

Abstract
The IKS K+ channel plays a major role in repolarizing the cardiac action potential. It consists of an assembly of two structurally distinct α and β subunits called KCNQ1 and KCNE1, respectively. Using two different expression systems, Xenopus oocytes and Chinese hamster ovary cells, we investigated the effects of external protons on homomeric and heteromeric KCNQ1 channels. External acidification (from pH 7.4 to pH 5.5) markedly decreased the homomeric KCNQ1 current amplitude and caused a positive shift (+25 mV) in the voltage dependence of activation. Low external pH (pHo) also slowed down the activation and deactivation kinetics and strongly reduced the KCNQ1 inactivation process. In contrast, external acidification reduced the maximum conductance and the macroscopic inactivation of the KCNQ1 mutant L273F by only a small amount. The heteromeric IKS channel complex was weakly affected by low pHo, with minor effects on IKS current amplitude. However, substantial current inhibition was produced by protons with the N-terminal KCNE1 deletion mutant Δ11-38. Low pHo increased the current amplitude of the pore mutant V319C when co-expressed with KCNE1. The slowing of IKS deactivation produced by low pHo was absent in the KCNE1 mutant Δ39-43, suggesting that the residues lying at the N-terminal boundary of the transmembrane segment are involved in this process. In all, our results suggest that external acidification acts on homomeric and heteromeric KCNQ1 channels via multiple mechanisms to affect gating and maximum conductance. The external pH effects on IKrversus IKS may be important determinants of arrhythmogenicity under conditions of cardiac ischaemia and reperfusion.
Two main K+ conductances are responsible for the late repolarization phase of the cardiac action potential, the IKr and IKs delayed-rectifier K+ currents (Noble & Tsien, 1969; Sanguinetti & Jurkiewicz, 1990). IKr is encoded by the KCNH2 (HERG) and KCNE2 (MIRP1) genes, while IKs is encoded by the KCNQ1 (KvLQT1) and KCNE1 (Mink) genes, whose protein products assemble, respectively, as α and β subunits of the corresponding channel complexes (Kurokawa et al. 2001; Pond & Nerbonne, 2001; Tseng, 2001). Mutations in each of the four genes produce long-QT (LQT) syndrome, a human genetically heterogeneous cardiovascular disease that is characterized by abnormal ventricular repolarization (Keating & Sanguinetti, 2001). Because of their importance in shaping the cardiac action potential under both normal and pathophysiological conditions, IKr and IKs are crucial targets for different regulatory processes such as modulation by external cations (Po et al. 1999; Numaguchi et al. 2000), nitric oxide (Taglialatela et al. 1999), protein kinase C (Barros et al. 1998), protein kinase A (Kiehn et al. 1998; Thomas et al. 1999) and pH (Berube et al. 1999; Jiang et al. 1999; Terai et al. 2000; Vereecke & Carmeliet, 2000).
Changes in pH markedly affect cardiac excitability. Extracellular and intracellular acidification of cardiac myocytes occurs within minutes following myocardial ischaemia. Acidosis results in a fall in the resting potential, a reduction in upstroke velocity, a prolongation of the action potential duration and the occurrence of early afterdepolarizations (Carmeliet, 1999). All of the acidosis-induced changes are arrhythmogenic and may explain why the threshold for ventricular fibrillation is reduced in acidosis. External pH (pHo) has been described as changing rapidly from 7.4 to values as low as 5.9 and thus to act on various ion channels (Axford et al. 1992; Carmeliet, 1999). Most of the ion channels that contribute to the cardiac action potential are inhibited by extracellular acidosis, including voltage-gated Na+ channels (INa), L-type Ca2+ channels (ICa L) and inward-rectifier (IK1), transient (Ito) and delayed-rectifier (IKr, IKs) K+ channels (Carmeliet, 1999). The mechanisms underlying this proton-mediated channel inhibition are diverse and could involve a decrease in unitary conductance, a reduction in open probability or voltage shifts in the activation-inactivation kinetics. In most cases, acidosis increases the cardiac action potential duration, suggesting that K+ channel-mediated repolarization is impaired. Recent studies have attempted to evaluate the impact of external protons on the molecular correlates of the cardiac K+ currents. Kv1.5 channels, which contribute to IK currents in phase 3 repolarization of the cardiac action potential, show enhanced C-type inactivation at low pHo (Steidl & Yool, 1999). HERG channels, which underlie IKr currents, exhibit a faster rate of deactivation and in some cases a positive shift in the voltage dependence of activation upon external acidification (Anumonwo et al. 1999; Berube et al. 1999; Jo et al. 1999). Fewer data are available for the impact of pHo on native and recombinant IKs currents. It was found that external acidification reduces the maximal conductance (Gmax) of IKs in a voltage-independent fashion, suggesting that the proton binding site is located outside the membrane electric field (Yamane et al. 1993). A recent study reported that glycosylation influences the pH sensitivity of IKs channels (Freeman et al. 2000). When expressed in a glycosylation-permissive Chinese hamster ovary (CHO) cell line, IKs channels are sensitive to external acidification, while they remain unaffected if expressed in Lec-1 glycosylation-deficient CHO cells (Freeman et al. 2000).
In the present study, we have compared systematically the effects of external acidification on homomeric KCNQ1 and heteromeric IKs channel properties, using two different expression systems, Xenopus oocytes and CHO cells. In this regard, it was shown recently that depending on the cell type, the degree of glycosylation of the KCNE1 β subunit could affect not only the gating properties but also the pH sensitivity of IKs channels (Freeman et al. 2000). We found that low pHo produces similar effects on both expression systems. External acidification (from pH 7.4 to pH 5.5) markedly inhibited the amplitude of KCNQ1 currents but weakly affected that of IKs. Low pHo slowed down the activation and deactivation kinetics and strongly reduced the KCNQ1 inactivation process. An N-terminal deletion of KCNE1 (Δ11-38) led to a substantial current inhibition by protons that is absent in wild-type (WT) IKs channels. We have also shown that KCNE1 residues 39-43, lying at the N-terminal boundary of the transmembrane segment are involved in slowing of the IKs deactivation produced by low pHo. Our data suggest that external acidification acts on homomeric and heteromeric KCNQ1 channels via multiple mechanisms to affect gating and Gmax.
Methods
Molecular biology
All KCNQ1 and KCNE1 mutants were produced by Pfu DNA polymerase (Promega) and mutagenic primers using PCR-based site-directed mutagenesis. For each mutant, DNA sequence analysis was performed prior to expression. For Xenopus oocyte expression, human KCNE1 and its mutants were linearized by BamH1, while WT KCNQ1 was linearized by Not1. Capped complementary RNAs (cRNAs) were transcribed from WT and mutants of human KCNE1, and WT human KCNQ1 by T3 and T7 RNA polymerases, respectively, using the mMessage mMachine transcription kit (Ambion). cRNA size and integrity were confirmed by formaldehyde-agarose gel electrophoresis.
RNA injection into Xenopus oocytes
Xenopus laevis frogs were purchased from the USA (Xenopus 1, Dexter, Michigan, USA). All procedures used and described herein conform with the UK Animals (Scientific Procedures) Act 1986. The procedures followed for surgery and maintenance of frogs were approved by the animal research ethics committee of Tel Aviv University. Frogs were anaesthetized with 0.2 % tricaine (Sigma). Pieces of the ovary were surgically removed and digested with 1 mg ml−1 collagenase (type IA, Sigma) in Ca2+-free ND96 (mm: 96 NaCl, 2 KCl, 1 MgCl2 and 5 Hepes titrated to pH 7.5 with NaOH) for 1 h, to remove follicular cells. Oocytes at stages V and VI were used for cRNA injection and maintained at 18 °C in ND96 (1.8 mm CaCl2), supplemented with 1 mm pyruvate and 50 μg ml−1 gentamycin. Human KCNE1 and its mutants were injected at 1 ng cRNA oocyte−1 together with WT KCNQ1 at 2 ng cRNA oocyte−1. Homomultimeric expression of WT KCNQ1 was performed by injecting 2 ng cRNA oocyte−1.
Electrophysiology in Xenopus oocytes
Standard two-electrode voltage-clamp measurements were performed 3-5 days following cRNA microinjection into oocytes, as described previously (Abitbol et al. 1999). Oocytes were bathed in a modified ND96 solution containing (mm) 96 NaCl, 2 KCl, 1 MgCl2, 0.1 CaCl2 and 5 Hepes titrated to pH 7.4 with NaOH under constant perfusion using a peristaltic pump (Gilson) at a flow rate of 0.4 ml min−1. Hepes buffer was used to titrate pH 7.5 and 6.5 and Mes buffer for pH 6.5, 6 and 5.5. CaCl2 was reduced to 0.1 mm to virtually eliminate the contribution of endogenous Ca2+-activated Cl− currents. Whole-cell currents were recorded at room temperature (20-22 °C) using a GeneClamp 500 amplifier (Axon Instruments). Glass microelectrodes (A-M Systems) were filled with 3 M KCl and had tip resistances of 0.5-1.5 MΩ. Stimulation of the preparation, data acquisition and analyses were performed using the pCLAMP 6.02 software (Axon Instruments) and a 586 personal computer interfaced with a Digidata 1200 interface (Axon Instruments). Current signals were filtered at 0.2-0.5 kHz and digitized at 1-2 kHz. Unless specified otherwise, the holding potential was -80 mV. Leak subtraction was performed off-line using the Clampfit program of the pCLAMP 6.02 software.
Electrophysiology in CHO cells
CHO cells were plated on poly-d-lysine-coated glass coverslips in a 24-multiwell plate and grown in Dulbecco's modified Eagle's medium supplemented with 2 mm glutamine, 10 % fetal calf serum (FCS) and antibiotics. Transfection was performed using 1.5-2 μl of lipofectamine (Gibco-BRL) according to the manufacturer's protocol and with 0.5 μg of the respective channel cDNA plasmids together with 0.5 μg of pIRES-CD8 (kindly provided by Dr A. Patel) as a marker for transfection. Transfected cells were visualized 48 h following transfection, using the anti-CD8 antibody-coated beads method (Jurman et al. 1994). Current measurements were performed 48 h following transfection, using the whole-cell configuration of the patch-clamp technique (Hamill & Sakmann, 1981). Currents were acquired at steady state after stabilization of run-down (if any). Signals were amplified using an Axopatch 200B patch-clamp amplifier (Axon Instruments), sampled at 1 kHz and filtered at 400 Hz via a four-pole Bessel low-pass filter. Data were acquired using pCLAMP 8.1 software (Axon Instruments) and an IBM-compatible computer in conjunction with a DigiData 1322A interface (Axon Instruments). The patch pipettes were pulled from borosilicate glass (with filament) and had a resistance of 3-7 MΩ and were filled with (mm) 130 KCl, 1 MgCl2, 5 K-ATP, 5 EGTA, 20 KOH and 10 Hepes at pH 7.4. The external solution contained (mm) 140 NaCl, 4 KCl, 1.8 CaCl2, 1.2 MgCl2, 11 glucose and 5.5 Hepes at pH 7.4. Hepes buffer was used to titrate to pH 7.5 and 6.5 and Mes buffer for pH 6.5, 6 and 5.5.
Data analyses
Data analysis was performed using the Clampfit program (pCLAMP 8.1, Axon Instruments), Microsoft Excel 5.0 (Microsoft), Axograph 4.0 (Axon Instruments) and CA-Cricket Graph III (Computer Associates International). The simplex algorithm was used to fit exponential functions to current traces in order to determine the time constants and their amplitudes. To analyse the voltage dependence of channel activation, a single exponential fit was applied to the tail currents and extrapolated to the beginning of the repolarizing step. Chord conductance (G) was calculated by using the following equation:
where I corresponds to the extrapolated tail current and Vrev, the measured reversal potential, which was taken to be -97 mV (-97 ± 2 mV; n = 7). G was estimated at various test voltages, V, and then normalized to Gmax (+60 mV in CHO cells and +30 mV in Xenopus oocytes). Activation curves were fitted by a Boltzmann distribution:
where V50 is the voltage at which the current is half-activated and s is the slope factor. To analyse the voltage dependence of KCNQ1 channel activation, G was deduced either from steady-state currents or from tail currents as above. For quantification of inactivation in the triple-pulse protocol, the current decay during the third test pulse (reinduction of inactivation) was best fitted by a single exponential function. The relative percentage of inactivation was calculated by dividing the current measured at the end of the third pulse to that extrapolated from the single exponential fit at the beginning of the third pulse. All data are expressed as the mean ± s.e.m. Statistically significant differences between paired groups were assessed by Student's t test.
Results
The homomeric KCNQ1 current amplitude and voltage dependence are altered by external acidification
The effect of external acidification on homomeric KCNQ1 currents was studied using two different expression systems, Xenopus oocytes and CHO cells, by means of the two-electrode voltage clamp technique and the whole-cell configuration of the patch-clamp technique, respectively. At pH 7.4, the KCNQ1 current was evoked by step depolarizations above a threshold of about -40 mV in transfected CHO cells (Fig. 1A and B) and -60 mV in Xenopus oocytes (Fig. 2A and B). In CHO cells, external acidification (from pH 7.4 to pH 5.5) reduced the current amplitude by 51 % at +60 mV (Fig. 1A and B, Table 1). The decrease in the KCNQ1 Gmax induced by protons was reversible (Fig. 1A, inset) and concentration-dependent, with a 30 % inhibition at pH 6.5 and a maximum block of about 50 % that was already reached at pH 6 (Fig. 1C). Similar results were obtained in Xenopus oocytes (at pH 5.5) with a 30 % inhibition of KCNQ1 current at +30 mV, (Fig. 2A and B). In addition to its inhibition of Gmax, external acidification also produced a rightward gating shift of homomeric KCNQ1 channels. In transfected CHO cells, lowering pHo progressively shifted the voltage dependence of activation of KCNQ1 with V50 = -16.0 ± 1.5 mV (s = -8.7 ± 0.4 mV, n = 42), V50 = -11.8 ± 2.4 mV (s = -10.6 ± 0.8 mV, n = 9), V50 = -8.0 ± 1.9 mV (s = -12.4 ± 0.7 mV, n = 5) and V50 = 9.4 ± 3.9 mV (s = -10.6 ± 0.6 mV, n = 8) at pH 7.4, 6.5, 6 and 5.5, respectively (Fig. 1D). Similarly, in Xenopus oocytes, acidification from pH 7.4 to pH 5.5 shifted the normalized conductance curve by more than +25 mV (Fig. 2C, Table 1). No shift in voltage sensitivity was observed following external alkalinization at pH 9 with V50 = -25.2 ± 3.3 mV and s = -17.2 ± 2.1 mV (n = 4). In contrast to external acidification, internal acidification at pH 6 did not significantly affect the KCNQ1 current (data not shown).
Figure 1. External acidification reduces the KCNQ1 current density and shifts the activation curve in transfected Chinese hamster ovary (CHO) cells.
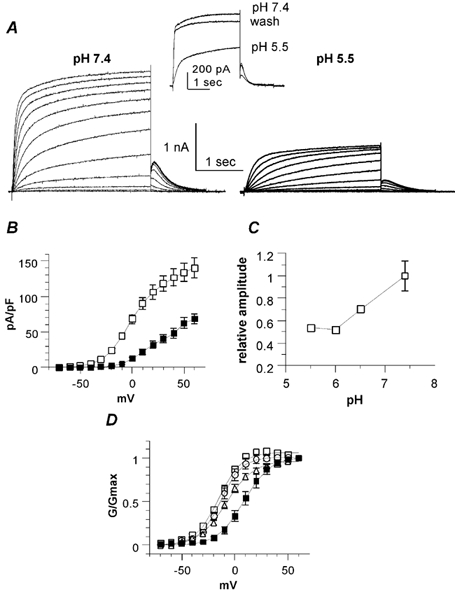
A, representative whole-cell current traces of KCNQ1 channels recorded from the same transfected CHO cell in an external solution at pH 7.4 (left) and pH 5.5 (right). The activation protocol was the following: from a holding potential of -85 mV, cells were stepped for 3 s from -50 to +60 mV in 10 mV increments and then repolarized for 1 s at -60 mV tail potential. Inset, typical traces showing washout of the effects of pH 5.5 using a train protocol, where the cells were stepped from a holding potential (-85 mV) to +30 mV for 3 s. B, current density (pA pF−1) plotted as a function of the membrane potential (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 12). C, the relative amplitude plotted against external pH (the reference is the current amplitude measured at +60 mV and at pH 7.4; n = 8-15). D, the normalized conductance (G/Gmax) plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares), pH 6.5 (open circles), pH 6 (open triangles) and pH 5.5 (filled squares; n = 8-15).
Figure 2. External acidification reduces the current amplitude, shifts the activation curve and slows down the activation and deactivation kinetics of KCNQ1 channels expressed in Xenopus oocytes.
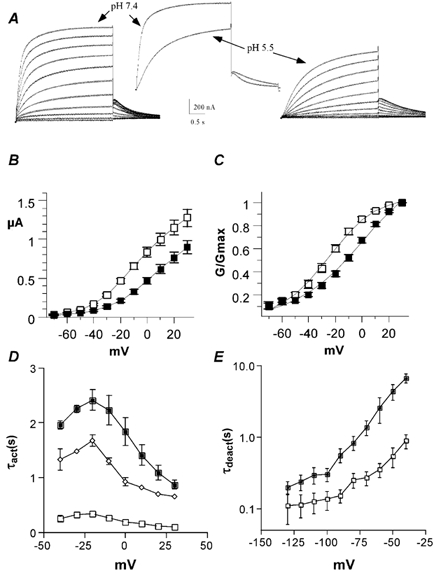
A, representative macroscopic current traces of KCNQ1 channels recorded from the same Xenopus oocyte in an external solution at pH 7.4 (left) and pH 5.5 (right). The activation protocol was the following: from a holding potential of -80 mV, oocytes were stepped for 3 s from -70 to +30 mV in 10 mV increments and then repolarized for 1.5 s at -60 mV tail potential. Inset, superimposed traces showing the inhibition of the current amplitude and the slowing of the activation and deactivation kinetics at +30 and -60 mV test and tail potentials, respectively. B, current-voltage relationship of KCNQ1 currents recorded at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 8). C, the normalized conductance (G/Gmax) plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 8). D, activation time constants (τact) were plotted against test potential in mV (n = 5). At pH 7.4, the KCNQ1 activation kinetics were best fitted by a double-exponential function, with fast (open squares) and slow (open diamonds) time constants. At pH 5.5, the KCNQ1 activation kinetics were best fitted by a single exponential function, with one time constant (filled squares). E, deactivation time constants (τdeact) are plotted against test tail potential in mV at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 5). τdeact data were obtained from the monoexponential fit of the tail currents measured from -130 to -40 mV in 10 mV increments following a prepulse to +20 mV.
Table 1.
Effect of external acidification on KCNQ1 and Iks parameters in Chinese hamster ovary (CHO) cells and Xenopus oocytes
| CHO cells (n = 9–42) | Xenopus oocytes (n = 5–12) | ||||||
|---|---|---|---|---|---|---|---|
| Current density at + 60mV (pA pF−1) | V50 (mV) | Slope (mV) | Current amplitude at +30mV (μA) | V50 (mV) | Slope (mV) | ||
| WT KCNQ1 | pH 7.4 | 140.2 ± 13.7 | −16.0 ± 1.5 | −8.7 ± 0.4 | 1.28 ± 0.11 | −24.8 ± 2.4 | −17.9 ± 1.3 |
| pH 5.5 | 68.3 ± 7.5 | 9.4 ± 3.9 | −10.6 ± 0.6 | 0.9 ± 0.08 | 0.8 ± 1.6 | −25.2 ± 1.45 | |
| L273F KCNQ1 | pH 7.4 | 64.3 ± 14.7 | −20.9 ± 1.7 | −18.2 ± 0.9 | ND | ND | ND |
| pH 5.5 | 50.5 ± 10.8 | −11.9 ± 3.1 | −17.5 ± 0.9 | ND | ND | ND | |
| WTKCNQ1+WTKCNE1 | pH 7.4 | 463.7 ± 49.3 | 22.4 ± 2.8 | −17.4 ± 1.2 | 2.97 ± 0.39 | −1.2 ± 0.9 | −14.2 ± 0.7 |
| pH 5.5 | 340.0 ± 54.2 | 21.4 ± 2.2 | −14.9 ± 1.5 | 2.80 ± 0.42 | −4.9 ± 1.6 | −16.5 ± 0.9 | |
| WTKCNQ1 +Δ 39-43 KCNE1 | pH 7.4 | 301.4 ± 66.2 | +34.1 ± 5.7 | −18.9 ± 1.3 | 0.81 ± 0.06 | −24.9 ± 7.7 | −11.9 ± 0.5 |
| pH 5.5 | 259.0 ± 53.6 | +29.9 ± 3.1 | −15.6 ± 0.5 | ND | ND | ND | |
| WTKCNQ1 + Δ 11–38 KCNE1 | pH 7.4 | ND | ND | ND | 1.92 ± 0.25 | −10.8 ± 1.8 | −13.6 ± 1.4 |
| pH 5.5 | ND | ND | ND | 1.20 ± 0.06 | −18.4 ± 2.5 | −15.1 ± 1.9 | |
| V319C KCNQ1 + WTKCNE1 | pH 7.4 | 226.5 ± 30.3 | 28.6 ± 1.6 | −13.2 ± 0.4 | 0.62 ± 0.06 | ND | ND |
| pH 5.5 | 274.5 ± 35.4 | 28.8 ± 4.1 | −15.1 ± 0.8 | ND | ND | ND | |
WT, Wild type; ND, not done; V50, voltage at which the current is half-activated
External acidification slows the activation and deactivation kinetics and inhibits the inactivation of KCNQ1 channels
Figure 2A and Figure 3A show that pH 5.5 slowed down the activation kinetics of homomeric KCNQ1 channels when expressed both in CHO cells and Xenopus oocytes. Low pHo significantly increased the activation time constants in a voltage-dependent manner, with a maximum slowing between -20 and 0 mV (Figs 2D, 3C and 3D). For example, in CHO cells the activation kinetics were well fitted by a double-exponential function, and the fast and slow time constants at 0 mV were increased from τ1 = 0.29 ± 0.02 s and τ2 = 1.28 ± 0.07 s at pH 7.4, to τ1 = 1.24 ± 0.13 s and τ2 = 1.99 ± 0.11 s at pH 5.5 (n = 10, Fig. 3C and D). The slowing effect of low pHo on KCNQ1 activation kinetics was less pronounced at depolarized potentials (Figs 2D, 3C and 3D). External acidification also slowed the KCNQ1 deactivation kinetics that could be described by a single exponential function (Fig. 2E and Fig. 3B). In Xenopus oocytes, the deactivation time constants (τdeact) increased at all potentials (Fig. 2E). For example, at a tail potential of -60 mV, τdeact = 0.34 ± 0.03 s at pH 7.4 and τdeact = 0.94 ± 0.04 s at pH 5.5 (n = 5, Fig. 2E).
Figure 3. External acidification slows down the activation and deactivation kinetics of KCNQ1 channels expressed in CHO cells.
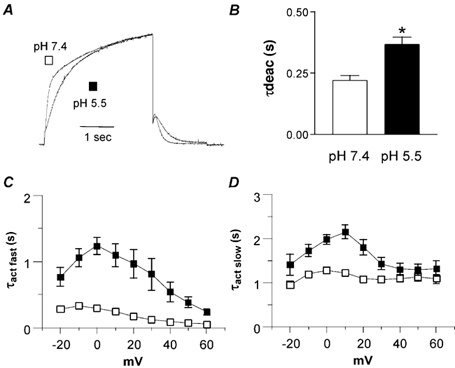
A, normalized KCNQ1 currents recorded from the same cell that was stepped to +30 mV at pH 7.4 and 5.5. B, τdeact data were obtained from the monoexponential fit of the tail currents measured at -60 mV from a prepulse potential of +30 mV (n = 16; *P < 0.01). C and D, KCNQ1 activation kinetics were best fitted by a double-exponential function, with fast (C, τact fast) and slow (D, τact slow) time constants. τact data are plotted against test potential in mV (n = 10) at pH 7.4 (open squares) and pH 5.5 (filled squares).
It has been shown previously that homomeric KCNQ1 channels undergo a time- and voltage-dependent inactivation process. This inactivation is incomplete, develops with a delay and is strongly inhibited by co-expression with KCNE1 (Pusch et al. 1998; Tristani-Firouzi & Sanguinetti, 1998). As suggested by Tristani-Firouzi & Sanguinetti (1998), KCNQ1 gating kinetics could be described by the following scheme:
where KCNQ1 channels populate multiple open states (e.g. O1 and O2) and where the slow transition from O1 to O2 accounts for the delay in inactivation (I; Tristani-Firouzi & Sanguinetti, 1998).
To study the impact of low pHo on KCNQ1 inactivation gating in CHO cells, we used a three-pulse protocol where the membrane potential is stepped for various durations to a +30 mV conditioning prepulse in order to activate and inactivate the channel; then, a brief (20 ms) hyperpolarizing interpulse to -130 mV is used to allow channel recovery from inactivation before a +30 mV test pulse is applied to reopen and reinactivate KCNQ1 channels (Fig. 4). The decaying current of the third test pulse (reinduction of inactivation) could be fitted by a single exponential function, and from its amplitude, which is estimated by extrapolating the fitted curve, one could deduce the relative percentage of inactivation (see methods and Fig. 4C). There was a longer delay in the onset of KCNQ1 inactivation at pH 5.5 when compared to pH 7.4 (Fig. 4A and B). The time course of KCNQ1 inactivation could be fitted by a single exponential function with time constants of 0.42 and 1.48 s at pH 7.4 and 5.5, respectively (Fig. 4C). As extrapolated from the fitted curves, the initial delays for inactivation were 198 and 370 ms at pH 7.4 and 5.5, respectively (Fig. 4C). Note that with conditioning prepulse durations of above 2 s, the percentage of inactivation at both pH values reached steady state; in other words, imposing a pH of 5.5 only delayed the inactivation process (Fig. 4C). In Xenopus oocytes, the effect produced by pH 5.5 on KCNQ1 inactivation was much stronger. Using similar three-pulse protocols, pH 5.5 totally suppressed the time- and voltage-dependent inactivation process (Fig. 5). The depressing effect probably results from a strong delay in the onset of inactivation, and one would probably need to generate a very long conditioning prepulse to produce a detectable inactivation.
Figure 4. External acidification delays the inactivation of the KCNQ1 current in CHO cells.
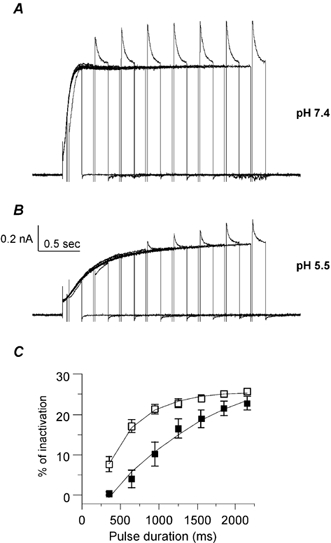
A and B, inactivation of KCNQ1 was revealed using a three-pulse protocol where the same cell was recorded at pH 7.4 (A) and then pH 5.5 (B). From a holding potential of -85 mV, the cell membrane was stepped for various durations (50-2150 ms) to a +30 mV conditioning prepulse to activate and inactivate KCNQ1 channels; then a brief (20 ms) hyperpolarizing interpulse to -130 mV was used to allow channel recovery from inactivation before a +30 mV test pulse (150 ms duration) is applied to reopen and reinactivate KCNQ1. C, the percentage of inactivation is plotted as a function of the prepulse duration at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 8). For quantification of inactivation, the decay of current during the third test pulse was best fitted by a single exponential function. The relative percentage of inactivation was calculated by dividing the current measured at the end of the third pulse to that extrapolated from the single exponential fit at the beginning of the third pulse.
Figure 5. External acidification inhibits the inactivation of KCNQ1 current in Xenopus oocytes.
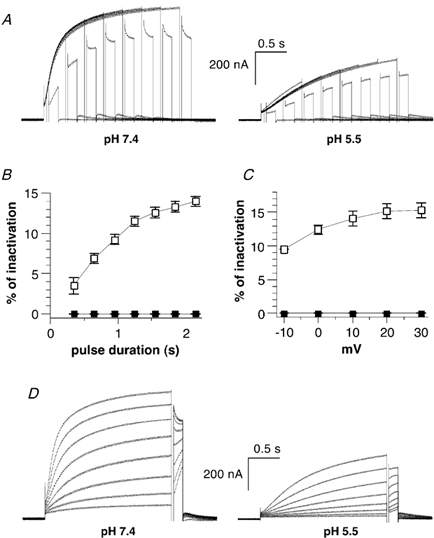
A, inactivation of KCNQ1 was revealed using a three-pulse protocol as in Fig. 4, except that the third test pulse was carried out at -10 mV. The same oocyte was recorded at pH 7.4 (left) and then pH 5.5 (right). B, the percentage of inactivation obtained at pH 7.4 (open squares) and pH 5.5 (filled squares) (n = 8) is plotted as a function of the prepulse duration and calculated as described in Fig. 4C. C, the percentage of inactivation obtained at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 6) is plotted as a function of the prepulse voltage and calculated as described in Fig. 4C. D, the same oocyte was recorded at pH 7.4 (left) and then pH 5.5 (right). A 2 s conditioning prepulse to varying potentials (from -40 to +30 mV, in 10 mV increments) was given, then a brief (20 ms) hyperpolarizing interpulse to -130 mV allowed recovery from inactivation before a test pulse of 150 ms to -10 mV was applied to reopen and reinactivate the channels.
External acidification poorly inhibits Gmax and the macroscopic inactivation of the KCNQ1 mutant L273F
To further investigate the effect of external acidification on KCNQ1 inactivation, we used the L273F KCNQ1 mutant. This LQT mutation is located in the transmembrane segment S5 and was recently found to produce macroscopic inactivation by stabilizing the KCNQ1 channel in the inactivated state (Shalaby et al. 1997; Seebohm et al. 2001). When expressed in transfected CHO cells, the L273F mutant exhibited a pronounced macroscopic inactivation as compared to WT KCNQ1 (Fig. 6A). A pHo value of 5.5 significantly decreased the L273F peak current density, from 64.3 ± 14.7 to 50.5 ± 10.8 pA pF−1 (n = 21, P < 0.01) at +60 mV (Fig. 6B). However, this inhibition (21 % at +60 mV) was weaker than that produced in WT KCNQ1 (51 % at +60 mV). Similarly, external acidification caused a significant (P < 0.01, n = 10) but smaller gating shift (+9 mV) of the L273F activation curve when compared to that of WT KCNQ1 (+25 mV; Fig. 6C, Table 1). As with WT KCNQ1, a low pHo slowed down the activation and deactivation kinetics of the L273F mutant (Fig. 6D). The time to peak was increased from 147.7 ± 10.5 to 217.2 ± 13.8 ms (P < 0.0001; n = 10) at pH 7.4 and pH 5.5, respectively. The tail currents also showed that acidification slowed down KCNQ1 deactivation kinetics, with τdeact increasing significantly from 0.63 ± 0.05 to 0.91 ± 0.09 s (n = 21; P < 0.001), for pH 7.4 and 5.5, respectively. In contrast to WT KCNQ1, the macroscopic inactivation produced by the L273F channel mutant was not very sensitive to pH 5.5. As determined by the ratio of the current amplitude measured at the peak versus the end of the test pulse, the fractional inactivation was decreased by only 13 %, from 0.79 ± 0.03 to 0.69 ± 0.03 at pH 7.4 and 5.5, respectively (n = 10; P < 0.016; Fig. 6E). This result suggests that the pronounced macroscopic inactivation produced by the L273F KCNQ1 mutant exhibits different properties to those displayed by the WT KCNQ1 channels. Low pHo slowed down the macroscopic inactivation kinetics, with the time constant of inactivation increasing from 0.64 ± 0.05 to 1.01 ± 0.13 s at pH 7.4 and 5.5, respectively (n = 21; P < 0.001; Fig. 6F).
Figure 6. Effects of low external pH (pHo) on the L273F KCNQ1 mutant expressed in CHO cells.
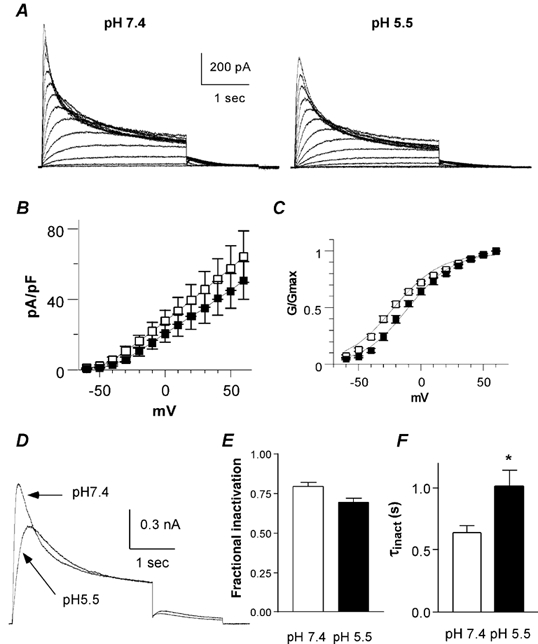
A, representative traces recorded from the L273F KCNQ1 mutant at pH 7.4 (left) and pH 5.5 (right), using the same activation protocol as described in Fig. 1A. B, the current density (pA pF−1) is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 21). C, the normalized conductance (G/Gmax) is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 10). D, superimposed traces taken from the same cell at pH 7.4 and 5.5 showing the L273F KCNQ1 current recorded at +30 and -60 mV test and tail potentials, respectively. E, fractional inactivation as determined by the ratio of the current amplitude measured at the peak versus the end of the test pulse at pH 7.4 and 5.5 (n = 10; *P < 0.016). F, the inactivation time constant τinact) was obtained from the monoexponential fit of the macroscopic inactivation measured at +60 mV at pH 7.4 and 5.5 (n = 20; *P < 001).
Low pHo weakly decreases the current amplitude of heteromeric IKs channels and alters the gating kinetics
Expressing the KCNQ1 α subunit together with the KCNE1 β subunit (IKs) changes the current characteristics dramatically, with a rightward gating shift, a slowing of the activation kinetics and an increase in current amplitude at both the macroscopic and microscopic levels (Barhanin et al. 1996; Sanguinetti et al. 1996; Pusch, 1998; Sesti & Goldstein, 1998; Yang & Sigworth, 1998). It was therefore important to check the sensitivity of IKs to low pHo. In CHO cells, the sensitivity of IKs current amplitude to external pH 5.5 was weaker than that of KCNQ1. Low pHo reversibly decreased the IKs current density by 27 % at +60 mV (n = 13, Fig. 7A and B, Table 1). In Xenopus oocytes, pH 5.5 produced a weak and non-significant inhibition (5 %) of IKs current amplitude (Fig. 8A and B, Table 1). In contrast to WT KCNQ1, low pHo produced no shift in the voltage dependence of activation of IKs (Fig. 8C, Table 1). Similar results were obtained in CHO cells (see Fig. 7C, Table 1 and data not shown).
Figure 7. Effects of low pHo on heteromeric IKs channels expressed in CHO cells.
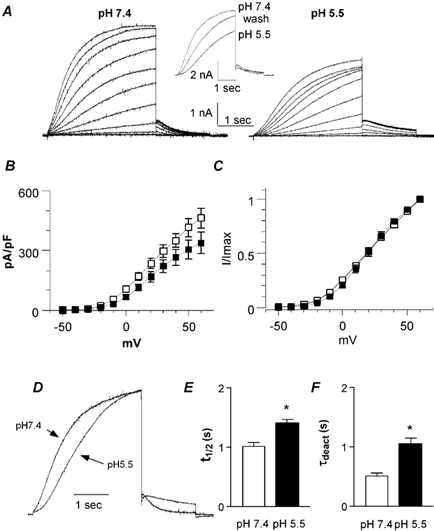
A, representative IKs current traces recorded at pH 7.4 (left) and pH 5.5 (right) using the activation protocol described in Fig. 1A. Inset, typical traces showing washout of the pH 5.5 effects using a train protocol, where the cells were stepped from the holding potential (-85 mV) to +30 mV for 3 s. B, the current density (pA pF−1) is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 13). C, normalized current is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; (n = 13). D, normalized IKs currents recorded from the same cell that was stepped to +30 mV at pH 7.4 and 5.5. E, t1/2, the time required to reach half of the maximal current amplitude at a given voltage, was measured at pH 7.4 and 5.5 at +60 mV (n = 27; *P < 0.0001). F, τdeact data were obtained at pH 7.4 and 5.5 from the monoexponential fit of the tail currents measured at -60 mV from a prepulse potential of +30 mV (n = 9, *P < 0.01).
Figure 8. Effects of low pHo on heteromeric IKs channels expressed in Xenopus oocytes.
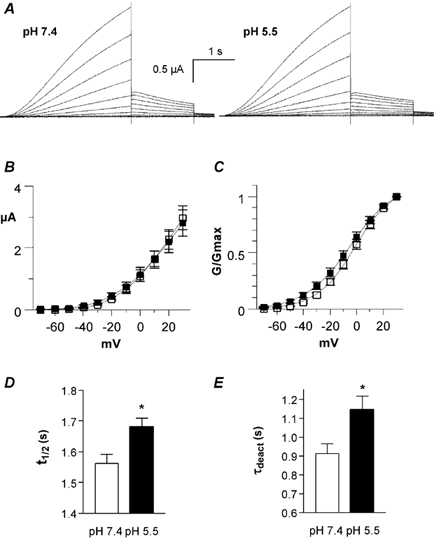
A, representative IKs current traces recorded at pH 7.4 (left) and pH 5.5 (right) using the activation protocol described in Fig. 2A. B, current-voltage relationship of IKs current recorded at pH 7.4 (open squares) and pH 5.5 (filled squares) (n = 8). C, the normalized conductance (G/Gmax) of the tail currents is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 5). D, t1/2 was measured at pH 7.4 and 5.5 at +30 mV (n = 7; *P < 0.0054). E, τdeact data were obtained at pH 7.4 and 5.5 from the monoexponential fit of the tail currents measured at -60 mV from a prepulse potential of +30 mV (n = 5, *P < 0.0027).
In spite of the weak effect of a pHo value of 5.5 on IKsGmax, low pHo still slowed down activation and deactivation kinetics of IKs, suggesting that multiple mechanisms are involved in these effects. In contrast to KCNQ1, the IKs activation process is complex and because of the sigmoidal delay it could not be fitted by exponential functions. Therefore, we used the t1/2 parameter as a rough estimate of IKs current activation. The t1/2 parameter is defined as the time required to reach half of the maximal current amplitude for a given potential. In CHO cells, t1/2 increased significantly from 1.01 ± 0.07 to 1.41 ± 0.05 s (n = 27; P < 0.0001; Fig. 7D and E). A similar slowing of IKs activation was observed in Xenopus oocytes, with an increased t1/2 (Fig. 8D).
In both expression systems, tail currents revealed that IKs deactivation is slowed down by pH 5.5, when compared to pH 7.4 (Fig. 7F and Fig. 8E). For example, in CHO cells, τdeact increased from 0.51 ± 0.05 to 1.05 ± 0.10 s at pH 7.4 and 5.5, respectively (n = 9, P < 0.01; Fig. 7F).
The Δ39-43 KCNE1 mutation prevents the slowing of deactivation but not that of activation produced by low pHo
To test whether the negatively charged residues located at the N-terminal boundary of the single transmembrane segment of KCNE1 play a role in the effects produced by external protons, we checked the effects of low pHo on the Δ39-43 KCNE1 mutant co-expressed with WT KCNQ1 in CHO cells. This KCNE1 deletion removes five amino acids facing the external solution, including the negatively charged residues Asp39 and Glu43. We have shown previously that the activation kinetics of Δ39-43 KCNE1 are distinct from those of both WT KCNQ1 and WT IKs (Abitbol et al. 1999). In contrast to WT KCNQ1, the current flowing through Δ39-43 KCNE1 does not reach a steady state following 2.5 s depolarizing pulses (Fig. 9A). The kinetics are slow, however. In contrast to WT IKs, no sigmoidal delay in activation is observed (Fig. 9A). As for WT IKs, lowering pHo from 7.4 to 5.5 weakly affected the current density that was reduced by only 14 % at +60 mV (n = 10, Fig. 9B, Table 1). Similarly, the voltage dependence of activation of Δ39-43 KCNE1 did not change significantly in response to acidification (Fig. 9C, Table 1). The activation kinetics of this mutant were also slowed down by a low pHo. The t1/2 parameter increased significantly from 0.57 ± 0.09 to 1.00 ± 0.12 s (n = 16; P < 0.001; Fig. 9E). The slowing of activation kinetics elicited by a low pHo was often reflected by the existence of a sigmoidal delay in activation (Fig. 9D). In general, external acidification affected the currents produced by Δ39-43 KCNE1 and WT IKs to a similar extent, except for the deactivation kinetics. In contrast to WT IKs, acidification did not change the Δ39-43 KCNE1 deactivation kinetics (Fig. 9A and F). The decay time constants of the tail current were τdeact = 0.28 ± 0.02 and 0.29 ± 0.02 s (at +60 mV test pulse and -60 mV tail potential; n = 16), for pH 7.4 and 5.5, respectively (Fig. 9F).
Figure 9. Effects of low pHo on the KCNE1 deletion mutant Δ39-43 co-expressed with wild-type (WT) KCNQ1 in CHO cells.
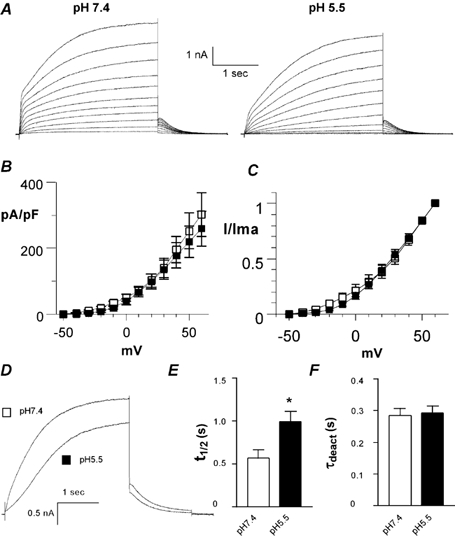
A, representative current traces recorded at pH 7.4 (left) and pH 5.5 (right) using the activation protocol described in Fig. 1A. B, the current density (pA pF−1) is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 10). C, normalized current is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and 5.5 (filled squares; n = 10). D, superimposed traces taken from the same cell at pH 7.4 and pH 5.5, showing the current recorded at +60 and -60 mV test and tail potentials, respectively. E, t1/2 was measured at pH 7.4 and 5.5 at +60 mV (n = 16; *P < 0.001). F, τdeact data were obtained at pH 7.4 and 5.5 from the monoexponential fit of the tail currents measured at -60 mV from a prepulse potential of +30 mV (n = 16).
Low pHo increases the current amplitude of the V319C KCNQ1 mutation
Recently, a residue located in the external vestibule of the Kv1.4 (K532) and Kv1.5 channels (R487) was shown to dramatically affect external proton block (Claydon et al. 2000; Kehl et al. 2002). This site has also been implicated in the regulation of inactivation of Shaker K+ channels (at residue T449; Lopez-Barneo et al. 1993). In KCNQ1 channels, the homologous residue at the external vestibule corresponds to valine 319. Thus, we examined the impact of external acidification on the V319C KCNQ1 mutant in CHO cells. When expressed alone, the V319C KCNQ1 mutant α subunit produced very small currents (not shown) and it was difficult to check the effects of low pHo under these conditions. Thus, we investigated the impact of pH 5.5 on the V319C KCNQ1 mutant co-expressed with WT KCNE1 (Fig. 10A). At pH 7.4, the current produced by V319C KCNQ1/WT KCNE1 exhibited different gating properties when compared to WT IKs, with slower activation kinetics and faster deactivation. In CHO cells, the t1/2 values were significantly higher for V319C KCNQ1/WT KCNE1 (1.37 ± 0.04 s) than for WT IKs (1.01 ± 0.07 s; n = 21-27, P < 0.001; compare Fig. 7E and Fig. 10E). Tail currents at pH 7.4 revealed that deactivation of V319C KCNQ1/WT KCNE1 is faster than that of WT IKs, with τdeact = 0.37 ± 0.02 s and τdeact = 0.51 ± 0.05 s, respectively (at +60 mV test pulse and -60 mV tail potential; Fig. 7F and Fig. 10F; n = 6-21, P < 0.01). V319C KCNQ1/WT KCNE1 also exhibited a small rightward gating shift (Table 1). External acidification did not further slow down the activation kinetics of V319C KCNQ1/WT KCNE1 (t1/2 = 1.35 ± 0.04 s at pH 5.5 versus t1/2 = 1.37 ± 0.04 s at pH 7.4; n = 21, Fig. 10E), suggesting that a maximum slowing of activation gating was already reached at pH 7.4. Similarly, low pHo did not alter the voltage dependence of activation (Fig. 10C). As for WT IKs, deactivation of V319C KCNQ1/WT KCNE1 was slowed down at pH 5.5, with τdeact of the tail current increasing significantly from 369 ± 15 to 559 ± 26 ms at pH 7.4 and 5.5, respectively (n = 21, Fig. 10B and F). In contrast to the current inhibition observed in WT KCNQ1, and to a lesser extent in WT IKs, the current amplitude of this mutant increased significantly (by ≈ 21 %) in response to external acidification (Fig. 10B and D, Table 1; n = 21, P < 0.0015).
Figure 10. Effects of low pHo on the V319C KCNQ1 mutant co-expressed with WT KCNE1 in CHO cells.
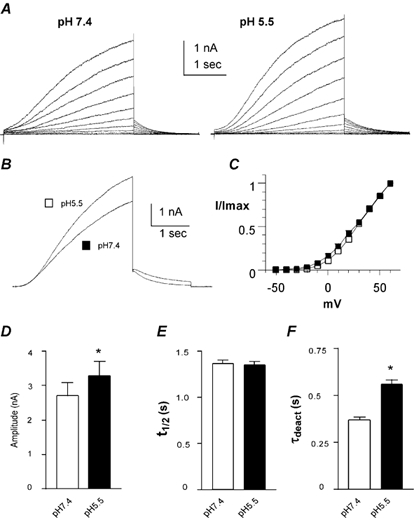
A, representative current traces recorded at pH 7.4 (left) and pH 5.5 (right) using the activation protocol described in Fig. 1A. B, superimposed traces taken from the same cell at pH 7.4 and 5.5, showing the current recorded at +60 and -60 mV test and tail potentials, respectively. C, normalized current is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 15). D, current amplitude measured at pH 7.4 and 5.5 and recorded at +60 mV (n = 21, *P < 0.0015). E, t1/2 was measured at pH 7.4 and pH 5.5 at +60 mV. F, τdeact were obtained at pH 7.4 and 5.5 from the monoexponential fit of the tail currents measured at -60 mV from a prepulse potential of +30 mV (n = 21; *P < 0.001).
Low pHo decreases the current amplitude of the Δ11-38 KCNE1 mutant
In both CHO cells and Xenopus oocytes, our results indicate that KCNE1 significantly weakens the inhibitory effect produced by external acidification on KCNQ1 currents. In addition, it was recently shown that glycosylation of KCNE1 at sites located in the extracellular N-terminus affects the pH sensitivity of WT IKs (Freeman et al. 2000). Thus, we checked in Xenopus oocytes the impact of low pHo on the N-terminal deletion mutant Δ11-38 KCNE1 that lacks one glycosylation site (N26). Co-expression of Δ11-38 KCNE1 with WT KCNQ1 produced large K+ currents that have gating characteristics very similar to those of WT IKs, including a sigmoidal delay and slow kinetics of activation and deactivation (Fig. 11A). However, Δ11-38 KCNE1 opened at more hyperpolarized potentials (V50 = -10.8 ± 1.8 mV, s = -13.6 ± 1.4 mV; from tail currents, n = 8) than WT IKs (V50 = -1.2 ± 1.0 mV, s = -14.2 ± 0.7 mV; from tail currents, n = 5). The Gmax of the Δ11-38 KCNE1 deletion mutant (co-expressed with WT KCNQ1) was substantially more sensitive to pH 5.5 block than WT IKs, with an inhibition at +30 mV of 38 % and 5 %, respectively (Fig. 11B). Following perfusion to pH 5.5, the voltage dependence of activation shifted slightly to more negative potentials (Table 1, Fig. 11C). Like WT KCNQ1 and WT IKs, external acidification slowed down the deactivation of Δ11-38 KCNE1/WT KCNQ1, with τdeact = 0.96 ± 0.06 s and τdeact = 1.31 ± 0.14 s (n = 5; P < 0.01, Fig. 11D).
Figure 11. Effects of low pHo on the KCNE1 deletion mutant Δ11-38 co-expressed with WT KCNQ1 in Xenopus oocytes.
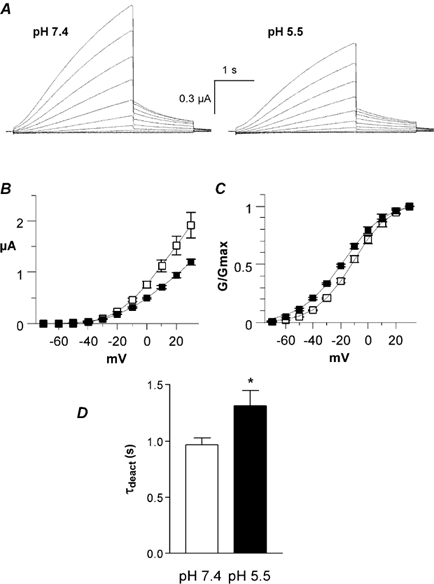
A, representative current traces recorded at pH 7.4 (left) and pH 5.5 (right) using the activation protocol described in Fig. 2A. B, current-voltage relationship of IKs current recorded at pH 7.4 (open squares) and pH 5.5 (filled squares) (n = 6). C, the normalized conductance (G/Gmax) of the tail currents is plotted as a function of the membrane voltage (mV) at pH 7.4 (open squares) and pH 5.5 (filled squares; n = 6). D, τdeact data were obtained at pH 7.4 and 5.5 from the monoexponential fit of the tail currents measured at -60 mV from a prepulse potential of +30 mV (n = 6; *P < 0.01).
Discussion
Using two complementary expression systems, Xenopus oocytes and CHO cells, we examined the effects of external acidification on homomeric KCNQ1 and heteromeric IKs channels. Our results show that acidification from pH 7.4 to pH 5.5 affects differentially KCNQ1 and IKs channels. Lowering pHo produces five main effects on homomeric KCNQ1 channels. It markedly decreases Gmax, causes a positive shift (+25 mV) in the voltage dependence of activation, slows down the activation and deactivation kinetics and reduces the inactivation. In contrast, external acidification poorly inhibits Gmax and the macroscopic inactivation of the KCNQ1 mutant L273F. Heteromeric IKs channels are weakly affected by low pHo, with minor effects on IKs current amplitude and no shift in the voltage dependence of activation. However, an N-terminal deletion of KCNE1 (Δ11-38) leads to significant current inhibition following exposure to pH 5.5. Low pHo slows down IKs activation and deactivation. We found that the KCNE1 mutant Δ39-43 that lacks five residues at the N-terminal boundary of the transmembrane segment does not exhibit slower deactivation kinetics upon exposure to pH 5.5.
Effects of low pHo on homomeric KCNQ1 channels
The present results demonstrate that extracellular acidification reduces KCNQ1 currents by decreasing Gmax, shifting the voltage dependence of activation rightward and slowing the activation kinetics. Our data suggest that low pHo acts on KCNQ1 channels via multiple mechanisms. For example, the small positive shift in V50 (4.25 mV) does not account well for the 30 % inhibition of the KCNQ1 currents obtained following exposure of CHO cells to pH 6.5. In addition, a low pHo reduces inactivation and slows down deactivation, two additional effects that oppose KCNQ1 current inhibition. Considering that WT KCNQ1 exhibits a very partial inactivation (Pusch et al. 1998; Tristani-Firouzi & Sanguinetti, 1998), the inhibition of KCNQ1 currents will dominate the effects produced by external acidification. The depressing effect produced by low pHo on KCNQ1 inactivation results mainly from a marked delay in the onset of inactivation. In Xenopus oocytes, the effect is so strong that there is no detectable inactivation under the experimental conditions we used. Although the mechanisms underlying this pHo action are not known, it is possible that the titration of a residue or the neutralization of negative surface charges produces a conformational change that leads to a slower transition rate from an open state to an inactivated state of the channel. According to the gating scheme of Tristani-Firouzi & Sanguinetti (1998), Cn⇔O1⇔O2⇔I, external protons may slow down the transition from O1 to O2, leading to the marked delay in the onset of inactivation observed at pH 5.5.
The depressing effect produced by protons on KCNQ1 inactivation is unique if one compares the impact of external acidification on other voltage-gated K+ channels. In most cases, external protons lead to enhanced channel inactivation. External acidification rightward shifts the steady state inactivation and accelerates the C-type inactivation kinetics of Shaker K+ channels (Perez-Cornejo, 1999). Likewise, it has been shown that C-type inactivation of Kv1.5 channels is faster and accumulates at acidic pH (Steidl & Yool, 1999). Recently, it was suggested that protonation of an extracellular histidine (H463) leads to the occurrence of a P-type inactivation in Kv1.5 channels (Kehl et al. 2002). Similarly, protonation of an extracellular histidine slows the recovery from N-type inactivation of Kv1.4 channels (Claydon et al. 2000). In contrast, low pHo alters neither the onset nor the recovery of the fast inactivation of recombinant HERG channels and native IKr currents (Anumonwo et al. 1999; Vereecke & Carmeliet, 2000).
Shifting of the voltage dependence of activation by external protons has been reported in many voltage-gated ion channels and is considered to be caused by non-specific surface charge effects. According to this mechanism, titration by protons of negative surface charges would lead the KCNQ1 voltage sensor to detect a decrease in outer negative charges as a hyperpolarization of the membrane, and thus produce a positive shift. However, we could not demonstrate a shift (negative) in voltage sensitivity when pHo was increased up to 9, suggesting that other mechanisms occur. Furthermore, our data show a substantial discrepancy between the effects of protons on activation and deactivation kinetics, which are in both cases slowed down. This is a rather surprising outcome. A more usual effect would imply a slowing of activation and an acceleration of deactivation, as we previously found for the modulation of a transient K+ current by the tyrosine kinase inhibitor genistein (Peretz et al. 1999). Likewise, WT IKs and the Δ39-43 KCNE1 mutant exhibited slower activation kinetics with no shift in the voltage dependence of activation (see below). In this regard, it is interesting to note that in another member of the KCNQ channel family, the KCNQ2 channel, external protons inhibit Gmax and markedly slow the activation kinetics, but do not produce a significant gating shift (data not shown).
Other possible mechanisms could account for the rightward gating shift produced by external acidification. For example, protons could bind to the channel external vestibule to inhibit KCNQ1 ionic currents and to restrict the movement of the voltage sensor. This mechanism may be consistent with the recent evidence for the close proximity of the external vestibule and the S4 voltage sensor segment (Cha & Bezanilla, 1998; Loots & Isacoff, 1998, 2000; Li-Smerin et al. 2000).
Effects of low pHo on the L273F KCNQ1 mutant channels
In contrast to WT KCNQ1, we found that low pHo inhibits Gmax only weakly and barely affects the macroscopic inactivation of the L273F KCNQ1 mutant. This LQT mutation is located in the transmembrane segment S5 and was shown to exhibit a pronounced macroscopic inactivation (Shalaby et al. 1997; Seebohm et al. 2001). The peculiar location of this mutation in the S5 transmembrane segment was suggested to stabilize the pore of KCNQ1 in an inactivated conformation. The lack of impact of protons we found on the macroscopic inactivation suggests that the L273F mutation generates a kind of inactivation that exhibits different properties to those displayed by the WT KCNQ1 channels. As shown by our present results, low pHo slightly slows down the kinetics of macroscopic inactivation of L273F KCNQ1 but does not alter its extent. The action of external protons on the inactivation of L273F KCNQ1 is clearly different to that produced on the inactivation of WT KCNQ1. Although the mechanism has yet to be elucidated, it is possible that this mutation affects the configuration of the pore (Seebohm et al. 2001) and consequently, external protons may have a different impact on the L273F mutant as compared to WT KCNQ1. Interestingly, low pHo still induces slower activation kinetics in L273F KCNQ1, further suggesting that the proton action involves multiple mechanisms.
Effects of low pHo on heteromeric IKs channels
Our data from both CHO cells and Xenopus oocytes indicate that external acidification exerts a much weaker inhibition (if any, see Xenopus oocytes) of Gmax of WT IKs channels, as compared to WT KCNQ1. Our results are in line with those of Freeman et al. (2000), who found that external protons (pH 6) reduce the WT IKs current amplitude by only 20 % (versus ≈ 65 % for WT KCNQ1) in glycosylation-permissive CHO cells. They also showed that a low pHo does not alter the amplitude of WT IKs currents when expressed in glycosylation-deficient CHO cells, suggesting that glycosylation of KCNE1 at its amino-terminus affects the pH sensitivity of IKs channels (Freeman et al. 2000). The quasi-insensitivity of WT IKs current amplitude to external protons in Xenopus oocytes could result from different levels of KCNE1 glycosylation in different expression systems (CHO cells versus Xenopus oocytes). Our results on recombinant IKs agree with those of Vereecke & Carmeliet (2000) on native IKs, who found no reduction in current amplitude and no shift in the voltage dependence of activation following exposure of guinea pig ventricular cells to acidic pH.
Beside its weak effect on WT IKs current amplitude, a low pHo still produces a significant slowing of the activation and deactivation kinetics. Our results show that the Δ39-43 KCNE1 mutation prevents the slowing of deactivation produced by external protons. This deletion removes five amino acids at the amino-terminal boundary of the KCNE1 transmembrane segment, including two negatively charged residues Asp39 and Glu43. We have shown previously that this KCNE1 mutation markedly alters the gating of IKs, including faster activation and regain of inactivation (Abitbol et al. 1999). The present data suggest that KCNE1 residues 39-43 are also important for controlling the proton effect on IKs deactivation.
By what mechanisms does KCNE1 reduce the inhibitory effect produced by external protons on KCNQ1 Gmax? Our data concerning the V319C KCNQ1 and the Δ11-38 KCNE1 mutations provide some hints. The suggest that the structure of the external vestibule is a crucial determinant of how external protons will affect current amplitude. In this regard, it is interesting to note that the V319C KCNQ1 mutant (co-expressed with KCNE1) may produce a conformational change of the external vestibule that leads to increased current amplitude following low pHo exposure. Our results with the Δ11-38 KCNE1 mutant suggest that the KCNE1 amino-terminus affects directly or allosterically the channel external vestibule to prevent access of protons and channel block.
In all, our results suggest that external protons act on KCNQ1 and IKs channels via multiple mechanisms to affect gating and Gmax. Low pHo exerts a much stronger inhibitory effect on KCNQ1 current as compared to IKs. Under conditions of cardiac ischaemia and reperfusion, the blockade of IKr by external acidification will probably predominate in comparison to the impact on IKs (Vereecke & Carmeliet, 2000). Overall, this will decrease the repolarization reserve, leading to a prolongation of action potential duration and an increase in the risk of arrhythmias.
Acknowledgments
This work is supported by the Israel Science Foundation (grant No: 540/01-1) and by a binational France-Israel AFIRST grant (No: 8026). We thank Dr Yael Uziyel for a critical reading of the manuscript.
References
- Abitbol I, Peretz A, Lerche C, Busch AE, Attali B. Stilbenes and fenamates rescue the loss of IKS channel function induced by an LQT5 mutation and other IsK mutants. EMBO Journal. 1999;18:4137–4148. doi: 10.1093/emboj/18.15.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anumonwo JMB, Horta J, Delmar M, Taffet SM, Jalife J. Proton and zinc effects on HERG currents. Biophysical Journal. 1999;77:282–298. doi: 10.1016/S0006-3495(99)76889-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axford TC, Dearani JA, Khait I, Park WM, Patel MA, Doursounian M, Neuringer L, Valeri CR, Khuri SF. Electrode-derived myocardial pH measurements reflect intracellular myocardial metabolism assessed by phosphorus 31-nuclear magnetic resonance spectroscopy during normothermic ischemia. Journal of Thoracic and Cardiovascular Surgery. 1992;103:902–907. [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current [see comments] Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Barros F, Gomez-Varela D, Viloria CG, Palomero T, Giraldez T, De La Pena P. Modulation of human erg K+ channel gating by activation of a G protein-coupled receptor and protein kinase C. Journal of Physiology. 1998;511:333–346. doi: 10.1111/j.1469-7793.1998.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube J, Chahine M, Daleau P. Modulation of HERG potassium channel properties by external pH. Pflügers Archiv. 1999;438:419–422. doi: 10.1007/s004240050930. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmais. Physiological Reviews. 1999;79:917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- Cha A, Bezanilla F. Structural implications of fluorescence quenching in the Shaker K+ channel. Journal of General Physiology. 1998;112:391–408. doi: 10.1085/jgp.112.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claydon TW, Boyett MR, Sivaprasadarao A, Ishii K, Owen JM, O'Beirne HA, Leach R, Komukai K, Orchard CH. Inhibition of the K+ channel Kv1. 4 by acidosis: protonation of an extracellular histidine slows the recovery from N-type inactivation. Journal of Physiology. 2000;526:253–264. doi: 10.1111/j.1469-7793.2000.00253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LC, Lippold JJ, Mitchell KE. Glycosylation influences gating and pH sensitivity of IsK. Journal of Membrane Biology. 2000;177:65–79. doi: 10.1007/s002320001100. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Sakmann B. Multiple conductance states of single acetylcholine receptor channels in embryonic muscle cells. Nature. 1981;294:462–464. doi: 10.1038/294462a0. [DOI] [PubMed] [Google Scholar]
- Jiang M, Dun W, Tseng GN. Mechanism for the effects of extracellular acidification on HERG-channel function. American Journal of Physiology. 1999;277:H1283–1292. doi: 10.1152/ajpheart.1999.277.4.H1283. [DOI] [PubMed] [Google Scholar]
- Jo SH, Youm JB, Kim I, Lee CO, Earm YE, Ho WK. Blockade of HERG channels expressed in Xenopus oocytes by external H+ Pflügers Archiv. 1999;438:23–29. doi: 10.1007/s004240050875. [DOI] [PubMed] [Google Scholar]
- Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Kehl SJ, Eduljee C, Kwan DC, Zhang S, Fedida D. Molecular determinants of the inhibition of human Kv1. 5 potassium currents by external protons and Zn(2+) Journal of Physiology. 2002;541:9–24. doi: 10.1113/jphysiol.2001.014456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn J, Karle C, Thomas D, Yao X, Brachmann J, Kubler W. HERG potassium channel activation is shifted by phorbol esters via protein kinase A-dependent pathways. Journal of Biological Chemistry. 1998;273:25285–25291. doi: 10.1074/jbc.273.39.25285. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Abriel H, Kass RS. Molecular basis of the delayed rectifier I(ks) in heart. Journal of Molecular and Cellular Cardiology. 2001;33:873–882. doi: 10.1006/jmcc.2001.1377. [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y, Hackos DH, Swartz KJ. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 2000;25:411–423. doi: 10.1016/s0896-6273(00)80904-6. [DOI] [PubMed] [Google Scholar]
- Loots E, Isacoff EY. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. Journal of General Physiology. 1998;112:377–389. doi: 10.1085/jgp.112.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots E, Isacoff EY. Molecular coupling of S4 to a K+ channel's slow inactivation gate. Journal of General Physiology. 2000;116:623–636. doi: 10.1085/jgp.116.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors and Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Noble D, Tsien RW. Outward membrane currents activated in the plateau range of potential in cardiac Purkinje fibers. Journal of Physiology. 1969;200:205–231. doi: 10.1113/jphysiol.1969.sp008689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numaguchi H, Johnson JPJ, Jetersen CI, Balser JR. A sensitive mechanism for cation modulation of potassium current. Nature Neuroscience. 2000;3:429–430. doi: 10.1038/74793. [DOI] [PubMed] [Google Scholar]
- Peretz A, Sobko A, Attali B. Tyrosine kinases modulate K+ channel gating in mouse Schwann cells. Journal of Physiology. 1999;519:373–384. doi: 10.1111/j.1469-7793.1999.0373m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cornejo P. H+ ion modulation of C-type inactivation of Shaker K+ channels. Pflügers Archiv. 1999;437:865–870. doi: 10.1007/s004240050856. [DOI] [PubMed] [Google Scholar]
- Po SS, Wang DW, Yang IC, Johnson JPJ, Jie L, Bennett PB. Modulation of HERG potassium channels by extracellular magnesium and quinidine. Journal of Cardiovascular Pharmacology. 1999;33:181–185. doi: 10.1097/00005344-199902000-00002. [DOI] [PubMed] [Google Scholar]
- Pond AL, Nerbonne JM. ERG proteins and functional I(Kr) channels in rat, mouse and human heart. Trends in Cardiovascular Medicine. 2001;11:286–294. doi: 10.1016/s1050-1738(01)00127-x. [DOI] [PubMed] [Google Scholar]
- Pusch M. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with mink. Pflügers Archiv. 1998;437:172–174. doi: 10.1007/s004240050765. [DOI] [PubMed] [Google Scholar]
- Pusch M, Magrassi R, Wollnik B, Conti F. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophysical Journal. 1998;75:785–792. doi: 10.1016/S0006-3495(98)77568-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current: differential sensitivity to block by class-III antiarrhythmic agents. Journal of General Physiology. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G, Scherer CR, Busch AE, Lerche C. Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. Journal of Biological Chemistry. 2001;276:13600–13605. doi: 10.1074/jbc.M008373200. [DOI] [PubMed] [Google Scholar]
- Sesti F, Goldstein SAN. Single-channel characteristics of wild-type IKs channels and channels formed with two mink mutants that cause long QT syndrome. Journal of General Physiology. 1998;112:651–663. doi: 10.1085/jgp.112.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby FY, Levesque PC, Yang WP, Little WA, Conder ML, Jenkins-West T, Blanar MA. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation. 1997;96:1733–1736. doi: 10.1161/01.cir.96.6.1733. [DOI] [PubMed] [Google Scholar]
- Steidl JV, Yool AJ. Differential sensitivity of voltage-gated potassium channels Kv1. 5 and Kv1.2 to acidic pH and molecular identification of pH sensor. Molecular Pharmacology. 1999;55:812–820. [PubMed] [Google Scholar]
- Taglialatela M, Pannaccione A, Iossa S, Castaldo P, Annunziato L. Modulation of the K(+) channels encoded by the human ether-a-gogo-related gene-1 (hERG1) by nitric oxide. Molecular Pharmacology. 1999;56:1298–1308. doi: 10.1124/mol.56.6.1298. [DOI] [PubMed] [Google Scholar]
- Terai T, Furukawa T, Katayama Y, Hiraoka M. Effects of external acidosis on HERG current expressed in Xenopus oocytes. Journal of Molecular and Cellular Cardiology. 2000;32:11–21. doi: 10.1006/jmcc.1999.1048. [DOI] [PubMed] [Google Scholar]
- Thomas D, Zhang W, Karle C, Kathofer S, Schols W, Kubler W, Kiehn J. Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. Journal of Biological Chemistry. 1999;274:27457–27462. doi: 10.1074/jbc.274.39.27457. [DOI] [PubMed] [Google Scholar]
- Tristani-Firouzi M, Sanguinetti MC. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. Journal of Physiology. 1998;510:37–45. doi: 10.1111/j.1469-7793.1998.037bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GN. I(Kr): the hERG channel. Journal of Molecular and Cellular Cardiology. 2001;33:835–849. doi: 10.1006/jmcc.2000.1317. [DOI] [PubMed] [Google Scholar]
- Vereecke J, Carmeliet E. The effect of external pH on the delayed rectifying K+ current in cardiac ventricular myocytes. Pflügers Archiv. 2000;439:739–751. doi: 10.1007/s004249900243. [DOI] [PubMed] [Google Scholar]
- Yamane TI, Furukawa T, Horikawa S, Hiraoka M. External pH regulates the slowly activating potassium current IsK expressed in Xenopus oocytes. FEBS Letters. 1993;319:229–232. doi: 10.1016/0014-5793(93)80552-6. [DOI] [PubMed] [Google Scholar]
- Yang Y, Sigworth FJ. Single-channel properties of IKs potassium channels. Journal of General Physiology. 1998;112:665–678. doi: 10.1085/jgp.112.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]