

Abstract
The regulation of a K+ current activating during oscillatory electrical activity (IK,slow) in an insulin-releasing β-cell was studied by applying the perforated patch whole-cell technique to intact mouse pancreatic islets. The resting whole-cell conductance in the presence of 10 mm glucose amounted to 1.3 nS, which rose by 50 % during a series of 26 simulated action potentials. Application of the KATP-channel blocker tolbutamide produced uninterrupted action potential firing and reduced IK,slow by ≈50 %. Increasing glucose from 15 to 30 mm, which likewise converted oscillatory electrical activity into continuous action potential firing, reduced IK,slow by ≈30 % whilst not affecting the resting conductance. Action potential firing may culminate in opening of KATP channels by activation of ATP-dependent Ca2+ pumping as suggested by the observation that the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor thapsigargin (4 μm) inhibited IK,slow by 25 % and abolished bursting electrical activity. We conclude that oscillatory glucose-induced electrical activity in the β-cell involves the opening of KATP-channel activity and that these channels, in addition to constituting the glucose-regulated K+ conductance, also play a role in the graded response to supra-threshold glucose concentrations.
Exposure of pancreatic β-cells to glucose and other stimulators of insulin secretion, is associated with the appearance of electrical activity (Henquin & Meissner, 1984; Ashcroft & Rorsman, 1989). Over the physiological range of glucose concentrations, this electrical activity consists of oscillations in membrane potential between depolarised plateaux, on which bursts of action potentials are superimposed, separated by repolarised electrically silent intervals. The oscillations in electrical activity are accompanied by changes in the cytoplasmic Ca2+ concentration (Santos et al. 1991), which in turn give rise to brief pulses (≈10 s) of insulin secretion (Gilon & Henquin, 1992; Bergsten, 1995; Barbosa et al. 1998). In addition to these rapid bouts of insulin secretion, there is also evidence for a much slower (10 min) oscillatory rhythm both systemically (Lang et al. 1979); and in isolated β-cells (Grapengiesser et al. 1991). The relationship between the rapid and slow oscillations is currently unclear.
Although the significance of the membrane oscillations is undisputed, the underlying mechanism remains elusive (review: Sherman, 1996). A problem has been the failure of isolated β-cells maintained in tissue culture (the standard preparation for patch-clamp experiments) to generate the characteristic bursting pattern (Smith et al. 1990; Ämmälä et al. 1991; Larsson et al. 1996; Kinard et al. 1999). We have developed a technique, which enables patch-clamp recordings on β-cells in intact mouse islets (Göpel et al. 1999a) and have thereby documented a K+ current, which develops gradually during simulated electrical activity (IK,slow; Göpel et al. 1999b). However, the mechanisms by which tolbutamide and high glucose concentration abolish oscillatory electrical activity and the significance of IK,slow in this context remains enigmatic. A recent study on isolated mouse β-cells, that did not exhibit any spontaneous membrane potential oscillations, revealed a glucose- and tolbutamide-sensitive current reminiscent of IK,slow (Rolland et al. 2002) but the magnitude of the current was small (0.2 nS) and its significance for bursting electrical activity in the intact islet is therefore unclear.
In this study we have extended the characterisation of IK,slow in β-cells within intact islets. We demonstrate that IK,slow is a mosaic of KATP channels (48 % of the current) and a tolbutamide-resistant K+ conductance (52 %). Additionally, we provide evidence that IK,slow in bursting β-cells is modulated by glucose in a range of concentrations associated with the conversion of bursting electrical activity into uninterrupted action potential firing. Finally, we outline a simple model by which electrical activity, via Ca2+ entry through voltage-gated Ca2+ channels, may result in activation of KATP channels and membrane repolarisation. Brief accounts of some of these observations have previously appeared in abstract form (Göpel et al. 1997, 2001; Galvanovskis et al. 2000).
Methods
All experiments were carried out on β-cells in intact islets isolated from Naval Medical Research Institute (NMRI) mice. The mice were stunned by a blow to the head and killed by cervical dislocation (procedures approved by the ethical committee at Lund University) and islets were isolated as described previously (Göpel et al. 1999b). The procedures for perforated patch whole-cell recordings and the criteria used for the functional identification of insulin-releasing β-cells have been described at length elsewhere (Göpel et al. 1999b). Pancreatic β-cells were functionally identified by the generation of bursting electrical activity in the presence of 10-15 mm glucose; the initial concentration of glucose was varied to elicit robust oscillatory electrical activity. Bursts of action potentials were simulated by a sequence of voltage-clamp pulses consisting of depolarisation to −40 from −70 mV for 5 s followed by a series of 26 100-ms voltage-clamp depolarisations between −40 and 0 mV (5 Hz). After the train, the cell was held at −40 mV for up to 20 s to facilitate the observation of outward K+ currents. The standard extracellular medium consisted of (mm): 140 NaCl, 3.6 KCl, 2 NaHCO3, 0.5 NaH2PO4, 0.5 MgSO4, 5 Hepes (pH 7.4 with NaOH), 2.5 CaCl2 and d-glucose as indicated. The pipette solution was composed of (mm): 76 K2SO4, 10 NaCl, 10 KCl, 1 MgCl2 and 5 Hepes (pH 7.35 with KOH). Tolbutamide was purchased from Sigma and dissolved in DMSO to produce a stock solution with a concentration of 100 mm. All experiments were conducted at 32-35 °C. During the experiments the islets were superfused with extracellular medium at a rate of 1-2 ml min−1. The effects of electrical activity on the membrane conductance is quoted relative to the pre-stimulatory level. Data are presented as mean values ± s.e.m. Statistical significances were evaluated using Student's t test.
Results
IK,slow hyperpolarises the β-cell
We have previously proposed that the repolarisation that terminates the bursts of action potentials results from the activation of an outward K+ current, which is turned on gradually during electrical activity (IK,slow; Göpel et al. 1999b). Figure 1A and B provides a direct demonstration that IK,slow is sufficient to hyperpolarise electrically active glucose-stimulated β-cells. The experiment commenced in 10 mm glucose to allow recording of glucose-induced electrical activity to ascertain the identity of the cell. The amplifier was subsequently switched into the voltage-clamp mode and IK,slow was elicited by the train of voltage-clamp depolarisations. The current thus elicited is shown on an expanded time base in Fig. 1B. The amplifier was finally returned to the current-clamp mode to record the membrane potential. The sustained increase in K+ conductance transiently hyperpolarised the β-cell (dashed horizontal line in Fig. 1A). In a series of 21 experiments conducted in the presence of 10 or 15 mm glucose, the most negative potential attained during the silent phase was −54 ± 2 mV under control conditions and −62 ± 2 mV (P < 0.0001) following the train. Electrical activity in the neighbouring cells spreading into the voltage-clamped cell via the gap junction, seen as upside-down burst-like current deflections (cf. Fig. 1F in Göpel et al. 1999b), was unaffected by the activation of IK,slow. The small-amplitude bursts observed shortly after electrical stimulation (arrows) we likewise attribute to current spread from neighbouring cells via the gap junctions.
Figure 1. Activation of IK,slow influences the β-cell membrane potential.
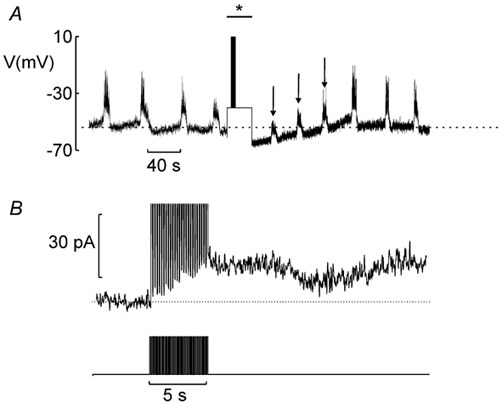
A, electrical activity recorded from a β-cell in an intact islet. During the period indicated by * and bar, the amplifier was switched into the voltage-clamp mode and the cell subjected to the train of simulated action potentials as indicated. The dashed horizontal line corresponds to the most repolarised potential during two successive bursts before going into the voltage-clamp mode. B, IK,slow (top) and stimulation protocol (lower) displayed on an expanded time base. The dotted lines indicate the pre-stimulatory current level.
Tobutamide-sensitive and tolbutamide-resistant IK,slow
Figure 2A shows bursting electrical activity recorded from a β-cell exposed to 10 mm glucose. All cells tested (n = 15) responded reversibly to 0.1 mm tolbutamide with the induction of continuous firing of action potentials. The action potentials seen in the presence of tolbutamide often tend to become grouped in short (2 s) bursts separated by brief and shallow (5 mV) repolarised intervals (Fig. 2B). At the time indicated by the letters a-c in Fig. 2A, the patch-clamp amplifier was switched from current- to voltage-clamp mode and electrical activity simulated by a train of voltage-clamp depolarisations. The evoked IK,slow responses are displayed in Fig. 2C. The peak IK,slow measured under control conditions at the end of the train of depolarisations to 0 mV averaged 61 ± 20 pA (n = 15). The effect of tolbutamide on electrical activity correlated with a reduction of IK,slow to 20 ± 2 pA (n = 15, P < 0.05). On average tolbutamide reduced IK,slow by 48 ± 7 % (P < 0.001). Thus, IK,slow reflects activation of both tolbutamide-sensitive channels (most likely KATP channels) and KATP-insensitive K+ channels. Following the removal of tolbutamide, the amplitude of IK,slow gradually returned to the control amplitude (Fig. 2Cc). The effect of tolbutamide was very variable and ranged between zero (cf. Göpel et al. 1999b) and 89 %. Thus, some unidentified cellular process (such as cell metabolism) determines the magnitude of KATP channel component of IK,slow. The role of the tolbutamide-resistant current is unclear but it is apparently insufficient for the generation of bursting electrical activity. Consistent with previous observations (Göpel et al. 1999a), there is little effect of tolbutamide on resting conductance when applied in the presence of ≥10 mm glucose (Fig. 2C; see current response when the membrane potential is stepped from −70 to −40 mV).
Figure 2. Tolbutamide-sensitive component of activity-dependent K+ current in bursting β-cells.
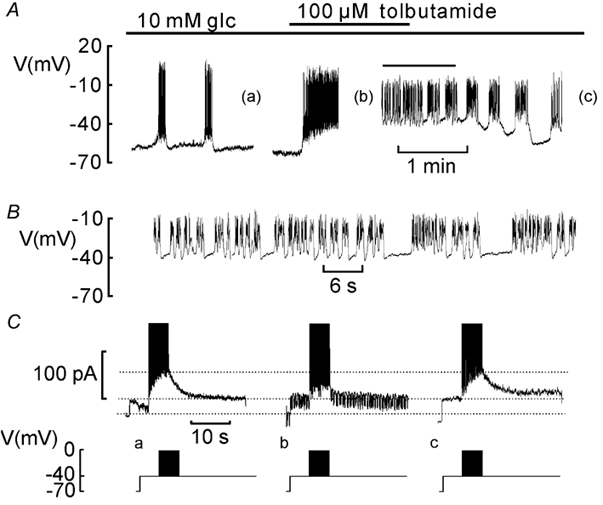
A, electrical activity recorded in the presence of 10 mm glucose alone (glc; left), in the presence of 0.1 mm tolbutamide (middle) and after withdrawal of the drug (right). At the times indicted by (a), (b) and (c), the membrane potential recording was interrupted to record IK,slow voltage-clamp currents. B, electrical activity in the presence of tolbutamide on an expanded time base. The expanded segment has been indicated in A by the horizontal line. C, lower, voltage-clamp stimulation protocol. C, upper, IK,slow recorded in the presence of 10 mm glucose alone (a), in the presence of 10 mm glucose and 0.1 mm tolbutamide (b) and following wash-out of tolbutamide and the resumption of oscillatory electrical activity (c). The horizontal dotted lines indicate (from top to bottom) the peak current amplitude at 10 mm glucose, the pre-stimulatory current level recorded at - 40 mV and the holding current at - 70 mV.
High glucose concentrations abolish the burst pattern by reducing IK,slow
Given that a significant fraction of IK,slow flows through KATP channels and that the KATP-channel blocker tolbutamide shares the ability of the sugar to produce continuous spiking (Fig. 2), it seemed possible that glucose modulates β-cell electrical activity by a concentration-dependent reduction of the KATP channel component of IK,slow. Figure 3A shows a membrane potential recording from a β-cell in an intact islet initially exposed to 15 mm glucose. Elevating the concentration of the sugar to 30 mm reversibly converted oscillatory electrical activity into continuous action potential firing. At the times indicated (a and b), the amplifier was switched into the voltage-clamp mode to record IK,slow. In the presence of 15 mm glucose, the amplitude of IK,slow was ≈50 pA (Fig. 3Ba). When the same protocol was repeated in the presence of 30 mm glucose, IK,slow only amounted to ≈10 pA (Fig. 3Bb). In a series of 11 experiments, peak IK,slow measured at 15 and 30 mm glucose averaged 45 ± 11 and 30 ± 8 pA, respectively (P < 0.05). The percentage decrease was 33 ± 10 %. The induction of uninterrupted action potential firing by high glucose was not associated with any reduction of the resting conductance (estimated from the current response when stepping from −70 to −40 mV), which averaged 0.97 ± 0.13 and 0.89 ± 0.13 nS (n = 10) at 15 and 30 mm glucose, respectively. We conclude that the KATP channels are nearly fully inhibited in the resting state and that electrical activity leads to (partial) reactivation of these channels.
Figure 3. Glucose modulation of IK,slow and β-cell electrical activity.
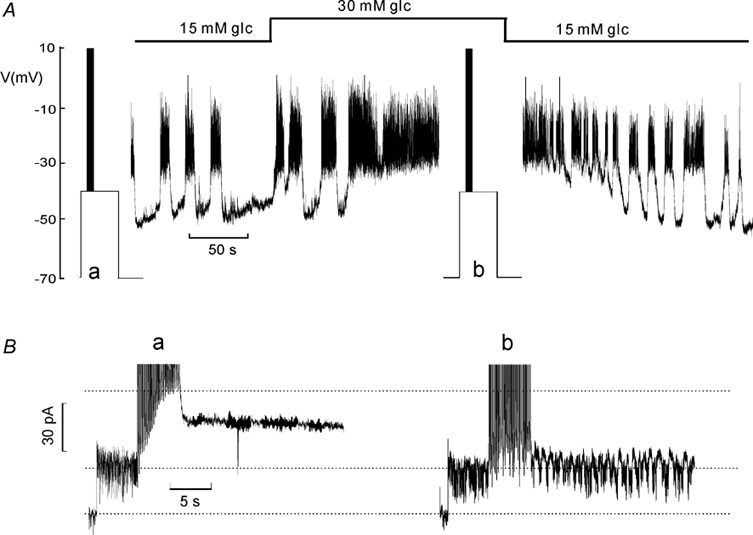
A, membrane potential recording. The glucose concentration (glc) was varied between 15 and 30 mm as indicated schematically above. At times indicated (a and b), the amplifier was switched from the current- to voltage-clamp mode to monitor IK,slow. B, IK,slow recorded at 15 mm (a) and 30 mm (b) glucose. The horizontal dotted lines indicate (from top to bottom) the peak current amplitude at 15 mm glucose, the pre-stimulatory current level recorded at −40 mV and the holding current at −70 mV.
Effects of thapsigargin and glucose on IK,slow
Thapsigargin, an inhibitor of the sarco-endoplasmatic reticulum Ca2+-ATPase (SERCA), shares the ability of glucose and tolbutamide to convert oscillatory electrical activity into continuous action potential firing (Fig. 4A, cf. Worley et al. 1994; Gilon et al. 1999). In a series of five experiments, exposure of islets, already exposed to 10 mm glucose, to thapsigargin (4 μm for 2 min) invariably increased burst duration and resulted in uninterrupted action potential firing in three (of five) cells. Accordingly, the fraction active phase (periods of electrical activity) averaged 37 ± 6 % in the presence of 10 mm glucose alone and increased to 88 ± 8 % (n = 5; P < 0.001) after exposure to thapsigargin. The latter effect associated with a moderate 24 ± 9 % (n = 5; P < 0.02) decrease in peak IK,slow (Fig. 4B), similar to the inhibition obtained when elevating glucose from 15 to 30 mm (Fig. 3). It was ascertained that the resting conductance, determined from the current response upon depolarisation from −70 to −40 mV, principally reflecting KATP channel activity and gap-junction conductance, was not affected by thapsigargin (indicated by the two lower dotted lines Fig. 4B). In the five experiments in which the effects of thapsigargin on β-cell electrical activity was tested, the resting conductance observed in the presence of the SERCA inhibitor averaged 109 ± 4 % of that seen in the absence of the compound making it unlikely that the stimulatory effect of thapsigargin on β-cell electrical activity results from closure of KATP channels (i.e. a tolbutamide-like effect) or activation of a depolarising cationic conductance (cf. Worley et al. 1994).
Figure 4. Modulation of IK,slow and β-cell electrical activity by thapsigargin.
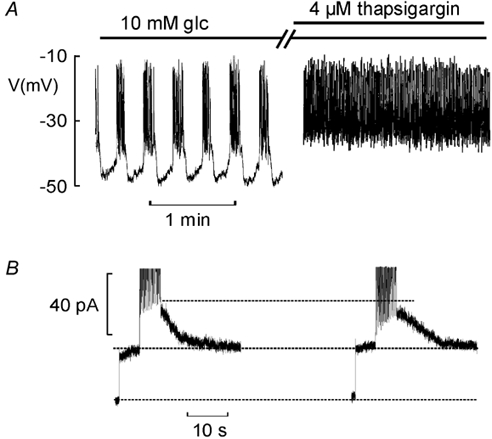
A, electrical activity recorded at 10 mm glucose before and after treatment of the islet for 2 min with 4 μm thapsigargin. B, IK,slow recorded under control conditions (left) and following treatment with thapsigargin (right). The horizontal lines indicate (from top to bottom) the peak amplitude observed under control conditions, the steady-state currents recorded at −40 mV pre- stimulation and the holding current at −70 mV.
Discussion
Glucose-induced insulin secretion is secondary to the induction of β-cell electrical activity. If we are to understand how glucose regulates insulin secretion in a concentration-dependent fashion, then it is essential to establish precisely how glucose modulates electrical activity in the pancreatic β-cell. Here we consider the generation of the β-cell bursts of action potentials at intermediate glucose concentrations and how glucose-dependent regulation of a K+ current elicited by electrical activity (IK,slow) may contribute to the graded insulin secretory response.
Based on the effects of tolbutamide, we conclude KATP channels contribute a significant fraction (48 %) of IK,slow. What is the link between electrical activity and opening of KATP channels? It has previously been demonstrated that IK,slow is dependent on Ca2+ influx (Göpel et al. 1999b; Rolland et al. 2002). Although elevation of [Ca2+]i has been postulated to stimulate mitochondrial metabolism and thus ATP production (Wollheim et al. 2000), the net effect of increased Ca2+ entry during electrical activity appears to be a reduction of the intracellular ATP:ADP ratio (Detimary et al. 1998). This could theoretically result from either stimulated ATP hydrolysis or inhibited ATP production. Indeed, the elevation of [Ca2+]i during electrical activity has been demonstrated to transiently depolarise the mitochondria and thereby (presumably) reduce ATP production (Krippeit-Drews et al. 2001). However, the observation that exposure of β-cells to thapsigargin, which increases basal [Ca2+]i (Chow et al. 1995), often results in permanent depolarisation and uninterrupted firing of action potentials (Fig. 4 and Roe et al. 1994) is difficult to reconcile with such a concept. Nevertheless, we believe that the ability of thapsigargin to stimulate electrical activity, whilst not affecting the resting conductance nor the holding current, provides a critical clue to the mechanism underlying the grouping of β-cell action potentials to bursts. Under control conditions, Ca2+ entry will be associated with rapid activation of Ca2+ pumping leading to ATP hydrolysis and a decreased cytoplasmic ATP:ADP ratio (Detimary et al. 1998), culminating in the opening of KATP channels and membrane repolarisation. By inhibition of SERCA, thapsigargin can be envisaged to exert an ATP-sparing action and under these conditions Ca2+ entry is no longer associated with a rapid depletion of sub-membrane ATP. This scenario is supported by the report that up to > 40 % of the total Ca2+-dependent ATPase activity in the β-cell is attributable to SERCA (Roe et al. 1994). In the presence of the SERCA inhibitor, Ca2+ is removed solely by the plasma membrane Ca2+-ATPase and the Na+-Ca2+ antiporter (Gall et al. 1999) resulting in a somewhat higher [Ca2+]i than under control conditions. Importantly, the rate of ATP hydrolysis under these conditions is sufficiently reduced so that it can (partially) be balanced by concomitant ATP production.
Even if the IK,slow exhibits several features implicating it in the generation of bursting electrical activity in β-cells (gradual turn-on, slow deactivation and sensitivity to thapsigargin), it remains to be established how glucose both increases the plateau phase and shortens the silent intervals between two successive bursts. It should be noted that in β-cells in situ, the relationship between resting conductance and glucose concentration is hyperbolic with a Km around 5 mm with little further inhibition at glucose concentrations >10 mm (Göpel et al. 1999a). Increasing glucose to 30 mm has little effect on resting conductance (Fig. 3B), so the concentration-dependent modulation of β-cell electrical activity by glucose cannot simply be accounted for by changes of the resting KATP channel activity. These considerations suggest that IK,slow itself must be regulated by glucose and we demonstrate here that its peak amplitude is decreased by one-third when glucose is elevated from 15 and 30 mm. Given that 48 % of IK,slow flows through KATP channels, it seems justifiable to assume that inhibition of these channels account for the metabolic regulation of IK,slow. Indeed, both the amplitude (15-20 pA) and deactivation kinetics (complete in < 5 s) of IK,slow observed at 30 mm glucose concentration are similar to the tolbutamide-resistant component of IK,slow (compare Figs. 2Cb and 3Bb) suggesting that glucose acts by suppressing the tolbutamide-sensitive component. We therefore propose that elevating glucose from an intermediate (e.g. 15 mm) to a maximally stimulatory concentration accelerates cellular metabolism sufficiently to balance the impact of stimulated Ca2+-ATPase activity on the sub-membrane ATP:ADP ratio and thus prevents activation of the KATP channels. Such a mechanism would easily account for the concentration-dependent increase in burst duration observed in response to increasing glucose concentrations and is perfectly compatible with the fact that β-cell oxidative metabolism and the amount of electrical activity increase in parallel (Atwater et al. 1979; Malaisse et al. 1984).
The finding that KATP channels contribute to the cyclic increase in K+ permeability occurring during oscillatory electrical activity has important functional implications. For example, the capacity of tolbutamide to produce continuous action potential firing becomes easy to understand if sulphonylurea-sensitive KATP channels participate in the generation of the bursts. The ability of every β-cell within the intact islet to respond in a graded fashion to elevated glucose levels with enhanced electrical activity, which in turn accounts for the sigmoidal relationship between plasma glucose concentrations and insulin release observed systemically, can also be explained by a concentration-dependent inhibition of KATP channels reactivated during electrical activity. Thus, the role of KATP channels in the β-cell extends beyond merely serving as the glucose-regulated resting conductance, they also contribute to the progressive stimulation of electrical activity and insulin release by supra-threshold glucose levels, i.e. in a range of concentrations where little metabolic modulation of the KATP channels was formerly believed to occur (Misler et al. 1986; Ashcroft et al. 1988; Göpel et al. 1999b). It remains to be explained why isolated β-cells lack bursting electrical activity but it is interesting that agents that increase intracellular cAMP levels (such as glucagon, normally secreted from neighbouring α-cells) induce both rapid membrane potential (Ämmälä et al. 1991) and [Ca2+]i oscillations (Grapengiesser et al. 1991). It should therefore be considered whether the absence of rapid glucose-induced membrane potential oscillations in isolated β-cells is a consequence of the loss of paracrine signalling normally operating in the intact islet, which in turn may affect cell metabolism and/or the copy number of Ca2+-ATPases and ion channels.
Acknowledgments
Dr T. Kanno's visit in Lund was supported by a grant from Hirosaki University. Dr S. O. Göpel holds a postdoctoral fellowship from the Swedish Association for Medical Research. Financial support was obtained from the Swedish Medical Research Council (grants no. 8647, 9890, 12234, 12708 and 13147), the Swedish Diabetes Association, the Juvenile Diabetes Foundation International and the Knut and Alice Wallenbergs Stiftelse, the Novo Nordisk Foundation, the Magnus Bergwalls Stiftelse and the Åke Wibergs Stiftelse.
References
- Ämmälä C, Larsson O, Berggren PO, Bokvist K, Juntti-berggren L, Kindmark H, Rorsman P. Inositol trisphosphate-dependent activation of a Ca2+-activated K+-conductance in glucose-stimulated pancreatic β -cells. Nature. 1991;353:849–852. doi: 10.1038/353849a0. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Ashcroft SJH, Harrison DE. Properties of single potassium channels modulated by glucose in rat pancreatic β-cells. Journal of Physiology. 1988;400:501–527. doi: 10.1113/jphysiol.1988.sp017134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Progress in Biophysics and Molecular Biology. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Atwater I, Ribalet B, Rojas E. Mouse pancreatic β-cells: tetraethylammonium blockage of the potassium permeability increase induced by depolarization. Journal of Physiology. 1979;288:561–574. [PMC free article] [PubMed] [Google Scholar]
- Barbosa RM, Silva AM, Tomé AR, Stamford JA, Santos RM, Rosário LM. Control of pulsatile 5-HT/insulin secretion from single mouse pancreatic islets by intracellular calcium dynamics. Journal of Physiology. 1998;510:135–143. doi: 10.1111/j.1469-7793.1998.135bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten P. Slow and fast oscillations of cytoplasmic Ca2+ in pancreatic islets correspond to pulsatile insulin release. American Journal of Physiology. 1995;268:E282–287. doi: 10.1152/ajpendo.1995.268.2.E282. [DOI] [PubMed] [Google Scholar]
- Chow RH, Lund PE, Loser S, Panten U, Gylfe E. Coincidence of early glucose-induced depolarization with lowering of cytoplasmic Ca2+ in mouse pancreatic β-cells. Journal of Physiology. 1995;485:607–617. doi: 10.1113/jphysiol.1995.sp020756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detimary P, Gilon P, Henquin JC. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochemical Journal. 1998;333:269–274. doi: 10.1042/bj3330269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall D, Gromada J, Susa I, Rorsman P, Herchuelz A, Bokvist K. Significance of Na/Ca exchange for Ca2+ buffering and electrical activity in mouse pancreatic β-cells. Biophysical Journal. 1999;76:2018–2028. doi: 10.1016/S0006-3495(99)77359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvanovskis J, Rorsman P, Gopel SO. Modelling of electrical activity in pancreatic islets: The role of a novel KCa -channel. Biophysical Journal. 2000;78:A210. [Google Scholar]
- Gilon P, Arredouani A, Gailly P, Gromada J, Henquin JC. Uptake and release of Ca2+ by the endoplasmic reticulum contribute to the oscillations of the cytosolic Ca2+ concentration triggered by Ca2+ influx in the electrically excitable pancreatic B-cell. Journal of Biological Chemistry. 1999;274:20197–20205. doi: 10.1074/jbc.274.29.20197. [DOI] [PubMed] [Google Scholar]
- Gilon P, Henquin JC. Inflence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin-secreting panrcreatic B-cell. Journal of Biological Chemistry. 1992;15:20713–20720. [PubMed] [Google Scholar]
- Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. Journal of Physiology. 1999a;521:717–728. doi: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renström E, Rorsman P. Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β cells. Journal of General Physiology. 1999b;114:759–770. doi: 10.1085/jgp.114.6.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpel SO, Kanno T, Rorsman P. Two components of activity-dependent transient K+-current (IKslow) in mouse pancreatic B-cells. Diabetologia. 2001;44:A20. [Google Scholar]
- Göpel SO, Rorsman P. Activation of a Ca2+ -activated K+ -conductance terminates the burst of action potentials in insulin-secreting pancreatic B-cells. Diabetologia. 1997;41:A540. [Google Scholar]
- Grapengiesser E, Gylfe E, Hellman B. Cyclic AMP as a determinant for glucose induction of fast oscillations in isolated pancreatic β-cells. Journal of Biological Chemistry. 1991;266:12207–12210. [PubMed] [Google Scholar]
- Henquin JC, Meissner HP. Significance of ionic fluxes and changes in membrane potential for stimulus-secretion coupling in pancreatic B-cells. Experientia. 1984;40:1043–1052. doi: 10.1007/BF01971450. [DOI] [PubMed] [Google Scholar]
- Kinard TA, De Vries G, Sherman A, Satin LS. Modulation of the bursting properties of single mouse pancreatic β-cells by artificial conductances. Biophysical Journal. 1999;76:1423–1435. doi: 10.1016/S0006-3495(99)77303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krippeit-Drews P, Dufer M, Drews G. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochemical and Biophysical Research Communications. 2000;267:179–183. doi: 10.1006/bbrc.1999.1921. [DOI] [PubMed] [Google Scholar]
- Lang DA, Matthews DR, Peto J, Turner RC. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. New England Journal of Medicine. 1979;301:1023–1027. doi: 10.1056/NEJM197911083011903. [DOI] [PubMed] [Google Scholar]
- Larsson O, Kindmark H, Branstrom R, Fredholm B, Berggren P-O. Oscillations in KATP channel activity promote oscillations in cytoplasmic free Ca2+ concentration in pancreatic β cell. Proceedings of the National Academy of Sciences of the USA. 1996;93:5161–5165. doi: 10.1073/pnas.93.10.5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaisse WJ, Malaisse-Lagae F, Sener A. Coupling factors in nutrient-induced insulin release. Experientia. 1984;40:1035–1043. doi: 10.1007/BF01971449. [DOI] [PubMed] [Google Scholar]
- Misler S, Falke LC, Gillis K, McDaniel ML. A metabolite-regulated potassium channel in rat pancreatic β-cells. Proceedings of the National Academy of Sciences of the USA. 1986;83:7110–7123. doi: 10.1073/pnas.83.18.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe MW, Philipson LH, Frangakis CJ, Kuznetsov A, Mertz RJ, Lancaster ME, Spencer B, Worley JF, III, Dukes ID. Defective glucose-dependent endoplasmic reticulum Ca2+-sequestration in diabetic mouse islets of Langerhans. Journal of Biological Chemistry. 1994;269:18279–18282. [PubMed] [Google Scholar]
- Rolland JF, Henquin JC, Gilon P. Feedback control of the ATP-sensitive K+ current by cytosolic Ca2+ contributes to oscillations of the membrane potential in pancreatic β-cells. Diabetes. 2002;51:376–384. doi: 10.2337/diabetes.51.2.376. [DOI] [PubMed] [Google Scholar]
- Santos RM, Rosario LM, Nadal A, Garcia-Sancho J, Sorio B, Valdeolmillos M. Widespread synchronous [Ca2+]i oscillations due to bursting electrical activity in single pancreatic islets. Pflügers Archiv. 1991;418:417–422. doi: 10.1007/BF00550880. [DOI] [PubMed] [Google Scholar]
- Sherman A. Contributions of modeling to understanding stimulus-secretion coupling in pancreatic β-cells. American Journal of Physiology. 1996;271:E362–372. doi: 10.1152/ajpendo.1996.271.2.E362. [DOI] [PubMed] [Google Scholar]
- Smith PA, Ashcroft FM, Rorsman P. Simultaneous recordings of glucose dependent electrical activity and ATP-regulated K+-currents in isolated mouse pancreatic β-cells. FEBS Letters. 1990;261:187–190. doi: 10.1016/0014-5793(90)80667-8. [DOI] [PubMed] [Google Scholar]
- Wollheim CB. β-cell mitochondria in the regulation of insulin secretion: a new culprit in type II diabetes. Diabetologia. 2000;43:265–277. doi: 10.1007/s001250050044. [DOI] [PubMed] [Google Scholar]
- Worley JF, McIntyre MS, Spencer Mertz RJ, Roe MW, Dukes ID. Endoplasmic reticulum calcium store regulates membrane potential in mouse islet β-cells. Journal of Biological Chemistry. 1994;269:14359–14362. [PubMed] [Google Scholar]