

Abstract
The release of endothelium-derived relaxing factors, such as nitric oxide (NO), is dependent on an increase in intracellular calcium levels ([Ca2+]i) within endothelial cells. Endothelial cell membrane potential plays a critical role in the regulation of [Ca2+]i in that calcium influx from the extracellular space is dependent on membrane hyperpolarization. In this study, the effect of inhibition of vascular smooth muscle delayed rectifier K+ (KDR) channels by 4-aminopyridine (4-AP) on endothelium-dependent relaxation of rat basilar artery to acetylcholine (ACh) was assessed. ACh-evoked endothelium-dependent relaxations were inhibited by N-(Ω)-nitro-l-arginine (l-NNA) or 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), confirming a role for NO and guanylyl cyclase. 4-AP (300 μm) also suppressed ACh-induced relaxation, with the maximal response reduced from ≈92 to ≈33 % (n = 11; P < 0.01). However, relaxations in response to exogenous NO, applied in the form of authentic NO, sodium nitroprusside or diethylamineNONOate (DEANONOate), were not affected by 4-AP treatment (n = 3-11). These data are not consistent with the view that 4-AP-sensitive KDR channels are mediators of vascular hyperpolarization and relaxation in response to endothelium-derived NO. Inhibition of ACh-evoked relaxation by 4-AP was reversed by pinacidil (0.5-1 μm; n = 5) or 18β-glycyrrhetinic acid (18βGA; 5 μm; n = 5), indicating that depolarization and electrical coupling of the smooth muscle to the endothelium were involved. 4-AP caused depolarization of both endothelial and vascular smooth muscle cells of isolated segments of basilar artery (mean change 11 ± 1 and 9 ± 2 mV, respectively; n = 15). Significantly, 18βGA almost completely prevented the depolarization of endothelial cells (n = 6), but not smooth muscle cells (n = 6) by 4-AP. ACh-induced hyperpolarization of endothelium and smooth muscle cells was also reduced by 4-AP, but this inhibition was not observed in the combined presence of 4-AP and 18βGA. These data indicate that 4-AP can induce an indirect inhibition of endothelium-dependent relaxation in the rat basilar artery by electrical coupling of smooth muscle membrane depolarization to the endothelium via myo-endothelial gap junctions.
Endothelium-dependent relaxation of vascular smooth muscle has been attributed to release of nitric oxide (NO), prostaglandin I2 (prostacyclin), and a third mechanism involving hyperpolarization by as yet ill-defined factor(s) and/or gap junctional communication from the endothelium to smooth muscle (Félétou & Vanhoutte, 1999). The release of endothelium-derived NO by agonists, such as acetylcholine (ACh), is dependent on a rise in intracellular calcium levels ([Ca2+]i) within endothelial cells and activation of NO synthase (NOS). The entry of Ca2+ from the extracellular space is required to maintain production of NO (Lückhoff et al. 1988; Kruse et al. 1994) and several lines of evidence indicate that endothelial cell membrane potential plays a critical role in the regulation of Ca2+ entry (Nilius & Droogmans, 2001). Ca2+ entry is not dependent on voltage-gated Ca2+ channel activity, rather it occurs via a nonselective cation pathway and is dependent on membrane hyperpolarization to provide an inwardly directed electrochemical driving force for Ca2+ movement (Busse et al. 1988; Lückhoff & Busse, 1990a; Kamouchi et al. 1999; Nilius & Droogmans, 2001). Significantly, several studies show that membrane depolarization inhibits both agonist-induced increases in [Ca2+]i and NO release from cultured endothelial cells (Adams et al. 1989; Schilling, 1989; Lückhoff & Busse, 1990b). However, to date, there are no data available concerning the influence of vascular smooth muscle membrane potential on endothelial cell function within intact arteries.
Following its release from the endothelium, NO is thought to cause vasorelaxation, at least in part, by affecting the activity of K+ channels within vascular smooth muscle cells to elicit hyperpolarization and thereby a decline in L-type Ca2+ channel activity, [Ca2+]i and tone development (reviewed by Félétou & Vanhoutte, 1999). There is considerable evidence that NO stimulates large conductance Ca2+-activated K+ (BKCa) channel activity of vascular smooth muscle cells (Robertson et al. 1993) due to: (1) direct effects of NO on channel gating (Lang et al. 2000); (2) phosphorylation by cGMP-dependent protein kinase (PKG; Robertson et al. 1993); and/or (3) modulation of SR Ca2+ release leading to an increased frequency of Ca2+ sparks (Porter et al. 1998). However, other K+ channel types, including ATP-sensitive K+ channels (Murphy & Brayden, 1995) and delayed rectifier K+ (KDR) channels (Zhao et al. 1997; Sobey & Faraci, 1999; Lovren & Triggle, 2000), have also been suggested to play a role in mediating endothelium-dependent vasorelaxation due to NO. For example, in the rat basilar artery in vivo, endothelium-dependent relaxations in response to ACh were inhibited by 4-AP (200 μm), and thus a role for KDR channels in mediating the effects of endothelium-derived NO was proposed (Sobey & Faraci, 1999). The stimulation of KDR channel activity was postulated to be due to a signalling cascade involving activation of guanylyl cyclase, cGMP, PKG and phosphorylation of the channels or associated modulatory subunits (Sobey & Faraci, 1999). Previous studies provide compelling evidence for the modulation of vascular KDR channels by serine/threonine kinase-mediated phosphorylation, for example by cAMP-dependent (PKA) and Ca2+/phospholipid-dependent (PKC) protein kinase (Aiello et al. 1995, 1998; Clément-Chomienne et al. 1996). However, direct evidence for their modulation by a pathway involving cGMP and PKG is lacking.
In this study, the hypothesis that endothelium-dependent NO release produces relaxation of the rat basilar artery via a modulation of KDR channel activity was tested using wire myography and intracellular microelectrode recordings. Although we found that 4-AP depressed ACh-induced endothelium-dependent relaxation of rat basilar artery, the data were not consistent with the view that KDR channel activity was modulated by NO released from the endothelium. Rather, our findings suggest a novel, alternative explanation for the depression of endothelium-dependent relaxation by 4-AP. Specifically, our findings suggest that electrotonic communication of the depolarization due to 4-AP treatment from the smooth muscle cells to the endothelial layer via myo-endothelial gap junctions results in a depression of endothelium-dependent relaxation due to a suppression of NO synthesis and/or release.
Methods
Tissue preparation
Male Sprague-Dawley rats (300-350 g) were maintained and killed by halothane inhalation and exsanguination according to the standards of the Canadian Council on Animal Care and a protocol reviewed by the Animal Care Committee of the Faculty of Medicine, The University of Calgary. Basilar arteries were gently removed, placed in ice-cold Krebs buffer and carefully cleaned of adherent connective tissue. The Krebs solution contained (mm): NaCl, 120; KCl, 4.8; NaHCO3, 25; NaH2PO4, 1.2; MgSO4, 1.2; glucose, 11; and CaCl2, 1.8.
Wire myography
For wire myograph studies, basilar arteries were cut into segments (2 mm in length) and mounted between two tungsten wires (40 μm in diameter) in a Mulvany-Halpern myograph (J. P. Trading, Denmark) for recording of isometric tension. Tissues were maintained at 37 °C in 5 ml of oxygenated (95 % O2-5 % CO2) Krebs buffer and resting tension was set to approximately 2 mN. Cumulative concentration-response relations for ACh, authentic NO (applied as bolus doses), sodium nitroprusside (SNP) and diethylamineNONOate (DEANONOate) were determined using arterial segments preconstricted with 5-hydroxytryptamine (5-HT; 0.1-1 μm) at a concentration that produced 75 % of maximal contraction. Peak relaxations in response to ACh were determined for each concentration and normalized to developed tension to obtain the percentage relaxation. To determine the concentration of 5-HT that would produce 75 % level of peak contraction, complete dose-response curves were determined for 26 tissues with an average peak developed force of 6.9 ± 0.3 mN observed with 3 μm 5-HT; the 75 % value, 5.3 mN, was obtained on average with 0.1-0.3 μm 5-HT. In some instances, it was necessary to adjust the concentration of 5-HT to obtain a similar level of precontraction in the presence of inhibitors as observed under control conditions. Contractility data were recorded to hard disk and analysed via a Powerlab A/D convertor and software (ADInstruments, USA).
Membrane potential recordings
For measurement of smooth muscle and endothelial cell membrane potential, basilar arteries were cut open longitudinally and pinned to the bottom of a Sylgard chamber, endothelial surface uppermost. Tissues were maintained at 37 °C and constantly perfused with warmed Krebs buffer at a rate of 5 ml min−1. Measurements of membrane potential were made with sharp glass microelectrodes, back-filled with 3 m KCl and with resistances of 60-100 MΩ. All data were recorded through a Powerlab (ADInstruments) and stored on disk. Drugs were added either as bolus doses or to the perfusate as indicated.
Solutions and chemicals
All drugs were obtained from Sigma Chemical Co. (St Louis, MO, USA) except for DEANONOate (Cayman Chemical Company, USA). All drugs were dissolved in Krebs solution except for DEANONOate, which was dissolved in degassed distilled water and, pinacidil and 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ), which were dissolved in DMSO and then diluted in Krebs solution to the desired concentration (DMSO at the maximal final dilution of 0.1 % had no effect on 5-HT-induced contraction (control 6.9 ± 1.0 mN versus DMSO 7.2 ± 1.1 mN; n = 5; P > 0.05) or ACh-induced relaxation (control 95.4 ± 2.3 % versus DMSO 90.8 ± 3.5 % at 1 μm ACh; n = 3; P > 0.05)). All Krebs solutions containing 4-AP were checked for appropriate pH of 7.4. Solutions of authentic NO were prepared by injecting research grade NO gas (Praxair, Canada) into deoxygenated H2O. Authentic NO solutions were injected into the myograph close to the arterial segments and in volumes of less than 250 μl.
Statistics
For wire myograph studies, all data were expressed as means ± s.e.m. of n rats with only one tissue per rat employed for a given experimental group. Changes in membrane potential (in mV) were expressed as means ± s.e.m. of the number of rats. The statistical significance of all differences between mean values was calculated using Student's paired t test or repeated measures ANOVA followed by Bonferonni's post hoc test. A level of P < 0.05 was considered to be statistically significant.
Results
Effect of 4-AP on endothelium-dependent relaxations in response to ACh
As shown in Fig. 1, ACh (1 nm to 1 μm) evoked concentration-dependent relaxation of segments of basilar artery precontracted with 5-HT. Removal of the endothelium prevented relaxation in response to ACh (data not shown) and the relaxations were significantly inhibited in the presence of l-NNA (100 μm) or ODQ (10 μm; Fig. 1), but not indomethacin (data not shown). Mean cumulative concentration-relaxation curves for ACh in the absence and presence of l-NNA and ODQ are shown in Fig. 1. Maximal relaxation was reduced from ≈90 and ≈88 to ≈18 (n = 14; P < 0.01) and ≈26 % (n = 13; P < 0.01) in l-NNA and ODQ, respectively. Several tissues showed complete suppression with l-NNA alone (e.g. representative data of Fig. 1), but on average, relaxation in response to ACh was still observed. Complete suppression of endothelium-dependent relaxation was obtained, however, with the combination of l-NNA and NG-nitro-l-arginine methyl ester (l-NAME; 100 μm; Fig. 1).
Figure 1. Effect of l-NNA (100 μm) and ODQ (10 μm) on ACh-evoked relaxation of segments of rat basilar artery precontracted with 5-HT.

A, representative recordings showing ACh (1 nm to 3 μm)-evoked relaxations of 5-HT (0.1 μm)-induced tone in the absence and presence of l-NNA. B and C, cumulative concentration-relaxation curves for ACh in the absence and presence of l-NNA and l-NNA +l-NAME (100 μm), and ODQ, respectively. Data are the means of 14, 4 and 13 experiments, respectively, ± s.e.m. (in some cases the error bars in this and subsequent figures are within the size of the data symbols).
Application of 4-AP (300 μm) induced an increase in basal tone in 6 of 11 basilar artery segments studied (mean increase 2.6 ± 0.4 mN; n = 11 arteries) and regular oscillations in tone development following 4-AP exposure were observed in some tissues (Fig. 2). In the presence of 4-AP, relaxations in response to ACh were significantly inhibited, with the maximal response reduced from ≈92 to ≈33 % (n = 11; P < 0.01; Fig. 2). Pretreatment with tetrodotoxin (10 μm; n = 5) or selective inhibition of neuronal Kv1.1 subunit-containing KDR channels with κ-dendrotoxin (Akhtar et al. 2002; 10 nm; n = 3) were without effect on basal tone or ACh-induced relaxations in the absence or presence of 4-AP (data not shown). We also employed clofilium for comparison with 4-AP; this drug inhibits recombinant Kv1.2 and Kv1.5 channels (Malayev et al. 1995; Yamagishi et al. 1995) and these channel subunits are expressed by vascular myocytes (Thorneloe et al. 2001). Clofilium at 3 μm (n = 5) inhibited ACh-induced relaxation, in a similar way to the suppression produced with 4-AP (Fig. 2), but it had no effect on relaxations produced by the putative BKCa channel activator, NS1619 (30 μm; n = 4) up to the maximal concentration tested of 10 μm (data not shown).
Figure 2. Effect of 4-AP (300 μm) on ACh-evoked relaxation of segments of rat basilar artery precontracted with 5-HT.

A, representative recording showing oscillatory increase in basal tension induced by 4-AP in some tissues. B, representative recording showing ACh (1 nm to 3 μm)-evoked relaxations of 5-HT (0.1 μm)-induced tone in the absence and presence of 4-AP. C, cumulative concentration-relaxation curves for ACh in the absence and presence of 4-AP and clofilium (3 μm). Data are the average of 11 and 5 experiments ± s.e.m. with 4-AP and clofilium, respectively,
Effect of 4-AP on relaxations in response to exogenous NO
To test whether NO released from the endothelium modulated vascular KDR channel activity, exogenous NO donors were applied to vessel segments in the absence and presence of 4-AP (300 μm). If KDR channels contributed to the relaxation induced by endothelium-derived NO, we reasoned that relaxations in response to authentic NO, or a NO donor, would be similarly depressed by 4-AP. However, in contrast to ACh, relaxations of basilar artery segments in response to authentic NO (1 nm to 1 μm; n = 11), SNP (1 nm to 3 μm; n = 5) or DEANONOate (1 nm to 0.3 μm; n = 3) were not inhibited by 4-AP (300 μm). Figure 3 shows a representative recording for authentic NO and mean cumulative concentration- relaxation curves for authentic NO, SNP and DEANONOate in the absence and presence of 4-AP. Similarly, clofilium (3 μm) did not affect relaxations in response to authentic NO (n = 4; data not shown).
Figure 3. Effect of 4-AP (300 μm) on relaxations in response to NO in rat basilar artery.
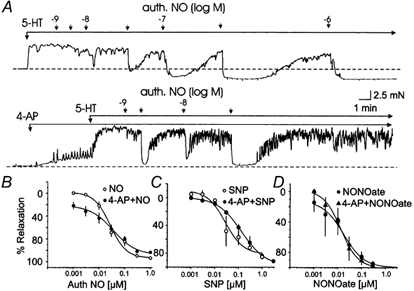
A, representative recordings showing authentic NO (1 nm to 1 μm)-induced relaxations of 5-HT (0.03 μm)-induced tone in the absence and presence of 4-AP. B, C and D, mean cumulative concentration-relaxation curves for authentic NO, SNP and DEANONOate (NONOate) in the absence and presence of 4-AP. Each point is the mean of 11, 5 and 3 experiments, respectively, ±s.e.m.
Prevention of 4-AP-induced inhibition of relaxation in response to ACh by pinacidil
To assess the role of membrane potential depolarization as a cause of the inhibition of ACh-induced endothelium-dependent relaxation induced by 4-AP, the K+ channel opener, pinacidil, was employed to activate ATP-sensitive K+ channels and evoke hyperpolarization within the vessel segments. Pinacidil was applied in increasing concentrations until reversal of 4-AP-induced (300 μm) increase in basal tension was observed (relaxation was noted at 1 μm pinacidil in most tissues). Significantly, in the combined presence of pinacidil and 4-AP, the inhibitory effect of KDR channel inhibition on ACh-evoked relaxations of segments of basilar artery was not apparent. A representative trace illustrating the lack of effect of 4-AP on ACh-induced relaxations in the presence of pinacidil and mean concentration-relaxation curves for ACh in the presence and absence of 4-AP and pinacidil (1 μm; n = 5) are shown in Fig. 4.
Figure 4. Effect of pinacidil (1 μm) on 4-AP-induced inhibition of relaxation in response to ACh in rat basilar artery.

A, representative recordings showing the prevention of 4-AP (300 μm)-induced inhibition of ACh (1 nm to 10 μm)-evoked relaxation of 5-HT (0.3 μm)-induced tone by pinacidil (1 μm). Note that relaxation of 4-AP-induced increase in basal tone was unaffected by 0.1, but it was reversed by 1 μm pinacidil. B, mean cumulative concentration-relaxation curves for ACh in the absence and presence of 4-AP and pinacidil (1 μm). Each point is the mean of 5 experiments ±s.e.m.
Prevention of 4-AP-induced inhibition of ACh-induced relaxation by 18βGA
Endothelial cells are not thought to express voltage-gated K+ channels (Himmel et al. 1993; Nilius & Droogmans, 2001) and for this reason we felt that the depolarization was not due to a direct effect of 4-AP on endothelial cell membrane potential. However, we reasoned that ACh-induced relaxation might be inhibited indirectly if the depolarization evoked by 4-AP inhibition of smooth muscle KDR channels was communicated to the endothelium via electrical coupling by myo-endothelial gap junctions. To test this hypothesis, ACh was applied in the combined presence of 4-AP and the gap junction uncoupling agent, 18βGA (Davidson & Baumgarten, 1988; Guan et al. 1996; Yamamoto et al. 1998). In preliminary experiments, 18βGA was found to depress 5-HT-induced contractions in a concentration-dependent fashion between 10 and 50 μm, probably as a result of nonspecific inhibition of tone development, as previously reported (Chaytor et al. 2000; Tare et al. 2002). However, at 5 μm, no effect on 5-HT-induced tone development (control 8.4 ± 0.7 versus 18βGA 8.3 ± 0.6 mN; n = 4; P > 0.05) or ACh-induced relaxation (control 78.4 ± 2.3 versus 18βGA 82.7 ± 9.8 % with 1 μm ACh; n = 3; P > 0.05) was observed, so this concentration was employed to determine the effect on endothelium-dependent relaxation in the presence of 4-AP. Treatment with 18βGA was found to prevent the inhibitory effect of 4-AP (300 μm) on ACh-induced relaxation. A representative recording and mean concentration- relaxation curves for ACh in the presence of 4-AP and 18βGA are shown in Fig. 5 (n = 5).
Figure 5. Effect of 18βGA (5 μm) on 4-AP-induced inhibition of relaxation in response to ACh in rat basilar artery.
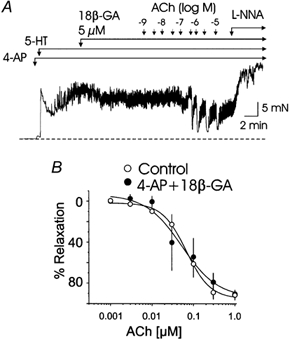
A, representative recordings showing the prevention of 4-AP (300 μm)-induced inhibition of ACh (1 nm to 10 μm)-evoked relaxations of 5-HT (0.3 μm)-induced tone by 18βGA (5 μm). B, mean cumulative concentration-relaxation curves for ACh in the absence and presence of 4-AP and 18βGA. Each point is the mean of 5 experiments ±s.e.m.
Effect of 4-AP on membrane potential of endothelial and vascular smooth muscle cells of rat basilar artery
To directly assess the effect of 4-AP on the membrane potential of vascular smooth muscle and endothelial cells of rat basilar artery and to determine if the gap junction uncoupling drug, 18βGA, would prevent depolarization of the endothelium by 4-AP, segments of rat basilar artery were opened and impaled with sharp microelectrodes from the luminal surface. In most cases, the endothelial cells were the first cells to be impaled using this approach and recordings of smooth muscle membrane potential were obtained by further advancing the pipette into the vessel wall. To confirm the specific cell type impaled during each recording, the response of membrane potential to BayK8644 (1 μm), an activator of L-type voltage-gated Ca2+ channels, was determined in the absence and presence 18βGA (5 μm). The rationale for the use of BayK8644 was as follows: L-type Ca2+ channels are expressed by vascular smooth muscle but not by endothelial cells (Nilius & Droogmans, 2001), so BayK8644 would be not expected to depolarize endothelial cells in the absence of electrical communication with the underlying smooth muscle cells. The representative recordings of Fig. 6 show that BayK8644 (1 μm) depolarized both endothelial and smooth muscle cells, but in the presence of the gap junction uncoupler, 18βGA (5 μm), only the membrane potential of smooth muscle cells was affected by the Ca2+ channel activator. The resting membrane potential of endothelial cells was depolarized compared to that of smooth muscle cells under control conditions (Table 1). BayK8644 caused depolarization of endothelial and smooth muscle cells by +10.4 ± 1.2 and +12.9 ± 1.5 mV, respectively (Table 1). Application of 18βGA (5 μm) induced a small, but significant depolarization of +4.1 ± 1.1 mV in endothelial cells (P < 0.01), but it had no effect on smooth muscle cell membrane potential (Table 1). In the presence of 18βGA, however, the membrane potential of endothelial cells was unaffected by BayK8644, but the membrane potential of vascular smooth muscle cells exhibited a depolarization of +14.2 ± 1.4 mV (P < 0.01; Table 1). In contrast to BayK8644, application of 5-HT (10 μm) had only a minimal effect on membrane potential of smooth muscle cells (+2.1 ± 0.9 mV; n = 15).
Figure 6. Effect of BayK8644 (1 μm) on membrane potential of endothelial and smooth muscle cells of rat basilar artery in the absence and presence of 18βGA (5 μm).

Representative microelectrode recordings of BayK8644-induced depolarization of endothelial (Endo) and smooth muscle cells (VSM) in the absence (left) and presence (right) of 18βGA. Note that the depolarization of the endothelial cell, but not the VSM cell, due to BayK8644 was prevented by 18βGA.
Table 1.
Effect of BayK8644 on endothelial and vascular smooth muscle cell membrane potential (mV) in the absence and presence of 18βGA
| Control | Endothelium -37±0.9* (n = 38) | Vascular smooth muscle -49.1±1.1 (n = 38) | ||||
|---|---|---|---|---|---|---|
| Before | After | Δ | Before | After | Δ | |
| BayK8644 (n = 14,14) | −37.4 ± 1.5 | −27.0 ± 2.0† | +10.4 ± 1.2 | −49.9 ± 2.2 | −37.0 ± 2.6† | +12.9 ± 1.5 |
| 18βGA (n = 38,38) | −37.8 ± 0.9 | −32.7 ± 0.7‡ | +4.1 ± 1.1 | −49.1 ± 1.1 | −48.8 ± 1.0 | +0.3 ± 1.0 |
| 18βGA + BayK8644 (n = 38,38) | −32.3 ± 1.0 | −29.0 ± 1.2 | +3.3 ± 0.9§ | −49.0 ± 1.7 | −34.8 ± 2.0‡ | +14.2 ± 1.4 |
Significantly different from smooth muscle cell membrane potential (P < 0.01).
Significantly different from value before Bay K8644 (P < 0.01).
Significantly different from value before 18βGA (endothelium) or 18βGA + BayK8644 (vascular smooth muscle; P < 0.011).
Significantly different from value in BayK8644 but absence of 18βGA (P < 0.01).
The representative recordings of Fig. 7 show that application of 4-AP (200 μm) evoked a similar level of depolarization in endothelial and vascular smooth muscle cells of rat basilar artery by +11.0 ± 1.0 (P < 0.05) and +10.0 ± 1.0 mV (P < 0.05), respectively. Significantly, however, the depolarization of endothelial cells induced by 4-AP was reversed by 18βGA (5 μm), whereas uncoupling gap junctions had no effect on the depolarization of the smooth muscle cells. Similarly, Fig. 7 shows that if 18βGA was added before the addition of 4-AP, then the magnitude of the depolarization of endothelial cells was reduced. Under control conditions, 4-AP depolarized endothelial cells by +12.4 ± 3.9 mV (n = 6), but after treatment with 18βGA, 4-AP treatment had no effect on membrane potential (Table 2). In contrast, application of 4-AP depolarized smooth muscle cells by +9.2 ± 1.1 mV before, and by a similar magnitude of +10.5 ± 1.1 mV after, treatment with 18βGA (Table 2).
Figure 7. Effect of 4-AP (200 μm) on membrane potential of endothelial and smooth muscle cells of rat basilar artery in the absence and presence of 18βGA (5 μm).
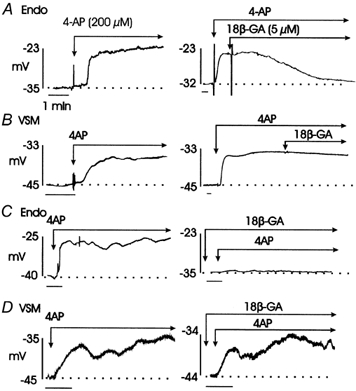
A, representative microelectrode recordings showing 4-AP-induced depolarization of an endothelial cell (left) and the ability of 18βGA to reverse the depolarization caused by 4-AP. B, representative microelectrode recordings showing 4-AP-induced depolarization of a vascular smooth muscle cell (left) and the inability of 18βGA to reverse the depolarization caused by 4-AP. C, representative microelectrode recordings showing 4-AP-induced depolarization of an endothelial cell (left) and the lack of depolarization in response to 4-AP in the presence of 18βGA. D, representative microelectrode recordings showing 4-AP-induced depolarization of a vascular smooth muscle cell in the absence (left) and presence of 18βGA (right).
Table 2.
Effect of 4-AP on endothelial and vascular smooth muscle cell membtane potential (mV) in the absence and presence of 18βGA
| Endothelium | Vascular smooth muscle | |||||
|---|---|---|---|---|---|---|
| Before | After | Δ | Before | After | Δ | |
| 4-AP (n = 15,11) | −38.9 ± 1.4 | −27.9 ± 0.7† | +11.0 ± 1.0 | −46.3 ± 1.8 | −36.4 ± 1.8† | +10.0 ± 1.0 |
| 4-AP (before 18βGA) (n = 6,6) | −40.2 ± 1.7 | −27.8 ± 2.0† | +12.4 ± 3.9 | −43.4 ± 2.5 | −34.2 ± 2.4† | +9.2 ± 1.1 |
| 4-AP (after 18βGA)* (n = 6,6) | −34.7 ± 1.7 | −33.0 ± 1.4 | +1.7 ± 1.0‡ | −47.4 ± 2.7 | −36.9 ± 3.3† | +10.5 ± 1.1 |
Paired impalements maintained during 4-AP application in the absence and presence of 18βGA.
Significantly different from value of membrane potential in the absence of 4-AP(P < 0.01)
Significantly different from changes in membrance potential due to 4-AP in the absence of 18βGA (P < 0.01).
Effect of 4-AP on ACh-induced hyperpolarization of endothelium and vascular smooth muscle cells of rat basilar artery
As shown in Fig. 8, bolus doses of ACh (10 μmol) caused transient hyperpolarizations of the membrane potential of endothelial and smooth muscle cells that were of a similar peak amplitude of approximately -10 mV (Table 3). The hyperpolarization of endotheliaI cells was unaffected by treatment with l-NNA, but hyperpolarization of smooth muscle cells was completely blocked (Table 3). These data indicate that the hyperpolarization of the smooth muscle cells in response to ACh was due to NO released from the endothelium. In the presence of 4-AP (200 μm), the membrane potential of both cell types was significantly depolarized and the hyperpolarizations induced by ACh did not reach the level of membrane potential attained in the absence of 4-AP. Additionally, 4-AP did not alter the magnitude of the ACh-induced hyperpolarization of endothelium, but the magnitude of smooth muscle hyperpolarization was significantly reduced (Fig. 8 and Table 3). To quantify the change in ACh-induced hyperpolarization in the presence of 4-AP, the amplitude and duration of the hyperpolarization were simultaneously considered by calculating the area under the curve for the endothelial and smooth muscle hyperpolarizations. Endothelial and smooth muscle cell hyperpolarization induced by ACh were significantly reduced from 2164 ± 141 to 1156 ± 80 mV s (n = 9; P < 0.01) and from 4427 ± 367 to 1982 ± 210 mV s (n = 9; P < 0.01), respectively by 4-AP. Thus, 4-AP reduced the extent of ACh-induced hyperpolarization of the endothelium and smooth muscle cells, as well as shifting the range over which the hyperpolarizations occurred to more positive potentials. Application of 18βGA did not alter hyperpolarization of endothelial or smooth muscle cells in response to ACh, indicating that myo-endothelial gap junctional coupling is not required for endothelium-dependent relaxation in rat basilar artery (n = 6). However, in the combined presence of 4-AP and 18βGA, ACh-induced hyperpolarization of smooth muscle cell membrane potential (-12.3 ± 1.6 mV) was significantly greater than in 4-AP alone (-6.5 ± 1.1 mV; Fig. 8 and Table 3). Additionally, the area under the curves for ACh-induced hyperpolarization of endothelial and smooth muscle cell membrane potential were 2297 ± 96 and 3645 ± 244 mV s (n = 4) in the presence of 4-AP and 18βGA, and significantly greater than the values obtained in 4-AP alone (P > 0.05), but not different from those determined for control conditions.
Figure 8. Effect of l-NNA (100 μm) and 4-AP (200 μm) on ACh-induced hyperpolarization of the membrane potential of endothelial and smooth muscle cells of rat basilar artery.

A, representative microelectrode recordings showing an ACh (bolus concentration 100 μmol)-induced hyperpolarization of a smooth muscle cell (VSM) in the absence (left) and lack of change in membrane potential following ACh application in the presence of l-NNA (right). B, representative microelectrode recordings of ACh-induced hyperpolarizations of an endothelial cell (Endo) in the absence (left) and in the presence of 4-AP alone (middle) and 4-AP + 18βGA (right). C, representative microelectrode recordings of ACh-induced hyperpolarization of a smooth muscle cell (VSM) in the absence (left) and in the presence of 4-AP (midde) and 4-AP + 18βGA (right).
Table 3.
Effect of 4-AP on ACh-induced hyperpolarization of endothelial and vascular smooth muscle cell membrane potential (mV) in the absence and presence of 18βGA
| Endothelium | Vascular smooth muscle | |||||
|---|---|---|---|---|---|---|
| Before | After | Δ | Before | After | Δ | |
| ACh (n = 3, 3) | −35.7 ± 1.8 | −47.3 ± 1.0† | −11.7 ± 1.1 | −43.3 ± 3.0 | −55.2 ± 2.0† | −11.8 ± 1.6 |
| ACh +l-NNA* (n = 3, 3) | −35.3 ± 2.4 | −44.3 ± 2.4† | −12.0 ± 1.8 | −40.7 ± 2.4 | −41.5 ± 3.1‡ | −0.8 ± 1.0‡ |
| ACh (n = 16, 16) | −39.8 ± 1.2 | −48.5 ± 1.6† | −9.5 ± 1.1 | −46.1 ± 1.4 | −56.4 ± 0.7† | −10.3 ± 1.1 |
| ACh + 4-AP (n = 9, 9) | −27.1 ± 1.1§ | −37.8 ± 1.1†§ | −7.9 ± 0.9 | −33.8 ± 2.1§ | −40.5 ± 2.1†§ | −6.5 ± 1.1§ |
| ACh + 4-AP and 18βGA (n = 4, 4) | −39.0 ± 2.1¶ | −52.0 ± 1.8† | −12.3 ± 1.0¶ | 35.3±2.0 | −47.5±1.9†¶ | −12.3 ± 1.6¶ |
Impalements maintained during 4-AP application in the absence and presence of l-NNA.
Significantly different from value of membrane potential in the absence of ACh (P < 0.01).
Significantly different from change in membrane potential due to ACh in the absence of L-NNA (P < 0.01).
Significantly different from value in the absence of 4-AP (P < 0.01).
Significantly different from value in the presence of 4-AP (P < 0.01).
Discussion
This study makes the novel observation that depolarization of vascular smooth muscle cells of the rat basilar artery due to inhibition of KDR channels with 4-AP, or the activation of L-type Ca2+ channels with BayK8644, leads to a corresponding depolarization of endothelial cell membrane potential due to electrical communication between the two cell types via myo-endothelial gap junctions. Significantly, depolarization of the endothelium in the presence of 4-AP was associated with a depression of ACh-induced endothelium-dependent relaxation mediated by NO. Based on our findings, it is unlikely that vascular smooth muscle KDR channels mediate NO-induced hyperpolarization and relaxation of the rat basilar artery. Rather, the results indicate that 4-AP-induced smooth muscle depolarization may indirectly inhibit ACh-induced relaxation via electrotonic depolarization of the endothelium and suppression of NO synthesis and/or release. The ability of endothelial cell hyperpolarization to affect smooth muscle membrane potential via myo-endothelial gap junctional communication is well recognized as a potential mechanism to account for non-NO, non-prostanoid endothelium-dependent relaxation, i.e. relaxation due to an endothelium-dependent hyperpolarizing factor (reviewed by Bény, 1999; Félétou & Vanhoutte, 1999). However, only limited evidence exists to indicate that electrical communication can occur in the reverse direction, from vascular smooth muscle cells to the endothelium, and the functional consequences of this coupling have not been assessed previously. The results of this study provide the first evidence that selective depolarization of vascular smooth muscle cells can influence the level of endothelial cell membrane potential and thereby endothelium function by reducing NO-dependent control of vascular tone.
A substantial body of evidence indicates that endothelium-dependent relaxation of arterial smooth muscle is mediated by at least three different factors/mechanisms, including NO, prostacyclin and an as-yet-unidentified endothelium-derived hyperpolarizing factor, which may be a P450-derived epoxide, K+ or myo-endothelial gap junctional electrical coupling (Edwards et al. 1998; Coleman et al. 2001; Fleming, 2001). Endothelium-dependent relaxation of the rat basilar artery evoked by ACh was shown to be completely suppressed by inhibition of NOS by the combination of l-NNA and l-NAME, and by the inhibitor of soluble guanylyl cyclase, ODQ, but not by indomethacin or 18βGA. This indicates that in this vessel, endothelium-dependent relaxation is exclusively due to the release of NO. There is compelling evidence that BKCa channels are important effectors of the action of NO in mediating vascular smooth muscle hyperpolarization and relaxation in many vessels (Homer & Wanstall, 2000). This role for BKCa was established in part by experiments showing that the specific BKCa inhibitor iberiotoxin suppressed endothelium-dependent relaxation induced by NO (Khan et al. 1993; Bialecki & Stinson-Fisher, 1995; Kitazono et al. 1997; Li et al. 1997; Satake et al. 1997). A similar approach has been employed to define a role for other K+ channels as effectors of endothelium-derived NO. For example, inhibition of ACh-evoked relaxation by 4-AP was interpreted to indicate that KDR channels mediate NO-induced relaxation of rat basilar arteries in vivo (Sobey & Faraci, 1999). In the light of the present data, however, this mechanism would not appear to be involved. This view is based on the lack of effect of 4-AP on relaxations evoked by exogenous sources of NO, including authentic NO, sodium nitroprusside and DEANONOate. Similar results with 1 mm 4-AP and DEANONOate were previously reported by Hemplemann et al. (2001). In the absence of a role for KDR channels in mediating the effect of NO on rat basilar arterial smooth muscle cells, an alternative explanation for the suppression of ACh-evoked endothelium-dependent relaxation by 4-AP is required.
This study indicates that membrane potential depolarization underlies the suppression of endothelium-dependent relaxation by 4-AP. Pinacidil was employed to activate ATP-sensitive K+ channels and hyperpolarize vascular smooth muscle cells (Nelson & Quayle, 1995) and was found to prevent the suppression of ACh-evoked relaxation by 4-AP. This indicates that depolarization of the vascular smooth muscle cells and/or the endothelium by 4-AP was responsible for the suppression of endothelium-dependent relaxation by NO. This is consistent with previous observations showing that depolarization of pulmonary arteries with elevated extracellular potassium (25 mm) blocked endothelium-dependent relaxation (Seiden et al. 2000). Depolarization of the basilar arterial smooth muscle cells by 4-AP per se did not block the actions of endothelium-derived NO on the myocytes because 4-AP-induced-depolarization had no effect on relaxations in response to exogenous NO. Alternatively, depolarization of the endothelium is known to reduce NO synthesis and/or release. Previous studies show that NO synthesis and/or release is dependent on a rise in intracellular [Ca2+]i in endothelial cells, that Ca2+ influx is required for sustained endothelium-dependent relaxation, and that this Ca2+ influx is decreased by endothelial cell depolarization (Adams et al. 1989; Lückhoff & Busse, 1990b; Wang & Van Breeman, 1999). Endothelial Ca2+ influx is not mediated by voltage-dependent Ca2+ channels and is not activated by depolarization. Rather, hyperpolarization appears to be required, in part to maintain an appropriate electrochemical driving force for ACh-induced Ca2+ influx through a nonselective cation conductance (Schilling, 1989; Kamouchi et al. 1999; Wang & Van Breeman, 1999), but it may also prevent Ca2+ entry by channel inactivation and/or reduction in unitary conductance. The latter is suggested by the fact that the decline in Ca2+ influx with depolarization positive to ≈-35 mV exceeds that which can be explained by a reduction in driving force (Oike et al. 1994; Wang & Van Breeman, 1999). According to these observations, a reduction in basal and agonist-evoked NO release would accompany endothelial depolarization from -39 to -27 mV under resting conditions and reduction in ACh-induced peak hyperpolarization from -49 to -38 mV in the presence of 4-AP. A limitation in this study is the lack of direct measurements to confirm the suppression of ACh-induced NO release from the basilar arterial endothelium by 4-AP treatment. However, the present results provide indirect evidence that NO release was reduced. First, the ACh-induced hyperpolarization of smooth muscle cells was blocked by l-NNA, indicating that it was induced by NO. Second, both the amplitude and the area under curve for the ACh-induced smooth muscle hyperpolarization were significantly reduced in the presence of 4-AP, suggesting that the magnitude of NO release was reduced.
It is unlikely that a direct effect of 4-AP on endothelial K+ channels was responsible for the depolarization observed in this study. This is based on: (1) the lack of effect of 4-AP on the membrane potential of endothelial cells in the presence of 18βGA; and (2) the lack of evidence of 4-AP-sensitive KDR channels in endothelial cells (Nilius & Droogmans, 2001) despite immunocytochemical evidence of voltage-gated K+ channel subunit expression in the endothelium of some resistance arteries and capillaries (Fan & Walsh, 1999; Cheong et al. 2001) and reports of their presence in cultured endothelial cells (Takeda et al. 1987; Hogg et al. 1999). Nonspecific effects would also seem to be an unlikely explanation for the suppression of ACh-induced relaxation by 4-AP. Although 4-AP is widely employed as an inhibitor of voltage-gated K+ channels in smooth muscle and other cell types (Nelson & Quayle, 1995), actions unrelated to channel block were previously reported and its selectivity and suitability for use in intact tissue and/or cell preparations has been questioned (e.g. Wood & Gillespie, 1998). However, the concentration of 4-AP (200 or 300 μm) used in this study is consistent with the IC50 for inhibition of KDR currents of vascular myocytes (Kerr et al. 2001), and significantly lower than that associated with reduced Ca2+ release, intracellular pH and/or BKCa channel activity (i.e. 5-20 mm; Petkova-Kirova et al. 2000; Quignard et al. 2000; Ghisdal & Morel, 2001). Additionally, clofilium (3 μm) was also employed to selectively inhibit KDR channels and found to mimic the actions of 4-AP treatment. For this reason, it is unlikely that 4-AP affected endothelial or vascular smooth muscle cell function via a mechanism(s) unrelated to an inhibition of smooth muscle KDR channels.
The electrophysiological data obtained in this study are consistent with the view that the inhibition of endothelium-dependent relaxation by 4-AP was the result of an indirect effect on endothelial cell membrane potential mediated by electrical coupling through myo-endothelial gap junctions. We employed 18βGA to uncouple gap junctions and to prevent myo-endothelial gap junctional communication based on the previous findings showing a phosphatase-mediated dephosphorylation of connexins and loss of cell-to-cell coupling via disassembly of gap junction plaques with this agent (Guan et al. 1996). 18βGA was found to prevent and/or reverse endothelial cell depolarization induced by 4-AP or BayK8644, but it did not affect smooth muscle cell depolarization by these treatments. These results indicate that uncoupling myo-endothelial gap junctions can prevent endothelial cell depolarization as a result of the selective depolarization of smooth muscle cells by two distinct mechanisms, inhibition of KDR channels and activation of L-type Ca2+ channels. Moreover, in the presence of 18βGA, we found that the inhibition of ACh-induced hyperpolarization and relaxation by 4-AP was significantly reduced. This indicates that uncoupling myo-endothelial gap junctions permitted a recovery of endothelium function despite the presence of maintained smooth muscle depolarization due to 4-AP. We do not attribute the effect of 18βGA to the nonspecific effects on membrane potential (Tare et al. 2002) and tone development (Chaytor et al. 2000) that were noted when this drug was employed at 50-100 μm. We approached the use of 18βGA with caution and used a concentration of 5 μm that did not: (1) cause relaxation of arterial segments precontracted with 5-HT; (2) alter the extent of ACh-induced relaxation or sensitivity to ACh; or (3) change smooth muscle membrane potential. 18βGA did cause a slight depolarization of basilar arterial endothelial cells, but at approximately +4 mV, the change in membrane potential was much smaller than the +30 mV depolarization observed with higher concentrations (Coleman et al. 2001). We attribute this +4 mV depolarization to the uncoupling of the endothelium from the hyperpolarizing influence of the underlying smooth muscle cells, as previously reported (Popp et al. 1996; Yamamoto et al. 1998).
The role of the endothelium in control of vascular smooth muscle contractility in health is well established and dysfunctional regulation of arterial tone development due to abnormal endothelial function and reduced NO release is recognized to be a important cause of abnormal contractility associated with vascular disease (Vanhoutte, 1997; Schiffrin, 2001; Baron, 2002). This study shows for the first time that vascular smooth muscle cell membrane depolarization can alter endothelial function. The presence of myo-endothelial gap junctions between endothelial and smooth muscle cells of several arteries has been identified (e.g. Sandow & Hill, 2000), and these structures are present in the rat basilar artery (S. L. Sandow & C. E. Hill, unpublished observations). Moreover, previous studies have shown that changes in arterial smooth muscle membrane potential can be communicated to endothelial cells in several vessels (von der Weid & Bény, 1993; Bény & Pacicca, 1994; Marchenko & Sage, 1994; Murai et al. 1999; White & Hiley, 2000). For example, noradrenaline-induced depolarization (rat aorta; Marchenko & Sage, 1994) and levcromakalim-induced hyperpolarization (rabbit and rat mesenteric arteries; Murai et al. 1999; White & Hiley, 2000) were reported to be transmitted from smooth muscle to endothelial cells. However, the physiological and pathophysiological significance of smooth muscle control of endothelial function via myo-endothelial gap junction communication has not been addressed previously. In the present study, 4-AP was found to cause an impairment of agonist-induced, endothelium-dependent regulation of tone development in the rat basilar artery. Based on this novel finding, it would seem worthwhile to consider the possibility that abnormal smooth muscle depolarization in disease may contribute to a loss of endothelium-dependent control of arterial tone development, for example, as a result of depressed smooth muscle K+ channel, or enhanced Ca2+ channel, activity and/or expression (Smirnov et al. 1994; Platoshyn et al. 2001). NO is also known to have additional roles within blood vessels, including control of proliferation and phenotype, and abnormal NO synthesis and/or release due to smooth muscle depolarization and electrotonic depolarization of the endothelium could also contribute to dysfunctional regulation of vasculogenesis, atheroscelerosis and retinopathy. To address these possibilities, further studies will be required to determine: (1) the electrical properties, such as the ratio of electrical impedance between the endothelium and smooth muscle layers, as well as the level of myo-endothelial gap junctional conductance, that are required for control of endothelial membrane potential by vascular smooth muscle; and (2) whether the properties of the endothelium, smooth muscle cells and myo-endothelial gap junctions of other arteries are appropriate for smooth muscle to endothelium communication in health and/or disease states.
Acknowledgments
This work was supported by the Alberta Heritage Foundation for Medical Research (AHFMR), Canadian Institutes of Health Research (MT-13505) and The Wellcome Trust (UK).
References
- Adams DJ, Barakeh J, Laskey R, Van Breeman C. Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB Journal. 1989;3:2389–2400. doi: 10.1096/fasebj.3.12.2477294. [DOI] [PubMed] [Google Scholar]
- Aiello EA, Malcolm AT, Walsh MP, Cole WC. Beta-adrenoceptor activation and PKA regulate delayed rectifier K+ channels of vascular smooth muscle cells. American Journal of Physiology. 1998;275:H448–459. doi: 10.1152/ajpheart.1998.275.2.H448. [DOI] [PubMed] [Google Scholar]
- Aiello EA, Walsh MP, Cole WC. Phosphorylation by protein kinase A enhances delayed rectifier K+ current in vascular smooth muscle cells. American Journal of Physiology. 1995;268:H924–934. doi: 10.1152/ajpheart.1995.268.2.H926. [DOI] [PubMed] [Google Scholar]
- Akhtar S, Shamotienko O, Papakosta M, Ali F, Dolly JO. Characteristics of brain Kv1 channels tailored to mimic native counterparts by tandem linkage of alpha subunits. Implications for K+ channelopathies. Journal of Biological Chemistry. 2002;277:16376–16383. doi: 10.1074/jbc.M109698200. [DOI] [PubMed] [Google Scholar]
- Baron AD. Insulin resistance and vascular function. Journal of Diabetes Complications. 2002;16:92–102. doi: 10.1016/s1056-8727(01)00209-4. [DOI] [PubMed] [Google Scholar]
- Bény JL. Information networks in the arterial wall. News in Physiological Sciences. 1999;11:68–73. doi: 10.1152/physiologyonline.1999.14.2.68. [DOI] [PubMed] [Google Scholar]
- Bény JL, Pacicca C. Bidirectional communication between smooth muscle and endothelial cells in the pig coronary artery. American Journal of Physiology. 1994;266:H1465–1472. doi: 10.1152/ajpheart.1994.266.4.H1465. [DOI] [PubMed] [Google Scholar]
- Bialecki RA, Stinson-Fisher C. KCa channel antagonists reduce NO donor-mediated relaxation of vascular and tracheal smooth muscle. American Journal of Physiology. 1995;268:L152–159. doi: 10.1152/ajplung.1995.268.1.L152. [DOI] [PubMed] [Google Scholar]
- Busse R, Fichtner H, Lückhoff A, Kohlhardt M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cells. American Journal of Physiology. 1988;255:H965–969. doi: 10.1152/ajpheart.1988.255.4.H965. [DOI] [PubMed] [Google Scholar]
- Chaytor AT, Marsh WL, Hutcheson IR, Griffith TM. Comparison of glycyrrhetinic acid isoforms and carbenoxolone as inhibitors of EDHF-type relaxations mediated via gap junctions. Endothelium. 2000;7:265–278. doi: 10.3109/10623320009072213. [DOI] [PubMed] [Google Scholar]
- Cheong A, Dedman AM, Xu SZ, Beech DJ. Kvα1 channels in murine arterioles: differential cellular expression and regulation of diameter. American Journal of Physiology. 2001;281:H1057–1065. doi: 10.1152/ajpheart.2001.281.3.H1057. [DOI] [PubMed] [Google Scholar]
- Clément-Chomienne O, Walsh MP, Cole WC. Angiotensin II activation of protein kinase C decreases delayed rectifier K+ current in rabbit vascular myocytes. Journal of Physiology. 1996;495:689–700. doi: 10.1113/jphysiol.1996.sp021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HA, Tare M, Parkington HC. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. Journal of Physiology. 2001;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap junctional intracellular communication. Structure-activity relationships. Journal of Pharmacology and Experimental Therapeutics. 1988;246:1104–1107. [PubMed] [Google Scholar]
- Edwards GA, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Fan J, Walsh KB. Mechanical stimulation regulated voltage-gated potassium currents in cardiac microvascular endothelial cells. Circulation Research. 1999;84:451–457. doi: 10.1161/01.res.84.4.451. [DOI] [PubMed] [Google Scholar]
- Félétou M, Vanhoutte PM. The third pathway: endothelium-dependent hyperpolarization. Journal of Physiology and Pharmacology. 1999;50:525–534. [PubMed] [Google Scholar]
- Fleming I. Cytochrome P450 enzymes in vascular homeostasis. Circulation Research. 2001;89:753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- Ghisdal P, Morel N. Cellular target of voltage and calcium-dependent K+ channel blockers involved in EDHF-mediated responses in rat superior mesenteric artery. British Journal of Pharmacology. 2001;134:1021–1028. doi: 10.1038/sj.bjp.0704348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Wilson S, Schlender KK, Ruch RJ. Gap-junction disassembly and connexin 43 dephosphorylation induced by 18 beta-glycyrrhetinic acid. Molecular Carcinogenesis. 1996;16:157–164. doi: 10.1002/(SICI)1098-2744(199607)16:3<157::AID-MC6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Hempelmann RG, Seebeck J, Kruse ML, Ziegler A, Mehdorn HM. Role of potassium channels in the relaxation induced by the nitric oxide (NO) donor DEA/NO in the isolated rat basilar artery. Neuroscience Letters. 2001;313:21–24. doi: 10.1016/s0304-3940(01)02225-x. [DOI] [PubMed] [Google Scholar]
- Himmel HM, Wharton AR, Strauss HC. Intracellular calcium currents and stimulus response coupling in endothelial cells. Hypertension. 1993;21:112–127. doi: 10.1161/01.hyp.21.1.112. [DOI] [PubMed] [Google Scholar]
- Hogg DS, Albarwani S, Davis AR, Kozlowski RS. Endothelial cells freshly isolated from resistance-sized pulmonary arteries possess a unique K+ current profile. Biochemical and Biophysical Research Communications. 1999;24:405–409. doi: 10.1006/bbrc.1999.1338. [DOI] [PubMed] [Google Scholar]
- Homer KL, Wanstall JC. Cyclic GMP-independent relaxation of rat pulmonary artery by spermine NONOate, a diazeniumdiolate nitric oxide donor. British Journal of Pharmacology. 2000;131:673–682. doi: 10.1038/sj.bjp.0703613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamouchi M, Droogmans G, Nilius B. Membrane potential as a modulator of the free intracellular Ca2+ concentration in agonist-activated endothelial cells. General Physiology and Biophysics. 1999;18:199–208. [PubMed] [Google Scholar]
- Kerr PM, Clément-Chomienne O, Thorneloe KS, Chen TT, Ishii K, Sontag DP, Walsh MP, Cole WC. Heteromultimeric Kv1. 2-Kv1.5 channels underlie 4-aminopyridine-sensitive delayed rectifier K+ current of rabbit vascular myocytes. Circulation Research. 2001;89:1038–1044. doi: 10.1161/hh2301.100803. [DOI] [PubMed] [Google Scholar]
- Khan SA, Mathews WR, Meisheri KD. Role of calcium-activated K+ channels in vasodilation induced by nitroglycerine, acetylcholine and nitric oxide. Journal of Pharmacology and Experimental Therapeutics. 1993;267:1327–1335. [PubMed] [Google Scholar]
- Kitazono T, Ibayashi S, Nagao T, Fujii K, Fujishima M. Role of Ca2+-activated K+ channels in acetylcholine-induced dilatation of the basilar artery in vivo. British Journal of Pharmacology. 1997;120:102–106. doi: 10.1038/sj.bjp.0700880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse HJ, Grunberg B, Siess W, Weber PC. Formation of biologically active autacoids is regulated by calcium influx in endothelial cells. Arteriosclerosis Thrombosis. 1994;14:1821–1828. doi: 10.1161/01.atv.14.11.1821. [DOI] [PubMed] [Google Scholar]
- Lang RJ, Harvey JR, McPhee GJ, Klemm MF. Nitric oxide and thiol reagent modulation of Ca2+ activated (BKCa) channels myocytes of the guinea-pig taenia caeci. Journal of Physiology. 2000;525:363–376. doi: 10.1111/j.1469-7793.2000.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PL, Zou AP, Campbell WB. Regulation of potassium channels in coronary arterial smooth muscle by endothelium-derived vasodilators. Hypertension. 1997;29:262–267. doi: 10.1161/01.hyp.29.1.262. [DOI] [PubMed] [Google Scholar]
- Lovren F, Triggle C. Nitric oxide and sodium nitroprusside-induced relaxation of the human umbilical artery. British Journal of Pharmacology. 2000;131:521–529. doi: 10.1038/sj.bjp.0703588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lückhoff A, Busse R. Activators of potassium channels enhance calcium influx into endothelial cells as a consequence of potassium currents. Naunyn-Schmiedebergs Archives of Pharmacology. 1990a;342:94–99. doi: 10.1007/BF00178979. [DOI] [PubMed] [Google Scholar]
- Lückhoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflügers Archiv. 1990b;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- Lückhoff A, Pohl U, Mülsch A, Busse R. Differential role of extra- and intracellular calcium in the release of EDRF and prostacyclin from cultured endothelial cells. British Journal of Pharmacology. 1988;95:189–196. doi: 10.1111/j.1476-5381.1988.tb16564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malayev AA, Nelson DJ, Philipson LH. Mechanism of clofilium block of the human delayed rectifier potassium channel. Molecular Pharmacology. 1995;47:198–205. [PubMed] [Google Scholar]
- Marchenko SM, Sage SO. Smooth muscle cells affect endothelial membrane potential in rat aorta. American Journal of Physiology. 1994;267:H804–811. doi: 10.1152/ajpheart.1994.267.2.H804. [DOI] [PubMed] [Google Scholar]
- Murai T, Muraki K, Imaizumi Y, Watanabe M. Levcromakalim causes indirect endothelial hyperpolarization via a myo-endothelial pathway. British Journal of Pharmacology. 1999;128:1491–1496. doi: 10.1038/sj.bjp.0702956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy ME, Brayden JE. Nitric oxide hyperpolarises rabbit mesenteric arteries via ATP-sensitive potassium channels. Journal of Physiology. 1995;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. American Journal of Physiology. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiological Reviews. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- Oike M, Gericke M, Droogmans G, Nilius B. Calcium entry activated by store depletion in human umbilical vein endothelial cells. Cell Calcium. 1994;16:367–376. doi: 10.1016/0143-4160(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Petkova-Kirova P, Gagov H, Krien U, Duridanova D, Noack T, Schubert R. 4-Aminopyridine affects rat arterial smooth muscle BK(Ca) currents by changing intracellular pH. British Journal of Pharmacology. 2000;131:1643–1650. doi: 10.1038/sj.bjp.0703742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ, Yuan X-J. Chronic hypoxia decreases KV channel expression and function in pulmonary artery myocytes. American Journal of Physiology. 2001;280:L801–812. doi: 10.1152/ajplung.2001.280.4.L801. [DOI] [PubMed] [Google Scholar]
- Popp R, Bauersachs J, Hecker M, Fleming I, Busse J. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. Journal of Physiology. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter VA, Bonev AD, Knot HJ, Heppner TJ, Stevenson AS, Kleppisch T, Lederer WJ, Nelson MT. Frequency modulation of Ca2+ sparks is involved in regulation of arterial diameter by cyclic nucleotides. American Journal of Physiology. 1998;274:C1346–1355. doi: 10.1152/ajpcell.1998.274.5.C1346. [DOI] [PubMed] [Google Scholar]
- Quignard JF, Félétou M, Thollon C, Vilaine JP, Duhault J, Vanhoutte PM. Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. British Journal of Pharmacology. 2000;129:1103–1112. doi: 10.1038/sj.bjp.0703175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson BE, Schubert R, Hescheler J, Nelson MT. CGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. American Journal of Physiology. 1993;265:C299–303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circulation. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- Satake N, Shibata M, Shibata S. The involvement of KCa, KATP and KV channels in vasorelaxing responses to acetylcholine in rat aortic rings. General Pharmacology. 1997;28:453–457. doi: 10.1016/s0306-3623(96)00238-8. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL. A critical review of the role of endothelial factors in the pathogenesis of hypertension. Journal of Cardiovascular Pharmacology. 2001;38(suppl. 2):S3–6. doi: 10.1097/00005344-200111002-00002. [DOI] [PubMed] [Google Scholar]
- Schilling WP. Effect of membrane potential on cytosolic calcium of bovine aortic endothelial cells. American Journal of Physiology. 1989;257:H778–784. doi: 10.1152/ajpheart.1989.257.3.H778. [DOI] [PubMed] [Google Scholar]
- Seiden JE, Platoshyn O, Bakst AE, McDaniel SS, Yuan J X-J. High K+-induced membrane depolarization attenuates endothelium-dependent pulmonary vasodilation. American Journal of Physiology. 2000;278:L261–267. doi: 10.1152/ajplung.2000.278.2.L261. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Robertson TP, Ward JPT, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. American Journal of Physiology. 1994;266:H365–370. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- Sobey CG, Faraci FM. Inhibitory effect of 4-aminopyridine on responses of the basilar artery to nitric oxide. British Journal of Pharmacology. 1999;126:1437–1443. doi: 10.1038/sj.bjp.0702439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Schini V, Stoeckel H. Voltage-activated potassium, but not calcium currents in cultured bovine aortic endothelial cells. Pflügers Archiv. 1987;410:385–393. doi: 10.1007/BF00586515. [DOI] [PubMed] [Google Scholar]
- Tare M, Coleman HA, Parkington HC. Glycyrrhetinic acid derivatives inhibit hyperpolarization in endothelial cells of guinea pig and rat arteries. American Journal of Physiology. 2002;282:H335–341. doi: 10.1152/ajpheart.2002.282.1.H335. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Chen TT, Kerr PM, Grier EF, Horowitz B, Cole WC, Walsh MP. Molecular composition of 4-aminopyridine-sensitive voltage-gated K+ channels of vascular smooth muscle. Circulation Research. 2001;89:1030–1037. doi: 10.1161/hh2301.100817. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM. Endothelial dysfunction and atherosclerosis. European Heart Journal. 1997;18(suppl. E):E19–29. doi: 10.1016/s0195-668x(97)90005-1. [DOI] [PubMed] [Google Scholar]
- Von Der Weid PY, Bény JL. Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cells. Journal of Physiology. 1993;471:13–24. doi: 10.1113/jphysiol.1993.sp019888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Van Breeman C. Depolarization-mediated inhibition of Ca2+ entry in endothelial cells. American Journal of Physiology. 1999;277:H1498–1504. doi: 10.1152/ajpheart.1999.277.4.H1498. [DOI] [PubMed] [Google Scholar]
- White R, Hiley CR. Hyperpolarisation of rat mesenteric endothelial cells by ATP-sensitive K+ channel openers. European Journal of Pharmacology. 2000;397:297–290. doi: 10.1016/s0014-2999(00)00271-5. [DOI] [PubMed] [Google Scholar]
- Wood PG, Gillespie JJ. In permeabilised endothelial cells IP3-induced Ca2+ release is dependent on the cytoplasmic concentration of monovalent cations. Cardiovascular Research. 1998;37:263–270. doi: 10.1016/s0008-6363(97)00207-1. [DOI] [PubMed] [Google Scholar]
- Yamagishi T, Ishii K, Taira N. Antiarrhythmic and bradycardic drugs inhibit currents of cloned K+ channels, KV1. 2 and KV1.4. European Journal of Pharmacology. 1995;281:151–159. doi: 10.1016/0014-2999(95)00240-l. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Fukata H, Nakahira Y, Suzuki H. Blockade by 18β-glycyrrhetinic acid of intercellular electrical coupling in guinea-pig arterioles. Journal of Physiology. 1998;511:501–508. doi: 10.1111/j.1469-7793.1998.501bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YJ, Wang J, Rubin LJ, Yuan XJ. Inhibition of Kv and KCa channels antagonizes NO-induced relaxation in pulmonary artery. American Journal of Physiology. 1997;272:H904–912. doi: 10.1152/ajpheart.1997.272.2.H904. [DOI] [PubMed] [Google Scholar]