

Abstract
A non-genomic antisecretory role for dexamethasone at low concentrations (0.1 nm to1 μm) is described in monolayers of human bronchial epithelial cells in primary culture and in a continuous cell line (16HBE14o- cells). Dexamethasone produced a rapid decrease of [Ca2+]i (measured with fura-2 spectrofluorescence) to a new steady-state concentration. After 15 min exposure to dexamethasone (1 nm), [Ca2+]i was reduced by 32 ± 11 nm (n = 7, P < 0.0001) from a basal value of 213 ± 36 nm (n = 7). We have shown previously that aldosterone (1 nm) also produces a rapid fall in [Ca2+]i; however, after the decrease in [Ca2+]i induced by dexamethasone, subsequent addition of aldosterone did not produced any further lowering of [Ca2+]i. The rapid response to dexamethasone was insensitive to pretreatment with cycloheximide and unaffected by the glucocorticoid type II and mineralocorticoid receptor antagonists RU486 and spironolactone, respectively. The rapid [Ca2+]i decrease induced by dexamethasone was inhibited by the Ca2+-ATPase pump inhibitor thapsigargin (1 μm), the adenylate cyclase inhibitor MDL hydrochloride (500 μm) and the protein kinase A inhibitor Rp-adenosine 3′,5′-cyclic monophosphorothioate (200 μm), but was not affected by the protein kinase C inhibitor, chelerythrine chloride (0.1 μm). Treatment of 16HBE14o- cell monolayers with dexamethasone (1 nm) inhibited the large and transient [Ca2+]i increase induced by apical exposure to ATP (10−4m). Dexamethasone (1 nm) also reduced by 30 % the Ca2+-dependant Cl− secretion induced by apical exposure to ATP (measured as the Cl−-sensitive short-circuit current across monolayers mounted in Ussing chambers). Our results demonstrate, for the first time, that dexamethasone at low concentrations inhibits Cl− secretion in human bronchial epithelial cells. The rapid inhibition of Cl− secretion induced by the synthetic glucocorticoid is associated with a rapid decrease in [Ca2+]i via a non-genomic mechanism that does not involve the classical glucocorticoid or mineralocorticoid receptor. Rather, it is a result of rapid non-genomic stimulation of thapsigargin-sensitive Ca2+-ATPase, via adenylate cyclase and protein kinase A signalling.
In the present study, we investigated the rapid effect of dexamethasone on intracellular Ca2+ signalling and on the Cl− secretion induced by apical ATP in cultured human bronchial epithelial cell monolayers. The glucocorticoids are an important class of drug for their anti-inflammatory and immunosupressive activity and are used clinically at various doses. Low doses are used for basal immunosuppressive treatment, including the treatment of airway tract infections such as asthma and cystic fibrosis. High doses are used for the treatment of acute conditions such as inflammatory exacerbation in multiple sclerosis (Filipovic et al. 1997), acute spinal cord injury (Bracken et al. 1990) or severe attacks of asthma (Bousquet, 2000). It has been shown that glucocorticoids produce their pharmacological effects through a classical genomic pathway involving binding to a specific cytosolic receptor, translocation into the nucleus and subsequent activation or repression of protein synthesis. The genomic effects of steroids are characterised by a latency of onset lasting several hours. In contrast, many steroid hormones have been reported to induce rapid effects on various second messenger systems and ion transporters. These fast responses are incompatible with the involvement of the classical genomic pathway for steroid hormone action. The rapid effects of glucocorticoids have been described in several tissues such as endometrial cells (Koukouritaki et al. 1996), vascular smooth muscle (Steiner et al. 1988; Muto et al. 2000) and renal cortical collecting duct (Harvey & Higgins, 2000). However, the cellular mechanism and the physiological role of the non-genomic effects of glucocorticoids are not understood and have not been reported in human bronchial epithelia.
Modulation of [Ca2+]i mediates different physiological responses to glucocorticoids (Lewis et al. 1986; Han et al. 1999), and opposing effects of glucocorticoids on [Ca2+]i have been reported. An increase in [Ca2+]i is induced by glucocorticoids in cortical collecting duct (Harvey & Higgins, 2000) and in vascular smooth muscle (Steiner et al. 1988). In contrast, a decrease of [Ca2+]i following glucocorticoid treatment has been reported in other tissues such as rat thymocyte (Buttgereit et al. 1997), human leukocyte, airway smooth muscle (Chhabra et al. 1999) and in human lymphoblast (Gardner & Zhang, 1999).
Using Ca2+ imaging and short-circuit current (ISC) experiments, we have investigated the effects of low concentrations (1 nm to 1 μm) of dexamethasone on [Ca2+]i and the Ca2+-dependent Cl− secretion induced by apical ATP in human bronchial epithelial cells. Airway epithelial cells have been shown to respond to extracellular nucleotides including ATP by activation of a Cl− secretory pathway (Willumsen & Boucher, 1989; Stutts et al. 1992) and inhibition of Na+ absorption (Mall et al. 2000). More specifically, the stimulation of Cl− secretion through airway epithelia in response to apical nucleotides has been described as being mediated by purinoreceptor activation and a rapid transient [Ca2+]i increase (Mason et al. 1991; Walsh et al. 2000).
The findings reported here of rapid antisecretory responses to a glucocorticoid in human airway epithelial cells might have interesting clinical implications regarding the immediate consequences of corticotherapy. In addition, cystic fibrosis and asthma are two pathologies that are correlated with a dysfunction of the secretory properties of the airway epithelium that controls the fluid and electrolyte composition of the lumen. Cystic fibrosis is associated with a deficit of Cl− secretion arising from the mutation of the gene coding for the cystic fibrosis conductance regulator (CFTR) protein, which functions as a Cl− channel (Clarke et al. 1992). In asthma, it has been reported recently that mucus overproduction is associated with an increased expression of Ca2+-activated Cl− channels (Hoshino et al. 2002). The rapid antisecretory role of glucocorticoid is a newly discovered property of glucocorticoid activity in human bronchial epithelium, which may be distinct from its anti-inflammatory role, but is relevant to the treatment of airway infections.
Methods
Cell culture
The human bronchial epithelial 16HBE14o- cell line is derived from the surface epithelium of mainstream, second-generation bronchi (Cozens et al. 1994) and form polarised monolayers with intact tight junctions, retain the Cl− transport properties typical of freshly isolated surface airway epithelial cells and express other differentiated features characteristic of the native epithelium (Cozens et al. 1994). Cells were grown on a vitrogen/collagen/fibronectin coating, in Eagle's Minimal Essential Medium (EMEM, BioWhittaker) supplemented with 10 % fetal calf serum, 1 % penicillin G, 1 % streptomycin and 1 % l-glutamine, and incubated in a 37 °C, 5 % CO2 atmosphere.
For cultures of native bronchus epithelium, primary cultures were grown from human bronchial epithelial cells obtained from bronchial tube surgery biopsy samples. The bronchial samples were taken from a normal area of bronchi removed from patients suffering from lung cancer but who had normal lung function, with their consent (approved by local medical ethics committee, Comité Consultatif de Protection des Personnes se prêtant à des Recherches Biomédicales (CCPPRB), Montpellier). After excision, the bronchial tubes were washed and incubated either overnight at 4 °C or for 2 h at 37 °C with 0.38 mg ml−1 hyaluronidase, 0.75 mg ml−1 collagenase, 1 mg ml−1 protease and 0.3 mg ml−1 DNAse in RPMI 1640 medium (Gibco, Grand Island, NY, USA) and then filtered through a 70 mm mesh nylon strainer. Pieces of epithelium retained on the strainer were washed and resuspended in phosphate-buffered saline (PBS). After centrifugation (1200 g, 5 min), epithelial cells were resuspended in small airway epithelium basal medium (Clonetics, BioWhittaker, San Diego, CA, USA) supplemented with 0.5 μg ml−1 human recombinant epidermal growth factors, 7.5 mg ml−1 bovine pituitary extract, 0.5 mg ml−1 adrenaline, 10 mg ml−1 transferrin, 5 mg ml−1 insulin, 0.1 μg ml−1 retinoic acid, 6.5 μg ml−1 triiodothyronine, 50 mg ml−1 gentamicin, 50 μg ml−1 amphotericin B and 50 mg ml−1 fatty-acid-free bovine serum albumin. The epithelial cell suspension was then transferred to 12 mm diameter permeable filters (Costar Snapwell-Clear 3801, 0.4 mm pore size) or onto a glass coverslip and incubated at 37 °C in a 5 % CO2 humidified atmosphere.
Ca2+ imaging
[Ca2+]i was determined in confluent monolayers of 16HBE14o- cells or primary cultures of human bronchial epithelial cells grown on fibronectin/collagen/BSA-coated glass coverslips, as described previously (Urbach & Harvey, 2001). The cells were loaded with 5 μm of the Ca2+-sensitive fluorescent probe fura-2 acetoxy-methyl ester (fura-2/AM) for 30 min, in the dark at room temperature (22 °C). Cells were then washed twice in Hepes-buffered Krebs-Heinseleit solution (NaCl 140 mm, KCl 5 mm, CaCl2 2 mm, MgCl2 1 mm, Hepes 10 mm, Tris-HCl 10 mm, glucose 10 mm, pH 7.4, 280-290 mosmol l−1). The coverslip covered with a confluent monolayer of bronchial cells was mounted on the stage of an inverted microscope equipped for epifluorescence (Diaphot 200, Nikon, The Netherlands). The light from a xenon lamp (Osram, Germany) was filtered through alternating 340 and 380 nm filters (Nikon), which were mounted on a motorised chopper under computer control (Starwise Fluo system, Imstar, France). The emission fluorescence produced after fura-2/AM excitation was filtered at 510 nm. The transmitted light image was detected using an intensified CCD video camera (Darkstar, Photonics Sciences, UK) that was coupled to the microscope. The fluorescence obtained at each excitation wavelength (F340 and F380) depends upon the level of Ca2+ binding to fura-2/AM, according to an in vivo calibration performed using a range of EGTA-buffered Ca2+ solutions of the fura-2 free acid. [Ca2+]i was calculated automatically by a computer program (Starwise, Imstar) using the Grynkiewicz equation (Grynkiewicz et al. 1985).
Cells were exposed to various dexamethasone concentrations made up from a 10−2m stock solution of hormone dissolved in methanol. We verified that 0.01 % methanol, corresponding to the maximum amount of solvent at which the cells were exposed, had no effect on [Ca2+]i (Urbach & Harvey, 2001). We also verified that the resting [Ca2+]i levels of 16HBE14o- cell monolayers were not significantly altered 20 min after the beginning of the recording in non-treated cells (Urbach & Harvey, 2001).
I SC measurement
16HBE14o- cells or human bronchial cells in primary culture were grown on permeable filters (Costar Snapwell-Clear 3801, 12 mm diameter, 0.4 mm pore size). Monolayers of 1.2 cm2 exposed surface area were mounted in temperature-controlled horizontal Ussing chambers and bathed in Hepes-buffered Krebs-Heinseleit solution for between 0 and 3 days after the electrical resistance had reached a maximum value (800-1200 Ω). The ISC experiments were usually performed at 37 °C. However, since the Ca2+-imaging experiments were carried out at room temperature, we verified that at room temperature, ATP produced a similar ISC response. The spontaneous transmembrane potential was measured using a voltage-clamp model amplifier (EVC 4000, World Precision Instruments) and clamped to 0 mV by application of a ISC. The ISC was sampled digitally into the computer at 10 s intervals. Under these conditions, the ISC is a measure of electrogenic transepithelial ion transfer. The output from the amplifier was digitised using a PowerLab system (Chart for Windows v4.0, ADInstruments).
Chemicals
Most of the chemicals were provided by Sigma: dexamethasone, budesonide, triamcilone, hydrocortisone (10−2m stock solution in methanol), RU486 (10−2m stock solution in ethanol), cycloheximide (10−1m in ethanol), spironolactone (10−1m in chloroform), adenosine 3′, 5′-cyclic monophosphorothioate, Rp-isomer, triethylammonium salt (Rp-cAMP, 5 × 10−3m stock in water), chelerythrine chloride (10−2m stock in water), thapsigargin (10−2m stock in DMSO). BAPTA-AM (10−2m stock in DMSO) and MDL-12, 330A hydrochloride (MDL; 5 × 10−3m stock in water) were provided by Calbiochem. Fura-2/AM (5 × 10−3m stock in DMSO) and 4-bromo-A23187 (10−3m stock solution in ethanol) were provided by Molecular Probes.
Data analysis
The mean [Ca2+]i and ISC variations given in the results section correspond to the [Ca2+]i or ISC variations between the mean [Ca2+]i or ISC measured during the 2 min prior to treatment with dexamethasone and the [Ca2+]i or ISC measured between 10 and 15 min after hormone treatment. In each experiment, the mean [Ca2+]i was obtained from all cells in the microscope field, irrespective of their ability to respond. Data are shown as the means ± s.e.m. of n experiments. Measures of statistical significance were obtained using either Student's t tests for paired data, or one-way analysis of variance (ANOVA) for multiple testing. A P value of < 0.05 was deemed to be significant. All statistical operations were performed using Excel software (Microsoft, USA).
Results
Rapid effect of dexamethasone on [Ca2+]i
Exposure of human bronchial epithelial cell monolayers to dexamethasone produced a rapid and significant decrease in [Ca2+]i. [Ca2+]i levels decreased within 30 s of exposure to dexamethasone (1 nm), and reached a new lower plateau value within 5 min (Fig. 1). Following 10 min exposure of the 16HBE14o- cell monolayers to dexamethasone (1 nm), [Ca2+]i was reduced by 32 ± 11 nm (n = 7, P < 0.0001) from a basal value of 213 ± 36 nm (n = 7; Fig. 1A). In primary cultures of human bronchial epithelial cells, dexamethasone (1 nm) also produced a [Ca2+]i decrease of 30 ± 12 nm (n = 3, P < 0.001; Fig. 1B). The lower [Ca2+]i was sustained for at least 90 min post-dexamethasone exposure. As illustrated by the effect of three different concentrations of dexamethasone (10−9, 10−7 and 10−6m), the magnitude of the [Ca2+]i decrease in 16HBE14o- cells, was dependent on the hormone concentration (Fig. 2). The effects of other glucocorticoids (budesonide, triamcilone acetonide and hydrocortisone) were also tested at different concentrations. These three glucocorticoids had a reduced potency in the stimulation of the rapid [Ca2+]i decrease compared to dexamethasone. Budesonide did not produce any significant [Ca2+]i change and triamcilone acetonide stimulated a [Ca2+]i decrease only at the highest concentration tested (1 μm). As shown in Fig. 2, and as reported previously (Urbach & Harvey, 2001), aldosterone also produced a [Ca2+]i decrease. However, after the [Ca2+]i decrease induced by dexamethasone, aldosterone did not produce any further change in [Ca2+]i (Fig. 1C).
Figure 1. Dexamethasone effect on [Ca2+]i.
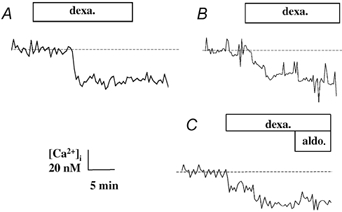
A, typical [Ca2+]i change upon apical exposure of monolayers of 16HBE14o- cells to dexamethasone (1 nm). B, typical [Ca2+]i change upon exposure of primary culture of human bronchial epithelial cells to dexamethasone (dexa.; 1 nm). C, typical [Ca2+]i response to apical exposure of monolayers of 16HBE14o- cells to dexamethasone (1 nm) followed by aldosterone (aldo; 1 nm).
Figure 2. Effect of different concentrations of a variety of glucocorticoids on [Ca2+]i.
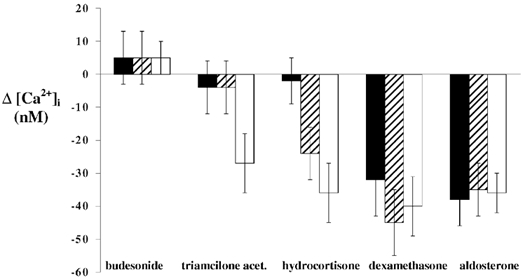
Mean [Ca2+]i variations in response to three different concentrations (10−6m, □; 10−7m, ; and 10−9m, ▪) of budesonide, triamcilone acetonide, hydrocortisone, dexamethasone and aldosterone in 16HBE14o- cell monolayers.
; and 10−9m, ▪) of budesonide, triamcilone acetonide, hydrocortisone, dexamethasone and aldosterone in 16HBE14o- cell monolayers.
Effect of RU486, spironolactone and cycloheximide
Since both dexamethasone and aldosterone stimulated a similar rapid [Ca2+]i response in human bronchial epithelium, we also tested the eventual involvement of glucocorticoid and mineralocorticoid receptors in the rapid response to dexamethasone on intracellular [Ca2+]i, using two antagonists of these receptors, RU486 and spironolactone, respectively (Fig. 3). The glucocorticoid receptor antagonist RU486 (1 μm) did not affect either the basal intracellular [Ca2+]i or the [Ca2+]i decrease induced by dexamethasone in 16HBE14o- cell monolayers (Fig. 3A). In the presence of RU486, dexamethasone (1 nm) still produced a [Ca2+]i decrease by 29 ± 8 nm (n = 6, P < 0.0001). As shown in Fig. 3B, spironolactone (10 μm) alone produced a transient [Ca2+]i increase by 22 ± 13 nm (n = 6, P < 0.0001) but did not affect the [Ca2+]i decrease induced by dexamethasone stimulation (Δ[Ca2+]i = -25 ± 13 nm, n = 6, P < 0.0001).
Figure 3. Effect of mineralocorticoid and glucocorticoid receptor antagonists and a protein synthesis inhibitor on the dexamethasone-induced decrease in [Ca2+]i.
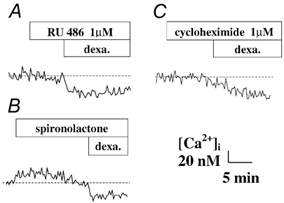
A and B, effect of the glucocorticoid receptor antagonist RU486 (A) and the mineralocorticoid receptor antagonist spironolactone (B) on the [Ca2+]i decrease induced by dexamethasone (1 nm) in 16HBE14o- cell monolayers. C, effect of cycloheximide, an inhibitor of protein synthesis, on the [Ca2+]i decrease induced by dexamethasone (1 nm) in 16HBE14o- cell monolayers.
The rapidity of the [Ca2+]i decrease induced by dexamethasone and the insensitivity of the response to inhibitors of the classical gluco- or mineralocorticoid receptors suggests strongly that the response does not involve a transcriptional pathway. In order to verify this hypothesis, we tested the effect of cycloheximide, a protein synthesis inhibitor. As shown in Fig. 3C, cycloheximide (1 μm) did not affect the basal [Ca2+]i or the [Ca2+]i response to dexamethasone (1 nm) (Δ[Ca2+]i = -27 ± 11 nm, n = 6, P < 0.0001).
Intracellular signalling pathway involved in the rapid response
In order to identify the intracellular signalling mechanism involved in the rapid Ca2+ response to dexamethasone we tested the effect of several inhibitors of signal transduction. MDL (500 μm), an inhibitor of adenylate cyclase, did not affect the basal [Ca2+]i but completely abolished the [Ca2+]i response usually observed with dexamethasone (1 nm; Δ[Ca2+]i = 0 ± 2 nm, n = 6, P < 0.0001), suggesting a role for adenylate cyclase activity in the response to dexamethasone (Fig. 4A). In order to test the role of cAMP-dependant protein kinase (PKA), we used Rp-cAMP (200 μm) to inhibit this pathway. Rp-cAMP did not affect the basal [Ca2+]i, but completely abolished the response to dexamethasone (Δ[Ca2+]i = 0 ± 3 nm, n = 6, P < 0.0001; Fig. 4B). In addition, we used the permeant form of cAMP, 8-bromo cAMP (5 μm) to stimulate PKA activity. After the initial stimulation of the PKA activity using 8-bromo cAMP, dexamethasone did not produce a further Ca2+ response (Δ[Ca2+]i = 0 ± 2 nm, n = 3, P < 0.05). Since dexamethasone did not cause an additive Ca2+ response under these conditions, it is proposed that the dexamethasone response occurs via cAMP. Taken together, these two results indicate a role for PKA activity in the response to dexamethasone.
Figure 4. Effect of inhibitors of adenylate cyclase, protein kinase A and protein kinase C on the [Ca2+]i decrease induced by dexamethasone in 16HBE14o- cell monolayers.
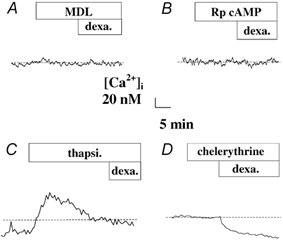
Effect of (A) 500 μm MDL 12, 230 hydrochloride, an inhibitor of adenylate cyclase, (B) 200 μm Rp-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt (Rp-cAMP), an inhibitor of protein kinase A, (C) 1 μm thapsigargin (thapsi), a Ca2+-ATPase pump inhibitor, and (D) 0.1 μm chelerythrine chloride, an inhibitor of protein kinase C, on the [Ca2+]i decrease induced by dexamethasone (1 nm) in 16HBE14o- cell monolayers.
Protein kinase C (PKC) does not appear to be involved in the signal transduction of the dexamethasone effect. The PKC inhibitor chelerythrine chloride (0.1 μm) did not affect the dexamethasone response (Δ[Ca2+]i = -23 ± 9 nm, n = 6, P < 0.0001), which was not significantly different from the dexamethasone-induced [Ca2+]i decrease observed under control conditions (P > 0.1).
These latter results suggest that the rapid [Ca2+]i response to dexamethasone may involve the same intracellular signalling pathway as for the [Ca2+]i decrease induced by aldosterone (1 nm) in airway epithelial cells (Urbach & Harvey, 2001). Given these similarities, we tested the effects of thapsigargin, an intracellular Ca2+-ATPase inhibitor, on the rapid response to dexamethasone. Thapsigargin (1 μm, in normal Ringer) produced a [Ca2+]i increase of 95 ± 20 nm (n = 6) followed by a slow return to a plateau value that was higher than basal levels. After thapsigargin treatment, dexamethasone (1 nm) did not produce any subsequent change in [Ca2+]i, suggesting that the rapid effect of dexamethasone to lower cytosolic Ca2+ involves the stimulation of thapsigargin-sensitive Ca2+ pumps (Fig. 4C). This conclusion is strengthened by the finding that pretreatment (5 min) of the cells with dexamethasone (1 nm) enhanced the effect of thapsigargin to increase [Ca2+]. Thapsigargin produced a [Ca2+]i increase of 132 ± 20 nm (n = 6) after dexamethasone exposure, which is significantly higher compared to the thapsigargin response obtained without dexamethasone (97 ± 21 nm, n = 6, P < 0.05). This result is compatible with an increased Ca2+ store following dexamethasone and is not compatible with an interpretation that dexamethasone reduces leakage of Ca2+ from stores.
We further verified that the latter results could not be explained by the inhibitory effect of dexamethasone on leakage of Ca2+ into the cell by testing the effect of dexamethasone after treatment by the Ca2+ ionophore 4-bromo-A23187 (5 μm). The 16HBE14o- monolayers treated with 4-bromo-A23187 still exhibited a Ca2+ decrease by 20 ± 11 nm (n = 3). Taken together, these results suggest that dexamethasone activates Ca2+ uptake into stores and does not affect the leakage pathways.
Apical ATP effect on [Ca2+]i
Exposure of 16HBE14o- monolayers to apical ATP produced an immediate increase in [Ca2+]i followed by a rapid decline towards a lower plateau value (Fig. 5). Upon apical addition of ATP (10−4m), [Ca2+]i increased by 377 ± 66 nm (n = 7) from a basal value of 112 ± 2 nm, with a return to a lower sustained value of 184 ± 8 nm (n = 7) within 5 min after ATP addition. As demonstrated in Fig. 5B, the [Ca2+]i response to apical ATP was dependent on the external ATP concentration, with a maximum response obtained at 10−4m ATP. Removal of external Ca2+ did not affect the [Ca2+]i response to apical ATP in 16HBE14o- monolayers. In the absence of external Ca2+, apical ATP (10−4m) produced a mean [Ca2+]i increase of 369 ± 29 nm (n = 6), which was not significantly different from the ATP-induced [Ca2+]i increase in the presence of external Ca2+. Furthermore, incubation of 16HBE14o- monolayers with thapsigargin (1 μm), produced a transient increase in [Ca2+]i of 89 ± 12 nm (n = 19), and completely abolished the [Ca2+]i response to subsequent apical ATP exposure. [Ca2+]i measured upon ATP (10−4m) exposure 20 min post-thapsigargin treatment was 123 ± 25 nm, which was not significantly different from basal levels of [Ca2+]i (n = 19, P < 0.001). Together, these results demonstrate that apical ATP stimulates a [Ca2+]i increase in 16HBE14o- monolayers due mainly to Ca2+ release from thapsigargin-sensitive intracellular stores.
Figure 5. External ATP effect on [Ca2+]i.
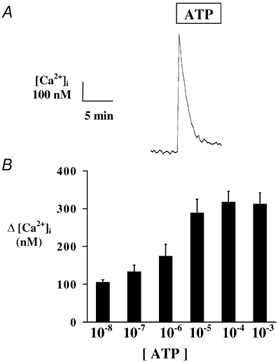
A, typical changes in [Ca2+]i upon apical exposure of 16HBE14o- monolayers to ATP (10−4m). B, concentration dependency of the [Ca2+]i response to apical ATP.
Dexamethasone effect on the ATP-stimulated [Ca2+]i response
In addition to the decrease of basal [Ca2+]i, pre-exposure of 16HBE14o- cell monolayers to dexamethasone (1 nm) for < 15 min caused a reduction in the [Ca2+]i response to apical ATP. As illustrated in Fig. 6, dexamethasone (1 nm) pretreatment largely reduced the amplitude of the ATP-induced [Ca2+]i increase. Following dexamethasone treatment, apical ATP (0.1 mm) produced a mean increase in [Ca2+]i of 214 ± 89 nm; (n = 7) from a basal value of 100 ± 13 nm. This response was significantly reduced compared to the ATP response produced in non-glucocorticoid-treated monolayers (Δ[Ca2+]i = 377 ± 66 nm; n = 7; P < 0.02). The duration of the dexamethasone exposure prior to ATP addition was between 5 and 15 min and the duration of the delay did not affect the amplitude of the inhibitory effect.
Figure 6. Dexamethasone effect on the [Ca2+]i response to ATP.
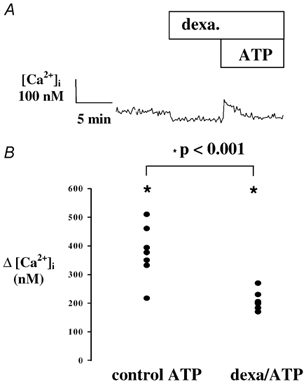
A, typical [Ca2+]i response to ATP (10−4m) following pretreatment with dexamethasone (10 nm). B, comparison of the [Ca2+]i variation produced by ATP (10−4m) under control conditions (control ATP 10−4m), and following pre-incubation with dexamethasone (10 nm, 5-15 min; dexa/ATP).
Stimulation of Cl− secretion by apical ATP
The transepithelial ISC measured across 16HBE14o- monolayers bathed in Krebs-Heinseleit solution on both sides of the epithelium, was 2.9 ± 1.6 μA cm−2 (n = 38). As shown in Fig. 7, apical application of ATP (10−4m) evoked a rapid and transient increase in ISC of 12.6 ± 3.8 μA cm−2 (n = 34). The ISC increase started without measurable latency, the peak was reached within 2 min of nucleotide application and was then followed by a rapid decline to baseline values in the continuous presence of external ATP (Fig. 7A). Since the ISC may be generated by either Na+ absorption or Cl− secretion (or both), we tested the effect of amiloride as an epithelial Na+ channel inhibitor. Amiloride did not significantly alter the basal or the ATP-stimulated ISC responses. In the presence of amiloride (10−4m), basal ISC was 3.23 ± 0.18 μA cm−2 (n = 12), and was not significantly different from the ISC value obtained in the absence of amiloride. During amiloride (10−4m) treatment, apical ATP (10−4m) exposure to 16HBE14o- cells, produced a ISC increase of 12.3 ± 3.1 μA cm−2 (n = 12), which was not significantly different from the ISC increase obtained in the absence of amiloride (P > 0.32).
Figure 7. ATP induced Cl− secretion.
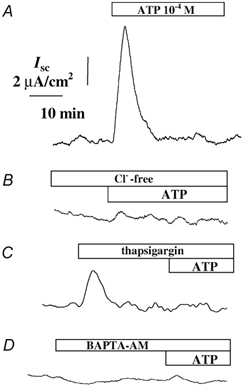
A, typical record of short-circuit current (ISC) during apical ATP (10−4m) exposure. B, inhibition of the ISC response to ATP (10−4m) in Cl−-free medium (Cl−-free). Inhibition of the ISC response to ATP by pretreatment with either thapsigargin (1 μm) (C) or BATA-AM (50 μm; D).
We further demonstrated that the ISC was primarily generated by Cl− secretion. Direct replacement of basolateral Cl− by gluconate completely abolished the ATP-induced ISC increase (ΔISC = 0.2 ± 0.1 μA cm−2n = 7, P < 0.05; Fig. 7B).
Role of [Ca2+]i in the ATP-induced Cl− secretion
Our previous results demonstrated that thapsigargin pretreatment completely abolished the ATP-induced [Ca2+]i increase. The effect of thapsigargin on ISC was recorded to determine the role of [Ca2+]i in apical ATP-induced Cl− secretion. Thapsigargin (1 μm) produced a transient increase in ISC of 3.09 ± 1.6 μA cm−2 (n = 6), and completely abolished the ISC response to subsequent apical ATP (10−4m) exposure (Fig. 7C). This result indicates an intracellular Ca2+ dependency of the apical ATP-induced Cl− secretion.
We further investigated the role of [Ca2+]i in apical ATP-induced Cl− secretion in 16HBE14o- monolayers, using the intracellular Ca2+ chelator BAPTA-AM. Following pretreatment of cells with BAPTA-AM (50 μm, 20 min), the ISC response to ATP (10−4m) was completely abolished (ΔISC = 0.5 ± 0.3 μA cm−2, P < 0.05, n = 4; Fig. 7D).
Rapid effect of dexamethasone on ATP-induced Cl− secretion
Since intracellular Ca2+ ions are a major signal transducer of secretion in epithelia, we tested the hypothesis that the reduction in [Ca2+]i induced by dexamethasone may antagonise the secretory event. Dexamethasone (1 nm, 5-15 min), applied to the apical side of 16HBE14o- monolayers did not significantly alter basal ISC. However, as illustrated Fig. 8, dexamethasone (1 nm), significantly inhibited apical ATP-induced Cl− secretion in 16HBE14o- cell monolayers. Pre-incubation with dexamethasone (10−9m, 10 min), reduced the mean ATP-induced ISC response to 0.86 ± 1.5 μA cm−2 (n = 7), significantly smaller than the increase obtained with ATP (10−4m) alone (ΔISC = 15.2 ± 5.6 μA cm−2, n = 7).
Figure 8. Dexamethasone effect on the Cl− secretory response to ATP.
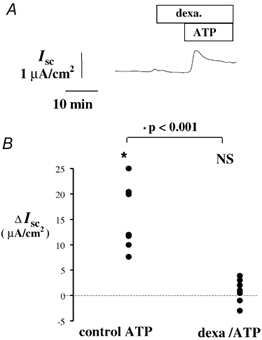
A, typical Cl− secretory response to ATP (10−4m) following pretreatment with dexamethasone (10 nm). B, comparison of the ISC variation produced by ATP (10−4m) under control conditions (control ATP 10−4m), and following pre-incubation with dexamethasone (10 nm, 5-15 min; dexa/ATP). NS = not significant.
Discussion
Taken together, our results provide evidence for a rapid non-genomic antisecretory effect of dexamethasone at low concentration (1 nm) involving an inhibition of intracellular Ca2+ signalling in the human airway epithelium. In a recent study, we provided evidence for a rapid decrease of [Ca2+]i induced by aldosterone (1 nm) in 16HBE14o- monolayers. This was the first report of a non-genomic effect of steroid hormones in human bronchial epithelium (Urbach & Harvey, 2001). Similarly to aldosterone, we show in the present paper that low concentrations (1 nm) of the glucocorticoid hormone dexamethasone, rapidly decreases basal [Ca2+]i in the immortalised 16HBE14o- cell line and in primary cultures of human bronchial epithelial cell monolayers An effect of steroid hormones (including glucocorticoids) on [Ca2+]i has previously been reported in different tissues. In some tissues, such as cortical collecting duct (Harvey & Higgins, 2000), proximal tubule (Han et al. 1999) and vascular smooth muscle (Steiner et al. 1988), glucocorticoids produce a rapid [Ca2+]i increase. In contrast, glucocorticoid treatment causes a decrease in Ca2+ influx via a non-genomic mechanism in myocytes (Passaquin et al. 1998), thymocytes (Buttgereit et al. 1997), human lymphoblast (Gardner & Zhang, 1999), human leukocytes and airway smooth muscle (Chhabra et al. 1999). We also reported previously that the glucocorticoids triamcilone acetonide and hydrocortisone, at high concentrations (10−6m) produce a rapid of [Ca2+]i in 16HBE14o- cells (Urbach & Harvey, 2001).
The fast (30 s) onset and the insensitivity of the dexamethasone-induced Ca2+ response to cycloheximide reported in the present study indicate a non-genomic mechanism. The nature of the eventual receptor(s) involved in the non-genomic effects of steroid hormone is still not clear. It has been suggested that at least some of the non-genomic effects of steroids are mediated by binding to a membrane receptor. Several membrane receptors or membrane binding sites for glucocorticoids have been described in various tissues. A receptor to cortisol has been characterised in neuronal membranes of the amphibian Tarisha granulosa (Moore et al. 1995). However, aldosterone and dexamethasone do not display a high affinity for this membrane receptor. Other putative membrane binding sites for glucocorticoids have been detected in mouse and rat liver, rat kidney, rat brain and calf adrenal cortex (Ibarrola et al. 1991; Trueba et al. 1991; Guo et al. 1995; Andres et al. 1997).
In our study, the similarity between the [Ca2+]i decrease induced by aldosterone in 16HBE14o- cells and the response to dexamethasone in the same cell preparation, suggest that dexamethasone acts as a mineralocorticoid-like agonist. In addition, the fact that subsequent addition of aldosterone did not stimulate a further response after the initial rapid response to dexamethasone suggests strongly that both hormones share a common receptor to produce the rapid response. However the insensitivity of the dexamethasone-induced [Ca2+]i decrease to spironolatone or RU486, indicates that the non-genomic effect of dexamethasone is not mediated via the classical mineralocorticoid or glucocorticoid receptor. In addition, we investigated the non-genomic effect of other glucocorticoids having a higher relative binding affinity than dexamethasone for the glucocorticoid receptor (Brattsand & Axelsson, 1997). Triamcylone acetonide only produced a detectable [Ca2+]i decrease at high pharmacological concentrations (10−6m) and budesonide did not produce any change in [Ca2+]i. These results support our conclusion of a non-genomic mechanism that is not mediated by the classical glucocorticoid receptor.
The difference in ability for the different glucocorticoids to produce the non-genomic response may be explained by their relative lipophilicity. The lipophilicity of dexamethasone is lower than that of triamcylone acetonide and budesonide, which may suggest that the non-genomic effect is the result of a direct interaction within the membrane. In addition, the rapidity of the onset of the [Ca2+]i decrease according to Buttgereit's classification, also suggests a non-specific binding of the hormone at the membrane (Buttgereit et al. 1999). It has been reported that the non-specific and non-genomic effects of glucocorticoids occur at high concentrations, > 10−4m (Buttgereit et al. 1999); however, we observed the rapid Ca2+ response to occur at low concentrations (10−9m) of dexamethasone.
The inhibition of the rapid response to dexamethasone by antagonists of adenylate cyclase and PKA suggests that the dexamethasone response occurs through PKA stimulation. The inhibition of the response to dexamethasone by an antagonist of Ca2+-ATPase and the stimulation by dexamethasone of the Ca2+ release following thapsigargin also suggests that dexamethasone acts to lower cytosolic Ca2+ via stimulation of the Ca2+-ATPase of intracellular stores. In addition, dexamethasone still produced an increase in [Ca2+]i after treatment with a Ca2+ ionophore, indicating that dexamethasone is not affecting the membrane Ca2+ leak pathways. There is evidence that Ca2+ ionophores also perforate intracellular organelles (Tarran et al. 2002). However, under our experimental conditions, such a possibility is incompatible with the dexamethasone effect of lowering cytosolic Ca2+ in the presence of the ionophore A23187. Other studies have also shown that very high concentrations of steroids decrease [Ca2+]i via stimulation of Ca2+-ATPase activity by affecting membrane fluidity (Massa et al. 1975; Whiting et al. 2000). The similarity of the aldosterone and glucocorticoid responses and the absence of an additive effect of the two hormones is most probably due to the sharing of an intracellular Ca2+ signalling pathway.
Extracellular nucleotides exert significant biological actions on different cell types in the upper airways and in the lung, and there are several reports of ATP release from epithelial tissues by physiological stimuli that are associated with the secretory event (Grygorzyk & Hanrahan, 1997; Sorensen & Novak, 2001). At least some of these diverse biological effects are mediated via cell-surface purino receptors that activate inositol 1,4,5-trisphosphate hydrolysis and Ca2+ mobilisation. Many studies substantiate the concept that ATP is a potent agonist with multiple receptor subtypes expressed ubiquitously on the cell surface, which are linked to diverse intracellular signal transduction pathways (Stutts et al. 1994). Evidence supporting the existence of a P2Y2 receptor in airway epithelia has emerged from functional studies of 16HBE14o- cells (Koslowsky et al. 1994), human tracheal epithelial cells (Yamaya et al. 1996), nasal epithelial cells (Mason et al. 1991), alveolar type II cells (Rice et al. 1995), CF/T43 cells (Brown et al. 1991), A549 cells (Clunes & Kemp, 1996) and rat tracheal epithelial cells (Hwang et al. 1996). Here we show that external ATP causes a transient increase in Ca2+ and ISC when added to the apical surface of 16HBE14o- cell monolayers. We showed the transient ISC increase induced by apical ATP was completely abolished in the absence of basolateral Cl−. We have also demonstrated that the source of the Ca2+ rise induced by apical ATP occurs from the mobilisation of Ca2+ sequestered within thapsigargin-sensitive intracellular stores. Finally, the Ca2+ dependency of the Cl− secretion induced by apical ATP was further verified by the inhibitory effect of thapsigargin and BAPTA-AM. These results support the conclusion that the apical ATP-induced Cl− secretory response in 16HBE14o- cells is mediated by cell-surface purinoceptors that respond by elevating [Ca2+]i and activating Ca2+-dependent Cl− channels.
In this study, we tested the hypothesis that the dexamethasone effect on [Ca2+]i might generate a rapid antisecretory effect. Dexamethasone applied to the apical side of the 16HBE14o- monolayer did not produce any significant effect on basal ISC. However, after exposure to dexamethasone, subsequent exposure to ATP resulted in a reduced Cl− secretory effect. In other tissues, dexamethasone has been reported to modulate Ca2+-dependent secretory mechanisms. Dexamethasone inhibited the nicotine-induced secretion of catecholamines in porcine adrenal medulla (Wagner et al. 1999) and the corticotropin-releasing-factor-induced ACTH secretion in rat (Hinz & Hirschelmann, 2000). Steroid hormones have also been described to exert an antisecretory effect on cAMP-dependent Cl− secretion. Inhibition of the cAMP-dependent Cl− secretion by 17β-oestradiol has also been reported to occur in T84 cells (Singh et al. 2000) and in rat distal colonic epithelium (Condliffe et al. 2001). In nasal epithelial cells, it has been reported that 4 days of treatment (for 30 min every 12 h) with the glucocorticoid fluticasone propionate stimulates an amiloride-sensitive ISC current and partially decreases cAMP-dependent Cl− secretion (Jepsen et al. 2000). Our study is the first report of a rapid response to glucocorticoid on Ca2+-dependent Cl− secretion.
Glucocorticoids are potent anti-inflammatory and immunosuppressive compounds that are used via systemic or inhaled delivery for the treatment of numerous diseases. The steroids are usually used at low doses for basal immunosuppressive treatment and their effects are not observed until after a few hours or days of treatment. It has been observed that a high-dose and short-term intravenous glucocorticoid therapy improves symptoms in acute exacerbation of multiple sclerosis or acute spinal cord injury (Bracken et al. 1990). It has been proposed that in some acute clinical situations the additional benefit of the use of high doses of glucocorticoids is due to their non-genomic effects (Buttgereit et al. 1997). The rapid non-genomic effect of glucocorticoids in human bronchial epithelia, as reported in this study, may have important clinical implications in the treatment of airway infections such as rhinitis, asthma and cystic fibrosis. In particular, the antisecretory effect of glucocorticoids used as anti-inflammatory compounds in the treatment of cystic fibrosis, may exacerbate the deficit in Cl− secretion associated with the disease.
Acknowledgments
This work was funded by The Wellcome Trust (055695/ Z/ 98/Z/CH/TG/JF), the French Institut National de la Santé et de la Recherche Medicale (INSERM) and the Regional Council of Languedoc Roussillon, France. The 16HBE14o- cell line was obtained as a gift from D. C. Gruenert (Gene Therapy Core Center, University of California, San Francisco, CA 94143, USA).
References
- Andres M, Marino A, Macarulla JM, Trueba M. Characterization of specific corticosterone binding sites in adrenal cortex plasma membrane and their localization by autoradiographic studies. Cellular and Molecular Life Science. 1997;53:673–680. doi: 10.1007/s000180050087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet J. Global Initiative for Asthma (GINA) and its objectives. Clinical and Experimental Allergy. 2000;30:2–5. doi: 10.1046/j.1365-2222.2000.00088.x. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard JM, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon J, Marshall LF, Perot PL, Piepmeier J, Sonntag VKH, Vagner FC, Wilberger JE, Winn HR. A randomized, controlled trial of methylprednisolone ornaloxane in the treatment of acute spinal cord injury. New England Journal of Medicine. 1990;322:1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- Brattsand R, Axelsson BI. Basis of airway selectivity of inhaled glucocorticoids. In: Schleimer RP, Busse WW, O'Byrne PM, editors. Inhaled Glucocorticoids in Asthma. New York: Dekker; 1997. pp. 351–380. [Google Scholar]
- Brown HA, Lazarowski ER, Boucher RC, Harden TK. Evidence that UTP and ATP regulate phospholipase C through a common extracellular 5′-nucleotide receptor in human airway epithelial cells. Molecular Pharmacology. 1991;40:648–655. [PubMed] [Google Scholar]
- Buttgereit F, Brand MD, Burmester GR. Equivalent doses and relative drug potencies for non-genomic glucocorticoid effects: a novel glucocorticoid hierarchy. Biochemical Pharmacology. 1999;58:363–368. doi: 10.1016/s0006-2952(99)00090-8. [DOI] [PubMed] [Google Scholar]
- Buttgereit F, Krauss S, Brand MD. Methylprednisolone inhibits uptake of Ca2+ and Na+ ions into concanavalin A-stimulated thymocytes. Biochemical Journal. 1997;1326:329–332. doi: 10.1042/bj3260329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra SK, Khanduja A, Jain D. Decreased sodium-potassium and calcium adenosine triphosphatase activity in asthma: modulation by inhaled and oral corticosteroids. Indian Journal of Chest Disease and Allied Sciences. 1999;41:15–26. [PubMed] [Google Scholar]
- Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC. Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science. 1992;257:1125–1128. doi: 10.1126/science.257.5073.1125. [DOI] [PubMed] [Google Scholar]
- Clunes MT, Kemp PJ. P2u purinoceptor modulation of intracellular Ca2+ in a human lung adenocarcinoma cell line: down-regulation of Ca2+ influx by protein kinase C. Cell Calcium. 1996;20:339–346. doi: 10.1016/s0143-4160(96)90039-1. [DOI] [PubMed] [Google Scholar]
- Condliffe SB, Doolan CM, Harvey BJ. 17beta-oestradiol acutely regulates Cl− secretion in rat distal colonic epithelium. Journal of Physiology. 2001;530:47–54. doi: 10.1111/j.1469-7793.2001.0047m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. American Journal of Respiratory Cellular and Molecular Biology. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- Filipovic SR, Drlovic J, Stojsavljevic N, Levic Z. The effects of high-dose intravenous methylprednisolone on event-related potentials in patients with multiple sclerosis. Journal of Neurosciences. 1997;152:147–153. doi: 10.1016/s0022-510x(97)00159-7. [DOI] [PubMed] [Google Scholar]
- Gardner JP, Zhang L. Glucocorticoid modulation of Ca2+ homeostasis in human B lymphoblasts. Journal of Physiology. 1999;514:385–396. doi: 10.1111/j.1469-7793.1999.385ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grygorczyk R, Hanrahan JW. CFTR-independent ATP release from epithelial cells triggered by mechanical stimuli. American Journal of Physiology. 1997;272:C1058–1066. doi: 10.1152/ajpcell.1997.272.3.C1058. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guo Z, Chen YZ, Xu RB, Fu H. Binding characteristics of glucocorticoid receptor in synaptic plasma membrane from rat brain. Functional Neurology. 1995;10:183–194. [PubMed] [Google Scholar]
- Han HJ, Kim DH, Park SH, Lee YS, Lee JH, Yang SI. Regulatory mechanism of polarized membrane transport by glucocorticoid in renal proximal tubule cells: involvement of [Ca2+]i. Journal of Veterinary and Medical Science. 1999;61:1197–1202. doi: 10.1292/jvms.61.1197. [DOI] [PubMed] [Google Scholar]
- Harvey BJ, Higgins M. Non-genomic effects of aldosterone on Ca2+ in M-1 cortical collecting duct cells. Kidney International. 2000;57:1395–1403. doi: 10.1046/j.1523-1755.2000.00981.x. [DOI] [PubMed] [Google Scholar]
- Hinz B, Hirschelmann R. Dexamethasone megadoses stabilize rat liver lysosomal membranes by non-genomic and genomic effects. Pharmacological Results. 2000;12:1489–1493. doi: 10.1023/a:1007652908104. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Morita S, Iwashita H, Sagiya Y, Nagi T, Nakanishi A, Ashida Y, Nishimura O, Fujisawa Y, Fujino M. Increased expression of the human Ca2+-activated Cl− channel 1 (CaCC1) gene in the asthmatic airway. American Journal of Respiratory and Critical Care Medicine. 2002;8:1132–1136. doi: 10.1164/ajrccm.165.8.2107068. [DOI] [PubMed] [Google Scholar]
- Hwang TH, Schwiebert EM, Guggino WB. Apical and basolateral ATP stimulates tracheal epithelial chloride secretion via multiple purinergic receptors. American Journal of Physiology. 1996;270:C1611–1623. doi: 10.1152/ajpcell.1996.270.6.C1611. [DOI] [PubMed] [Google Scholar]
- Ibarrola I, Ogiza K, Marino A, Macarulla JM, Trueba M. Steroid hormone specifically binds to rat kidney plasma membrane. Journal of Bioenergy and Biomembrane. 1991;23:919–926. doi: 10.1007/BF00786009. [DOI] [PubMed] [Google Scholar]
- Jepsen M, Graham S, Karp PH, Zabner J. Effect of topical nasal pharmaceuticals on sodium and chloride transport by human airway epithelia. American Journal of Rhinology. 2000;14:405–409. doi: 10.2500/105065800779954365. [DOI] [PubMed] [Google Scholar]
- Koslowsky T, Hug T, Ecke D, Klein P, Gruenert DC, Kunzelmann K. Ca(2+)- and swelling-induced activation of ion conductances in bronchial epithelial cells. Pflügers Archiv. 1994;428:597–603. doi: 10.1007/BF00374583. [DOI] [PubMed] [Google Scholar]
- Koukouritaki SB, Theodoropoulos PA, Margioris AN, Gravanis A, Stournaras C. Dexamethasone alters rapidly actin polymerization dynamics in human endometrial cells: evidence for nongenomic actions involving cAMP turnover. Journal of Cell Biology. 1996;62:251–261. doi: 10.1002/(SICI)1097-4644(199608)62:2%3C251::AID-JCB13%3E3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Lewis GD, Campbell WB, Johnson AR. Inhibition of prostaglandin synthesis by glucocorticoids in human endothelial cells. Endocrinology. 1986;119:62–69. doi: 10.1210/endo-119-1-62. [DOI] [PubMed] [Google Scholar]
- Mall M, Wissner A, Gonska T, Calenborn D, Kuehr J, Brandis M, Kunzelmann K. Inhibition of amiloride-sensitive epithelial Na(+) absorption by extracellular nucleotides in human normal and cystic fibrosis airways. American Journal of Respiratory Cellular and Molecular Biology. 2000;23:755–761. doi: 10.1165/ajrcmb.23.6.4207. [DOI] [PubMed] [Google Scholar]
- Mason SJ, Paradiso AM, Boucher RC. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. British Journal of Pharmacology. 1991;103:1649–1656. doi: 10.1111/j.1476-5381.1991.tb09842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa EM, Morero RD, Bloj B, Farias RN. Hormone action and membrane fluidity: effect of insulin and cortisol on the Hill coefficients of rat erythrocyte membrane-bound acetylcholinesterase and (Na++ K+)-ATPase. Biochemical and Biophysical Research Communications. 1975;66:115–122. doi: 10.1016/s0006-291x(75)80302-0. [DOI] [PubMed] [Google Scholar]
- Moore FL, Orchinik M, Lowry C. Functional studies of corticosterone receptors in neuronal membranes. Receptor. 1995;5:21–28. [PubMed] [Google Scholar]
- Muto S, Ebata S, Okada K, Saito T, Asano Y. Glucocorticoid modulates Na+/H+ exchange activity in vascular smooth muscle cells by nongenomic and genomic mechanisms. Kidney International. 2000;57:2319–2333. doi: 10.1046/j.1523-1755.2000.00092.x. [DOI] [PubMed] [Google Scholar]
- Passaquin AC, Lhote P, Ruegg UT. Calcium influx inhibition by steroids and analogs in C2C12 skeletal muscle cells. British Journal of Pharmacology. 1998;124:1751–1759. doi: 10.1038/sj.bjp.0702036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR, Burton FM, Fiedeldey DT. Cloning and expression of the alveolar type II cell P2u-purinergic receptor. American Journal of Respiratory Cellular and Molecular Biology. 1995;12:27–32. doi: 10.1165/ajrcmb.12.1.7811468. [DOI] [PubMed] [Google Scholar]
- Singh AK, Schultz BD, Katzenellenbogen JA, Price EM, Bridges RJ, Bradbury NA. Estrogen inhibition of cystic fibrosis transmembrane conductance regulator-mediated chloride secretion. Journal of Pharmacological Experimental Therapy. 2000;295:195–204. [PubMed] [Google Scholar]
- Sorensen CE, Novak I. Visualisation of ATP release in pancreatic acini in response to cholinergic stimuli. Use of fluorescent probes and confocal microscopy. Journal of Biochemical Chemistry. 2001;276:3295–3332. doi: 10.1074/jbc.M103313200. [DOI] [PubMed] [Google Scholar]
- Steiner A, Vogt E, Locher R, Vetter W. Stimulation of the phosphoinositide signalling system as a possible mechanism for glucocorticoid action in blood pressure control. Journal of Hypertension. 1988;6:S366–368. doi: 10.1097/00004872-198812040-00114. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Chinet TC, Mason SJ, Fullton JM, Clarke LL, Boucher RC. Regulation of Cl− channels in normal and cystic fibrosis airway epithelial cells by extracellular ATP. Proceedings of the National Academy of Sciences of the USA. 1992;89:1621–1625. doi: 10.1073/pnas.89.5.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts MJ, Fitz JG, Paradiso AM, Boucher RC. Multiple modes of regulation of airway epithelial chloride secretion by extracellular ATP. American Journal of Physiology. 1994;267:C1442–1451. doi: 10.1152/ajpcell.1994.267.5.C1442. [DOI] [PubMed] [Google Scholar]
- Tarran R, Loewen ME, Paradiso AM, Olsen JC, Gray MA, Argent BE, Boucher RC, Gabriel SE. Regulation of murine airway surface liquid volume by CFTR and Ca(2+)-activated Cl(-) conductances. Journal of General Physiology. 2002;120:407–418. doi: 10.1085/jgp.20028599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueba M, Ibarrola I, Ogiza K, Marino A, Macarulla JM. Specific binding sites for corticosterone in isolated cells and plasma membrane from rat liver. Journal of Membrane Biology. 1991;120:115–124. doi: 10.1007/BF01872394. [DOI] [PubMed] [Google Scholar]
- Urbach V, Harvey BJ. Rapid and non-genomic reduction of intracellular [Ca2+] induced by aldosterone in human bronchial epithelium. Journal of Physiology. 2001;537:267–275. doi: 10.1111/j.1469-7793.2001.0267k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PG, Jorgensen MS, Arden WA, Jackson BA. Stimulus-secretion coupling in porcine adrenal chromaffin cells: acute effects of glucocorticoids. Journal of Neurosciences. 1999;57:643–650. [PubMed] [Google Scholar]
- Walsh DE, Harvey BJ, Urbach V. CFTR regulation of intracellular calcium in normal and cystic fibrosis human airway epithelia. Journal of Membrane Biology. 2000;177:209–219. doi: 10.1007/s002320010004. [DOI] [PubMed] [Google Scholar]
- Whiting KP, Restall CJ, Brain PF. Steroid hormone-induced effects on membrane fluidity and their potential roles in non-genomic mechanisms. Life Sciences. 2000;67:743–757. doi: 10.1016/s0024-3205(00)00669-x. [DOI] [PubMed] [Google Scholar]
- Willumsen NJ, Boucher RC. Activation of an apical Cl− conductance by Ca2+ ionophores in cystic fibrosis airway epithelia. American Journal of Physiology. 1989;256:C226–233. doi: 10.1152/ajpcell.1989.256.2.C226. [DOI] [PubMed] [Google Scholar]
- Yamaya M, Sekizawa K, Kakuta Y, Ohrui T, Sawai T, Sasaki H. P2u-purinoceptor regulation of chloride secretion in cultured human tracheal submucosal glands. American Journal of Physiology. 1996;270:L979–984. doi: 10.1152/ajplung.1996.270.6.L979. [DOI] [PubMed] [Google Scholar]