. Early clinical descriptions identified a discrete subgroup of patients with dominantly inherited but phenotypically mild disease, a condition termed myotonia levior (DeJong, 1966). In two of these patients, an arginine-for-glutamine substitution at position 552 of the CLCN1 gene (Q552R) was identified (Lehmann-Horn et al. 1995). When expressed in the Xenopus oocyte system, the mutant alone did not produce current. Expression was achieved in concert with wild-type (WT) human ClC-1 (hClC-1) DNA, although the functional properties were more predictive of a strongly dominant phenotype (Pusch et al. 1995b). It was shown subsequently, however, that some mutations that appeared to be non-functional in the Xenopus expression system could be expressed successfully in mammalian cells (Fahlke et al. 1997a; Zhang et al. 2000a) and in the former case, illustrated an entirely new mechanism of myotonia generation. We therefore decided to investigate the functional properties of Q552R in a mammalian cell line.
As functional ClC channels are dimeric proteins (Middleton et al. 1994; Fahlke et al. 1997b; Dutzler et al. 2002), three classes of muscle chloride channels coexist in the sarcolemma of heterozygous patients: homodimeric WT channels, heterodimeric channels consisting of one WT and one mutant subunit, and homodimeric mutant channels. In order to achieve a homogeneous population of heterodimers and to be able to characterise the functional properties of heterodimeric WT-Q552R channels, we engineered a WT-mutant concatameric construct (Fahlke et al. 1997b). Assuming equal expression and coassembly, these channels represent the largest fraction (50 %) of functional chloride channels in heterozygous patients, and their functional properties thus play a critical role in determining the mode of inheritance and resting sarcolemmal gCl in these patients.
We report here the functional characterisation of homo- and heterodimeric Q552R channels under various ionic conditions. Our results demonstrate gating alterations of mutant channels that account fully for the clinical features of myotonia levior and its inheritance pattern. In addition, based on the location of this mutation, we speculate as to the underlying mechanism of hyperpolarisation-activated gating for this and other muscle chloride channel mutations.
Methods
Construction of mutants
Site-directed mutagenesis of the hClC-1 and construction of concatamers were performed as described previously (Fahlke et al. 1997b). Briefly, the mutation Q552R was introduced into hClC-1 using a single-step polymerase chain reaction mutagenesis strategy. Two independent recombinant mutant clones were then subcloned back into the full-length hClC-1 cDNA in the pRc/CMV vector for expression studies.
Cell culture and transfection
tsA201 cells are a simian virus 40 (SV40), T-antigen-expressing derivative of the human embryonic kidney cell line, HEK-293. Cells were grown in Earle's minimum essential medium supplemented with 10 % fetal calf serum and maintained at 37 °C with 5 % carbon dioxide. Transient transfection was performed by means of a lipid-assisted technique (Effectene Transfection Kit, Qiagen, Hilden, Germany) using ≈1.0 μg of cDNA. Cells were transiently transfected with either pRc/CMV-Q552R/hClC-1 or the concatameric pRc/CMV-hClC-1/WT-Q552R construct to study the properties of the mutant homodimer or WT-mutant heterodimer, respectively. Cotransfection with the reporter plasmid pEGFP-N1 (‘enhanced green fluorescent protein’, Clontech, Heidelberg, Germany) was used to identify successfully transfected cells.
Electrophysiology
Standard whole-cell recordings (Hamill et al. 1981) were performed at room temperature (20-22 °C) using an EPC9 amplifier and Pulse software (HEKA Elektronik, Lambrecht, Germany). Pipettes were pulled from borosilicate glass (Harvard Apparatus) and heat polished to a resistance of 1.2-2.0 MΩ. Linear leakage or capacitive currents were not subtracted. The series resistance was compensated by an analog procedure to ensure a calculated voltage error due to series resistance below 5 mV. The standard external (bath) solution contained (mm): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2 and 5 Hepes adjusted to pH 7.4 with NaOH. In experiments to test the relative anion permeability, an external solution containing (mm): 140 NaX (where X− + Br−, NO3−, I− or thiocyanate ion (SCN−)), 4 KCl, 2 CaCl2, 1 MgCl2, 5 Hepes, pH 7.4 was used.
The standard internal (pipette) solution contained (mm): 130 CsCl, 2 MgCl2, 5 EGTA and 10 Hepes adjusted to pH 7.4 with CsOH. In some experiments, NaCl was substituted for CsCl in the internal solution without appreciable difference in results. To obtain different chloride gradients, chloride in the external or internal solutions was substituted by equimolar concentrations of glutamate. No difference in results was observed when using sodium glutamate or caesium glutamate in the internal solution. For experiments with external pH (pHo) + 8.5, Hepes in the external solution was substituted with an equimolar concentration of 3-[(1,1-dimethyl-2-hydroxyethyl)amino]-2-hydroxypropanesulphonic acid, for those with pHo + 6.5 with an equimolar concentration of 2-(N-morpholino)ethanesulphonic acid. Cells were clamped to either 0 mV (for standard internal solution) or −85 mV (for low-chloride internal solution) for at least 5 s between two test sweeps. Standard solutions were used unless stated otherwise. Agar bridges were used to connect the bath solution and, when the low-chloride solution was used, the pipette solution to the amplifier. Junction potentials calculated using the JPCalc software (Barry, 1994) and offset potentials measured at the end of each experiment were used to correct results.
Data analysis
Data were analysed using a combination of Clampfit (Axon Instruments), Excel (Microsoft, Unterschleissheim, Germany) and Sigma Plot 4.0 (Jandel Scientific, San Raphael, CA, USA). To determine the voltage dependence of the relative open probability (Po) of a channel, the instantaneous current amplitudes at −105 mV were normalised to their extrapolated maximum value and then plotted versus the test potential. Where possible, a single Boltzmann function and a voltage-independent value of the form:
was fitted to the resulting curve, where Imax is the extrapolated maximum current, V1/2 is the voltage at half-maximal activation and kv is the slope. For data that a single Boltzmann function could not be fitted to, the instantaneous current amplitudes at a constant test pulse to −105 mV were determined following a 400 ms prepulse to different voltages. Current amplitudes were normalised to their maximum value and the normalised data were then plotted versus the preceding potential to yield the voltage dependence of Po. To determine the relative anion permeability, reversal potentials were measured in cells perfused with standard internal solution in an external solution in which NaCl was completely substituted with sodium salts of different anions. Permeability ratios PX/PCl were calculated using the Goldman-Hodgkin-Katz equation, as described previously (Fahlke et al. 1997a). All data are given as the mean ± s.e.m., and n = number of observations. Statistical analysis was performed using Student's paired t test, with the level of statistical significance set at P ≤ 0.05.
Results
Functional expression of Q552R hClC-1 channels in tsA201 cells
The Q552R mutation causes pronounced changes in voltage-dependent gating. Figure 1 illustrates representative macroscopic current recordings from cells expressing WT or Q552R hClC-1 channels. Chloride currents conducted by WT hClC-1 rose instantaneously during hyperpolarising as well as during depolarising voltage steps. At more negative potentials, a consecutive slower deactivation to a non-zero, steady-state level was observed, while depolarising voltages produced currents that were almost time independent (Fig. 1A). In contrast, membrane hyperpolarisation of Q552R hClC-1-expressing cells elicited a time-dependent increase in the current amplitude. Membrane depolarisation additionally caused current activation, resulting in prominent current deactivation at a subsequent voltage step to −105 mV (Fig. 1B, inset).
Figure 1. Whole-cell currents and current-voltage relationships for wild-type (WT) and Q552R human voltage-gated skeletal muscle chloride channels (hClC-1 channels) expressed in tsA201 cells.
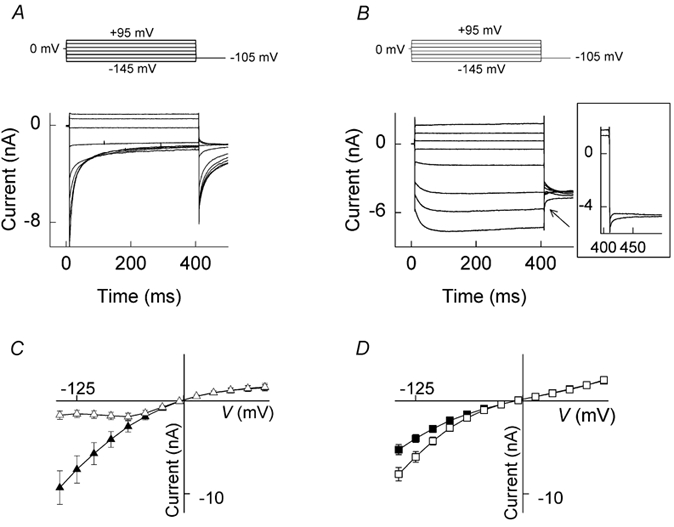
From a holding potential of 0 mV, currents were elicited in 20 mV intervals over the voltage range −145 to +95 mV followed by a fixed pulse of −105 mV for 90 ms. A, current traces from a cell expressing WT hClC-1 channels. B, current traces from a cell expressing Q552R hClC-1 channels (only alternate current traces are shown for clarity). The arrow points to a tail current deactivation following current activation at +95 mV. The inset shows the tail currents following pulses of +75 and +95 mV. C and D, the voltage dependence of the instantaneous (filled symbols) and late (open symbols) current amplitudes for WT and Q552R hClC-1 channels, respectively. Mean values ± s.e.m. are given for n = 6 (WT) and n = 15 (Q552R) cells.
Both WT and Q552R hClC-1 channels were expressed well, with a mean instantaneous current amplitude at −125 mV of 7.4 ± 1.5 nA for WT (n = 6) and a peak current amplitude of 4.1 ± 0.4 nA for Q552R (n = 15) under these experimental conditions (Fig. 1C and D). Pore properties appeared to be only minimally affected by the mutation. WT and mutant channels were both inwardly rectifying, although rectification appeared to be somewhat less pronounced in the case of Q552R (Fig. 1D). Permeability ratios (PX/PCl) determined from measurement of bi-ionic reversal potentials in the presence of foreign anions revealed a permeability sequence of: SCN− (2.1) > Cl− (1.0) > Br− (0.8) > NO3− (0.5) > I− (0.3) for Q552R. This differed from that of WT (Cl− (1.0) > SCN− (0.9) > Br− (0.7) > NO3− (0.6) > I− (0.3)) only in the selectivity of SCN− over Cl−.
Q552R channels exhibit a high resting Po at high [Cl−]i
Myotonia arises from a significant reduction of the macroscopic gCl of muscle at the resting membrane potential. This conductance is dependent upon (1) the number of functional chloride channels, (2) their unitary conductance and (3) the steady-state Po. We investigated the voltage dependence of the relative Po of WT and mutant channels in standard internal and external solutions by plotting the normalised instantaneous current amplitude obtained at a test pulse to −105 mV versus the prepulse potential. For WT channels, this activation curve can be fitted with a single Boltzmann function, assuming a minimum value at very negative potentials and rising steadily with increasingly positive potentials (Fig. 2). Under our standard conditions ([Cl−]i + 134 mm), the voltage dependence of the relative Po of Q552R channels exhibited a distinctly unusual curve of an inverted bell-shape, compatible with the superposition of two Boltzmann distributions with inverted voltage dependence (Fig. 2). No appreciable difference in results was obtained when using CsCl or NaCl in the internal solution. Unlike many of the dominant myotonia mutations analysed to date, which cause a rightward shift in the voltage-activation curve, thereby lowering the resting Po (Pusch et al. 1995b; Beck et al. 1996; Wagner et al. 1998; Zhang et al. 2000a), the hyperpolarisation-induced gating of Q552R channels actually caused the relative Po to be larger than that of WT at the resting membrane potential (-85 mV). This, however, would be expected to result in an increase rather than a decrease in sarcolemmal gCl and so could not account for the manifestation of myotonia.
Figure 2. Voltage dependence of the relative open probability (Po) for WT and Q552R hClC-1 channels.
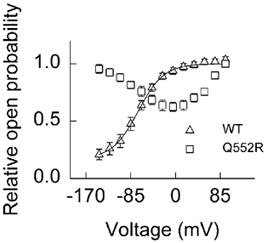
Currents were recorded from cells using an internal (pipette) solution containing a chloride concentration ([Cl−]) of 134 mm (i.e. standard internal solution). Values for relative Po were derived from normalised peak tail currents at −105 mV and plotted versus the test potential. Data from WT channels (▵, n = 11) can be fitted to a single Boltzmann distribution (continuous line). Data from Q552R channels (□, n = 15) produced an inverted bell-shaped curve with a relative Po higher than that of WT at a resting membrane potential of −85 mV.
Mutant channels exhibit a low Po at low [Cl−]i
Gating of ClC channels is known to be modulated by permeant anions (Richard & Miller, 1990; Pusch et al. 1995a). In particular, two myotonia-generating mutations have shown a profound reduction in Po under low (physiological) chloride conditions (Fahlke et al. 1995; Zhang et al. 2000b). We therefore investigated further the gating of Q552R hClC-1 channels under physiological conditions by substituting chloride in the internal solution with an equimolar concentration of glutamate in order to achieve a [Cl−]i of 5 mm. WT channels exhibited similar gating properties at high and low [Cl−]i (Fig. 3A, compare Fig. 1A). In contrast, a radical alteration in the voltage dependence of mutant channels was observed. The hyperpolarisation-induced activation was now abolished, while the depolarisation-induced activation remained unaltered under these conditions (Fig. 3B). This alteration of mutant gating resulted in a dramatic reduction in macroscopic gCl (Fig. 3C). The current amplitude at −125 mV was significantly reduced from 7.1 ± 0.9 nA for WT channels (n = 7) to 0.49 ± 0.13 nA for Q552R channels (n = 9) in low-chloride solution (P < 0.001).
Figure 3. Effect of low internal [Cl−] ([Cl−]i) on the gating of WT and Q552R hClC-1 channels.
Currents were recorded from cells using an internal (pipette) solution containing a [Cl−] of 5 mm. From a holding potential of 0 mV, currents were elicited in 20 mV intervals over the voltage range −145 to +95 mV followed by a fixed pulse of −105 mV. A, a representative current family from a cell expressing WT hClC-1 channels. B, the averaged current traces from six cells expressing Q552R hClC-1 channels. C, voltage dependence of the instantaneous current amplitude for Q552R hClC-1-expressing cells at high (▪, n = 7) and low (□, n = 9) [Cl−]i. D, voltage dependence of the relative Po for WT (▵, n = 8) and Q552R (□, n = 9) at a [Cl−]i of 5 mm. The continuous lines represent fits of a Boltzmann distribution.
The decrease in macroscopic current amplitude at low [Cl−]i was due to a substantial alteration in the activation curve of mutant channels. Under these ionic conditions, relative Po was conspicuously reduced, predominantly in the negative voltage range (Fig. 3D), and could now be described by a single Boltzmann function. When compared with WT hClC-1 channels under the same conditions, the value of V1/2 was shifted by +90 mV towards more positive potentials (Table 1). The relative Po at −85 mV for mutant channels (0.23 ± 0.03, n = 9) was now smaller than that of WT (0.59 ± 0.05, n = 8) and remained reduced throughout the physiological voltage range.
Table 1.
Boltzmann paramaters of mutant channels at high and low internal chloride concentrations([Cl−]i)
| high [Cl−]i | low [Cl−]i | |||||
|---|---|---|---|---|---|---|
| Channel type | n | V1/2(mV) | kv(mV) | n | V1/2(mV) | kv(mV) |
| WT | 11 | −71.1 ± 2.1 | 24.3 ± 1.8 | 8 | −92.5 ± 3.7 | 20.3 ± 3.1 |
| Q552R | 15 | — | — | 9 | 8.5 ± 5.9 | 42.3 ± 5.6 |
| WT-Q552R | 7 | −55.9 ± 9.7 | 51.1 ± 10.1 | 6 | −63.7 ± 5.6 | 41.2 ± 5.8 |
Values for V1/2 (half -maximal activation voltage) and kv (slope) were obtained as discribed in methods, and voltages have been corrected to account for liquid junction potentials. WT = wild-type channels. Empty fields are given where the could not be fitted with a single Boltzmann function. Results are expressed as means ± s.e.m.
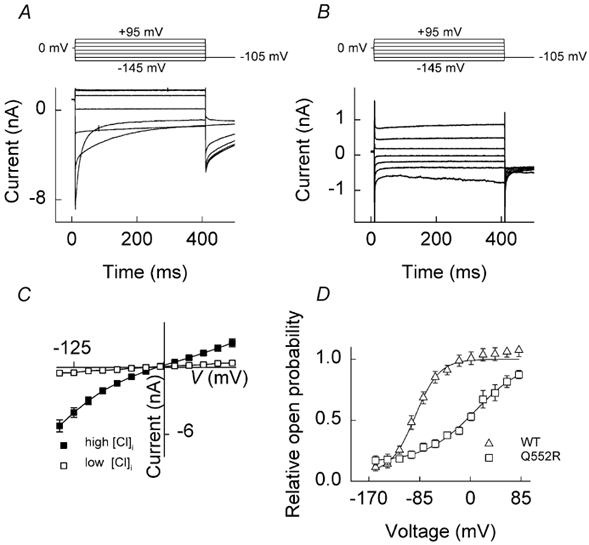
Mutant channels display two separate gating processes
The predominant effect of low [Cl−]i at negative voltages suggested the possibility of two separate gating processes with opposing voltage dependence in mutant channels. We investigated this further by measuring current responses to a fixed number of voltage steps following various conditioning pulses (Fig. 4). Currents were recorded from cells using an internal (pipette) solution containing 134 mm [Cl−] (i.e. standard internal solution). Channels were first conditioned to 0, −125 or +125 mV, then stepped from −145 to +95 mV, and finally held at a fixed pulse of −125 mV. After a conditioning pulse to 0 mV, hyperpolarising steps elicited a time-dependent increase in the current amplitude due to channel activation (Fig. 4A). Following a negative potential (-125 mV), no activation phase was visible at the onset of the hyperpolarising pulse. When, however, a positive conditioning pulse (+125 mV) was employed, membrane hyperpolarisation induced a fast deactivation, in clear contrast to the other two experimental protocols and quite similar to that of WT hClC-1 (Fig. 4C). These and the foregoing experiments with low [Cl−]i demonstrate that Q552R hClC-1 channels exhibit two discrete gating processes: a slow activation upon membrane hyperpolarisation and a fast activation upon membrane depolarisation. At high [Cl−]i, hyperpolarisation-activated gating is prominent, but mutant channels retain the ability to function in a WT (deactivating) fashion under certain conditions of voltage and [Cl−].
Figure 4. Current responses to different voltage steps determined after three distinct conditioning pulses in cells expressing Q552R hClC-1 channels.
Current responses of a single cell to a pulse protocol consisting of a fixed conditioning pulse and a variable pre-pulse (from −145 mV to +95 mV in 40 mV intervals), followed by a fixed step to −125 mV. The following conditioning potentials were applied: 0 mV (A), −125 mV (B) and +125 mV (C).
The separate gating processes of mutant channels are differentially modulated by external chloride and pHo
For WT channels, reducing the external chloride concentration ([Cl−]o) has been observed to cause a parallel shift in the voltage-activation curve to more positive potentials (Rychkov et al. 1996, 1998), demonstrating that binding of the chloride ion is required for depolarisation-induced activation. To examine the effect of [Cl−]o reduction on Q552R, we substituted chloride in the external solution with glutamate to achieve a [Cl−]o of 20 mm while keeping [Cl−]i at 134 mm. Currents recorded under these conditions were characterised by hyperpolarisation-induced activation. The voltage dependence of relative Po was reduced throughout the physiological voltage range, this time with the effect being most marked at positive potentials (Fig. 5A). This contrasts with the effect of low [Cl−]i, where a striking reduction in relative Po was observed at negative voltages, but curves tended to converge at more positive potentials. Therefore, whereas hyperpolarisation-activated gating appears to be dependent upon [Cl−]i, depolarisation-induced channel activation is, as for WT channels, dependent upon [Cl−]o.
Figure 5. Effect of extracellular chloride and pH on mutant channel Po.
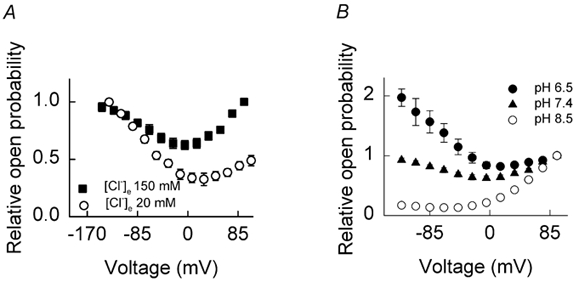
All currents were recorded from cells using an internal (pipette) solution containing a [Cl−] of 134 mm (i.e. standard internal solution). Following a 400 ms pre-pulse to different voltages, the instantaneous current amplitude at a constant test pulse of −105 mV was determined and normalised to its maximum value. The normalised data were then plotted versus the preceding potential to yield the voltage dependence of relative Po. Results are plotted as means ± s.e.m. A, currents recorded from cells transfected with Q552R hClC-1 and bathed in standard (150 mm) or low-chloride (20 mm) external solution. ▪, values obtained at 150 mm external [Cl−] (n = 15). ○, values obtained at 20 mm external [Cl−] (n = 5). B, cells were bathed in standard external solution. Values for external pH were: pH 6.5 (•), pH 7.4 (▴), pH 8.5 (○), n = 4 cells in each case.
External as well as internal protons are known to modulate WT hClC-1 gating (Fahlke et al. 1996; Rychkov et al. 1996). External acidification causes an increase in the minimum Po at negative potentials without altering the midpoint or slope factor of the activation curve (Fahlke et al. 1996). The influence of pHo on Q552R channels is illustrated in Fig. 5B. Lowering pHo from 7.4 to 6.5 increased the Po of the channel mostly in the negative voltage range. Elevating pHo to 8.5 had the opposite effect, decreasing the Po at predominantly negative potentials. As the voltage became more positive, modulation of pHo had less of an effect, and the three curves tended to converge. Just as [Cl−]o had a relatively selective effect upon the depolarising current component of mutant channels, pHo appears to selectively modulate the hyperpolarisation-induced gating.
Functional analysis of the WT-Q552R heterodimer
To study the properties of heterodimeric channels, we used a concatameric construct linking two hClC-1 coding sequences in a single open reading frame. This has been shown to result in a homogenous population of these channels in transfected cells, with no appreciable effect on channel function due to the peptide linker sequence (Fahlke et al. 1997b). A representative current family from a cell expressing the WT-Q552R concatameric construct is illustrated in Fig. 6A. Heterodimeric channels are inwardly rectifying and display gating properties that are similar, but not identical, to those of WT, displaying deactivation upon membrane hyperpolarisation and activation at more positive potentials. There are no indications of the hyperpolarisation-induced activation steps seen with the mutant. A plot of relative Po as a function of membrane voltage for heterodimeric channels revealed a shift in the voltage dependence towards more positive potentials (Fig. 6B, Table 1). In contrast to the homodimeric mutant, lowering [Cl−]i had only a slight effect upon the parameters of the heterodimer (Table 1). A comparison of voltage-activation curves for WT and WT-Q552R under physiological conditions illustrates a shift in the V1/2 of heterodimeric channels of +28 mV (Table 1). This results in a reduction of the Po at the resting membrane potential from 0.59 ± 0.05 (WT) to 0.37 ± 0.05 (WT-Q552R) and, consequently, a reduction in sarcolemmal gCl. Unlike mutant channels, which display hyperpolarisation-induced activation and a sensitivity to [Cl−]i, WT-Q552R hClC-1 channels more closely resemble WT, displaying a depolarisation-induced activation that is only slightly affected by a reduction in [Cl−]i. However, heterodimeric channels differ from WT channels in the effect of changing the pHo. External acidification to pH 6.5 led to a hyperpolarising shift of the voltage-activation curve (V1/2 + −70.7 ± 2.9 mV, n = 4) and an increase in the minimum Po at negative potentials (not illustrated). Raising pHo to 8.5 revealed a minimal shift of the voltage dependence to more positive potentials (V1/2 + −43.5 ± 4.2 mV, n = 4).
Figure 6. Functional properties of WT-Q552R heterodimeric channels.
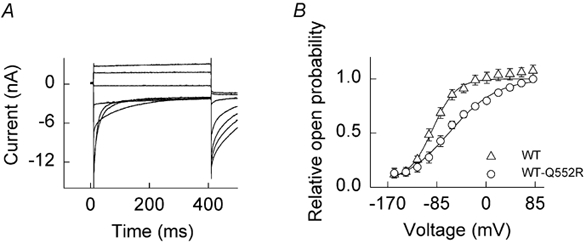
A, current responses of a Q552R-WT hClC-1-expressing cell. From a holding potential of 0 mV, currents were elicited in 20 mV intervals over the voltage range −145 to +95 mV followed by a fixed pulse of −105 mV. Only alternate traces are shown for clarity. B, voltage dependence of the relative open probability for WT hClC-1 channels (▵, n = 8) and WT-Q552R hClC-1 channels (○, n = 6) at a [Cl−]i of 5 mm. Continuous lines represent fits of a Boltzmann distribution.
Discussion
Until now, dominantly inherited myotonia mutations have been reported to alter the function of muscle chloride channels in just two ways. All but one of the investigated mutations cause a rightward shift in the activation curve (Pusch et al. 1995b; Beck et al. 1996; Wagner et al. 1998; Zhang et al. 2000a), thereby reducing sarcolemmal gCl with consequent myofibre hyperexcitability. The second functional effect, described for the G230E mutation, is an alteration of pore properties resulting in an increased cation permeability (Fahlke et al. 1997a). We investigated the functional alterations of Q552R channels and observed that under standard conditions, mutant channels display gating properties quite unlike those previously reported for Thomsen, or Becker, myotonia. Q552R channels in fact display two separate gating processes of opposite voltage dependence. A hyperpolarisation-activated and a depolarisation-activated component coexist, leading to a very abnormal voltage dependence of the relative Po with peak values at the extremes of the voltage scale and a minimum value close to 0 mV (Fig. 2). Hyperpolarisation-activated gating has already been described for two recessive ClC-1 mutations, D136G (Fahlke et al. 1995) and G499R (Zhang et al. 2000b). In both cases, these mutations only exhibited hyperpolarisation-activated gating with no evidence of activation upon depolarisation. The distinguishing feature of Q552R channels is that they exhibit both hyperpolarisation and depolarisation-activated gating, a finding not previously reported for mutant ClC-1 channels.
The hyperpolarisation-activated gating of mutation Q552R is strongly dependent upon [Cl−]i. At the low [Cl−]i present in muscle fibres (Dulhunty, 1978), this gate is closed and the voltage dependence of the depolarisation-activated component is shifted dramatically to the right when compared with that of WT channels (Fig. 3D). Under these conditions, homodimeric mutant channels therefore have a significantly reduced sarcolemmal gCl at the resting membrane potential. Similarly, it was shown that the conductance of D136G and G499R channels was markedly reduced at a physiological [Cl−]i. The properties of the myotonia levior mutation therefore appear to combine two of the gating alterations observed in myotonia congenita (i.e. rightward shift of the activation curve and the induction of hyperpolarisation-activated, chloride-dependent gating), as reported for the recessive mutations D136G and G499R.
Dominant and recessive mutations causing a similar alteration of gating (hyperpolarisation activation) in homodimeric channels differ, however, in the functional properties of their heterodimeric channels. WT-Q552R heterodimeric channels display a voltage dependence of the relative Po that is similar to that of WT but shifted to more positive potentials. In the sarcolemma of heterozygous patients, two classes of dysfunctional, dimeric chloride channels coexist (WT-mutant and mutant-mutant). The experimentally determined relative Po of Q552R homo- and heterodimeric channels allow a prediction regarding the reduction in gCl caused by these dysfunctional channels. If one assumes that WT and mutant subunits are equally expressed, our results predict a reduction in the sarcolemmal gCl to about 60 % of control levels. Decreasing mutant protein would raise this value, bringing the sarcolemmal gCl closer to that of WT. These results imply that the Q552R mutant imparts a weak though dominant effect, a finding entirely in keeping with the clinical description of myotonia levior. WT-D136G channels, on the other hand, have an increased Po at negative potentials (Fahlke et al. 1997b). As a result, heterodimeric channels do not reduce the sarcolemmal gCl and so cannot produce myotonic symptoms. This finding is fully compatible with the observed recessive pattern of inheritance. Thus, for both a dominant (Q552R) and a recessive (D136G) myotonia mutation, the functional properties of heterodimeric channels appear to correlate with the inheritance pattern and severity of the disease.
Q552R channels exhibited hyperpolarisation-activated gating only in the homodimeric state. The complete absence of this gating pattern in heterodimeric channels is in agreement with the notion that the appearance of this novel gating process requires the presence of the particular mutation in both subunits. Hyperpolarisation-activated gating occurring via a cooperative conformational change of both subunits has been described for several ClC WT and mutant channels (Thiemann et al. 1992; Fahlke et al. 1995; Rychkov et al. 1996; Pusch et al. 1997; Zhang et al. 2000b) as well as for two novel dominant mutations, S132C and T550M (Wu et al. 2002). At present, the mechanisms underlying hyperpolarisation-induced activation in hClC-1 are not understood.
Fahlke et al. (1995) suggested that the D136G mutation inactivates voltage-sensing processes that are coupled to the opening and closing of a gate that is common to both mutant and WT channels. In D136G channels, the opening of this gate is coupled to the [Cl−]i, resulting in hyperpolarisation-induced activation. In contrast, in WT channels a voltage-sensing channel component (whose function is abolished by the D136G mutation) controls the same gate, resulting in depolarisation-activated gating. This model cannot explain our results for Q552R channels, as the hyperpolarisation or depolarisation gating components can be selectively modified by alteration of the pH or anion composition on both sides of the membrane. These findings suggest that rather than having a single gate that is capable of both hyperpolarisation- and depolarisation-induced activation, mutant channels possess two separate gating processes that are under the control of discrete molecular mechanisms.
Zhang et al. (2000b), in their study on G499R, suggested that the introduction of a positive charge at this residue leads to the addition of a new chloride binding site at the intracellular site of the channel. Occupation of this novel binding site at negative voltages was suggested to open the channel and thus to cause hyperpolarisation-induced activation of G449R mutant channels. The recently reported high-resolution structure of a prokaryotic ClC channel (Dutzler et al. 2002) demonstrated that the corresponding residue (G372) in this homologue is located at the extracellular side of the membrane, thus refuting this possible explanation.
The atomic structure of a ClC channel allows predictions about the location of the currently known hClC-1 mutations that cause this particular type of gating (Fig. 7). All mutations that give rise to hyperpolarisation-induced activation are located on the extracellular side of the membrane. There are two clusters of residues visible in this diagram. One contains G50 (corresponding to the S132C mutation, Wu et al. 2002), E54 (D136G, Fahlke et al. 1995) and R147 (K231C, Fahlke et al. 1997c). At present, one can only speculate as to the role of this cluster in hClC-1 gating. The second cluster, comprising residues T416 (T550M, Wu et al. 2002) and N418 (Q552R) is located very close to the dimer interface. The finding that each of these mutants exhibits hyperpolarisation-activated gating only in the homodimeric state demonstrates that a subunit interaction is necessary in order to produce these gating steps. One might therefore speculate that the Q552R mutation, similar to the T550M mutation, interferes with a complex functional interaction between both halves of the channel. As depolarisation-induced gating exhibits gating steps that require an interaction between the two subunits (Fahlke et al. 1997b; Saviane et al. 1999), such an action would also predict the slight functional alterations observed in the WT-Q552R heterodimeric channels.
Figure 7. Location of residues at which mutations cause hyperpolarisation-induced activation.
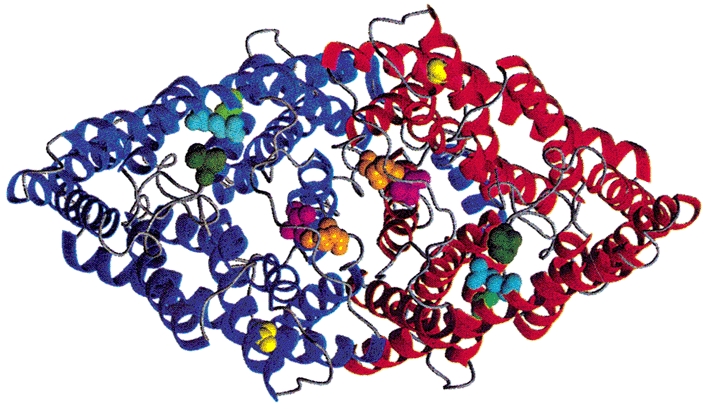
Backbone fold of the Salmonella typhimurium ClC dimer, StClC (Dutzler et al. 2002), shown from the extracellular side. The locations of corresponding hClC-1 mutations are shown as space-filling models: A52 (S132C), light green; E54 (D136G), cyan; R147 (K231C), dark green; G372 (G499R), yellow; T416 (T550M), magenta; N418 (Q552R), orange. This figure was generated with the program MOLMOL (Koradi et al. 1996).
Our results are in contrast to those obtained with Q552R channels in the Xenopus oocyte system. In the latter case, currents could not be recorded from homodimeric channels with the two-microelectrode voltage-clamp technique (Pusch et al. 1995b). In WT-mutant coexpression experiments, on the other hand, a large shift in Po (implying a large reduction in gCl and prominent myotonia) was observed. What is the reason for the discrepancy in these findings? There is no perfect heterologous expression system for the study of ion channels and each system has its limitations. There are, however, several arguments in favour of the mammalian system in this case. Mammalian cells have fewer endogenous chloride currents (Barish, 1983; Kowdley et al. 1994), channel expression in tsA cells is robust, and more importantly, several ClC-1 mutants that failed to express in oocytes have subsequently been expressed well in mammalian cells (Fahlke et al. 1997a; Zhang et al. 2000a). Finally, the functional properties of WT-mutant channels described here correlate closely with the clinical description, indicating our results to be truly representative of the in vivo situation.
In conclusion, the data demonstrate that Q552R is a functional mutation in the mammalian expression system and displays a novel mechanism of hClC-1 gating. The findings also provide further insights into the nature of hyperpolarisation-activated gating in hClC-1 channels and suggest that a degree of subunit interaction is necessary in order to produce these gating steps. Future work will lead to a more precise determination of the mechanisms involved.
Acknowledgments
We would like to thank Dr Patricia Hidalgo for helpful discussions and D. Van De Carre and Dr A. L. George Jr for providing the pRcCMV-Q552R hClC-1 expression clones. This work was supported by the IZKF, Ulm (to A.R.), the MDA (to C.F.) and the Deutsche Forschungsgemeinschaft (to C.F.: Fa 301/4-1, FOR450/1).
References
- Adrian RH, Bryant SH. On the repetitive discharge in myotonic muscle fibres. Journal of Physiology. 1974;240:505–515. doi: 10.1113/jphysiol.1974.sp010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barish ME. A transient calcium-dependent chloride current in the immature Xenopus oocyte. Journal of Physiology. 1983;342:309–325. doi: 10.1113/jphysiol.1983.sp014852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. Journal of Neuroscience Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Beck CL, Fahlke Ch, George AL., Jr Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proceedings of the National Academy of Sciences of the USA. 1996;93:11248–11252. doi: 10.1073/pnas.93.20.11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant SH. Muscle membrane of normal and myotonic goats in normal and low external chloride. Federal Proceedings. 1962;21:312. [Google Scholar]
- Bryant SH, Morales Aguilera A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. Journal of Physiology. 1971;219:367–383. doi: 10.1113/jphysiol.1971.sp009667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJong JGY. Myotonia levior. In: Kuhn E, editor. Progressive Muskeldystrophie Myotonie Myasthenie. Heidelberg: Springer; 1966. pp. 255–259. [Google Scholar]
- Dulhunty AF. The dependence of membrane potential on extracellular chloride concentration in mammalian skeletal muscle fibres. Journal of Physiology. 1978;276:67–82. doi: 10.1113/jphysiol.1978.sp012220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3. 0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Fahlke Ch, Beck CL, George AL., Jr A mutation in autosomal dominant myotonia congenita affects pore properties of the muscle chloride channel. Proceedings of the National Academy of Sciences of the USA. 1997a;94:2729–2734. doi: 10.1073/pnas.94.6.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke Ch, Knittle T, Gurnett CA, Campbell KP, George AL., Jr Subunit stoichiometry of human muscle chloride channels. Journal of General Physiology. 1997b;109:93–104. doi: 10.1085/jgp.109.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke Ch, Rosenbohm A, Mitrovic N, George AL, Jr, Rüdel R. Mechanism of voltage-dependent gating in skeletal muscle chloride channels. Biophysical Journal. 1996;71:695–706. doi: 10.1016/S0006-3495(96)79269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke Ch, Rüdel R, Mitrovic N, Zhou M, George AL., Jr An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron. 1995;15:463–472. doi: 10.1016/0896-6273(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Fahlke Ch, Yu HT, Beck CL, Rhodes TH, George AL., Jr Pore-forming segments in voltage-gated chloride channels. Nature. 1997c;390:529–532. doi: 10.1038/37391. [DOI] [PubMed] [Google Scholar]
- George AL, Jr, Crackower MA, Abdalla JA, Hudson AJ, Ebers GC. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita) Nature Genetics. 1993;3:305–310. doi: 10.1038/ng0493-305. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Koch MC, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmann-Horn F, Grzeschik KH, Jentsch TJ. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. Journal of Molecular Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Kowdley GC, Ackerman SJ, John JE, Jones LR, Moorman JR. Hyperpolarization-activated chloride currents in Xenopus oocytes. Journal of General Physiology. 1994;103:217–230. doi: 10.1085/jgp.103.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann-Horn F, Mailänder V, Heine R, George AL., Jr Myotonia levior is a chloride channel disorder. Human Molecular Genetics. 1995;4:1397–1402. doi: 10.1093/hmg/4.8.1397. [DOI] [PubMed] [Google Scholar]
- Middleton RE, Pheasant DJ, Miller C. Purification, reconstitution, and subunit composition of a voltage-gated chloride channel from Torpedo electroplax. Biochemistry. 1994;33:13189–13198. doi: 10.1021/bi00249a005. [DOI] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Jentsch TJ. Temperature dependence of fast and slow gating relaxations of ClC-0 chloride channels. Journal of General Physiology. 1997;109:105–116. doi: 10.1085/jgp.109.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Rehfeldt A, Jentsch TJ. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 1995a;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- Pusch M, Steinmeyer K, Koch MC, Jentsch TJ. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the ClC-1 chloride channel. Neuron. 1995b;15:1455–1463. doi: 10.1016/0896-6273(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Richard EA, Miller C. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 1990;247:1208–1210. doi: 10.1126/science.2156338. [DOI] [PubMed] [Google Scholar]
- Rüdel R, Lehmann-Horn F. Membrane changes in cells from myotonia patients. Physiological Reviews. 1985;65:310–356. doi: 10.1152/physrev.1985.65.2.310. [DOI] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Astill DS, Roberts ML, Jentsch TJ, Bretag AH. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. Journal of Physiology. 1996;497:423–435. doi: 10.1113/jphysiol.1996.sp021778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Roberts ML, Jentsch TJ, Bretag AH. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. Journal of General Physiology. 1998;111:653–665. doi: 10.1085/jgp.111.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C, Conti F, Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. Journal of General Physiology. 1999;113:457–467. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S, Jentsch TJ. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature. 1991;354:304–308. doi: 10.1038/354304a0. [DOI] [PubMed] [Google Scholar]
- Thiemann A, Gründer S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- Wagner S, Deymeer F, Kürz LL, Benz S, Schleithoff L, Lehmann-Horn F, Serdaroglu P, Özdemir C, Rüdel R. The dominant chloride channel mutant G200R causing fluctuating myotonia: clinical findings, electrophysiology and channel pathology. Muscle and Nerve. 1998;21:1122–1128. doi: 10.1002/(sici)1097-4598(199809)21:9<1122::aid-mus2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Wu FF, Ryan A, Devaney J, Warnstedt M, Korade-Mirnics Z, Poser B, Escriva MJ, Pegoraro E, Yee AS, Felice KJ, Giuliani MJ, Mayer RF, Mongini T, Palmucci L, Marino M, Rüdel R, Hoffman EP, Fahlke Ch. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain. 2002 doi: 10.1093/brain/awf246. in the Press. [DOI] [PubMed] [Google Scholar]
- Zhang J, Bendahhou S, Sanguinetti MC, Ptacek LJ. Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology. 2000a;54:937–942. doi: 10.1212/wnl.54.4.937. [DOI] [PubMed] [Google Scholar]
- Zhang J, Sanguinetti MC, Kwiecinski H, Ptacek LJ. Mechanism of inverted activation of ClC-1 channels caused by a novel myotonia congenita mutation. Journal of Biological Chemistry. 2000b;275:2999–3005. doi: 10.1074/jbc.275.4.2999. [DOI] [PubMed] [Google Scholar]