

Abstract
Muscle wasting is a prominent feature of several systemic diseases, neurological damage and muscle disuse. The contribution of calpain proteases to muscle wasting in any instance of muscle injury or disease has remained unknown because of the inability to specifically perturb calpain activity in vivo. We have generated a transgenic mouse with muscle-specific overexpression of calpastatin, which is the endogenous inhibitor of calpains, and induced muscle atrophy by unloading hindlimb musculature for 10 days. Expression of the transgene resulted in increases in calpastatin concentration in muscle by 30- to 50-fold, and eliminated all calpain activity that was detectable on zymograms. Muscle fibres in ambulatory, transgenic mice were smaller in diameter, but more numerous, so that muscle mass did not differ between transgenic and non-transgenic mice. This is consistent with the role of the calpain-calpastatin system in muscle cell fusion that has been observed in vitro. Overexpression of calpastatin reduced muscle atrophy by 30 % during the 10 day unloading period. In addition, calpastatin overexpression completely prevented the shift in myofibrillar myosin content from slow to fast isoforms, which normally occurs in muscle unloading. These findings indicate that therapeutics directed toward regulating the calpain-calpastatin system may be beneficial in preventing muscle mass loss in muscle injury and disease.
Muscle wasting is a common and clinically important outcome of several diseases including cancer (Baracos, 2001), sepsis (Ruff & Secrist, 1984), and neuromuscular diseases such as Duchenne muscular dystrophy (Emery, 2002). Reduced muscle loading also causes muscle wasting in which as much as 40-60 % of muscle mass can be lost within a 2 week period (Thomason & Booth, 1990). Investigations of the contribution of lysosomal proteins, calcium-dependent proteases and the ubiquitin- proteasome system to muscle wasting that occurs in disease, injury or muscle disuse indicate that the relative contributions of different proteolytic systems may be specific to the condition leading to the atrophy (Furuno et al. 1990; Tischler et al. 1990). However, a clear assessment of the relative roles of these proteolytic systems has been limited by the inability to specifically perturb the activity of any of these systems in muscle in vivo.
Calpains are a family of calcium-activated proteases that are ubiquitously expressed and cleave a great variety of substrates (Croall & DeMartino, 1991). Muscle expresses at least six isoforms of calpain although only three isoforms, calpains 1, 2 and 3, have been detected in muscle by immunoblotting for the most conserved epitope (Spencer et al. 1995; Sorimachi & Suzuki, 2001). Levels of calpain expression can vary during tissue injury (Belcastro, 1993) or disease (Spencer et al. 1995; Voisin et al. 1996), which has been typically interpreted as reflecting changes in calpain-mediated proteolysis. Calpastatin is a specific, endogenous inhibitor of calpain 1 and calpain 2, and shows a similar, ubiquitous expression. Interactions between calpain and calpastatin may be promoted by calpain association with membrane phospholipids (Imajoh et al. 1986), so that localization of calpain in the cell may reflect its stage of activation or inactivation. Thus, several mechanisms are available for regulating calpain-mediated proteolysis, which include the regulation of the expression and relative concentrations of calpains and calpastatin, regulation of interaction between calpains and calpastatin, and control of cytosolic free calcium concentrations.
The results of in vitro studies have led to conflicting conclusions concerning the role of the calpain-calpastatin system in regulating muscle mass loss during atrophy that is caused by muscle unloading (Furuno & Goldberg, 1966; Rodemann et al. 1982; Zeman et al. 1985; Baracos et al. 1986). However, the extent to which the in vitro assessment of excised muscle mimics the proteolytic process in unloaded muscle in vivo is unknown. In addition, the relatively brief periods of study of muscle response to unloading for a few hours in vitro are not similar to the duration of chronic wasting that occurs in vivo with long-term disease or disuse.
In vivo assessments of the role of the calpain-calpastatin system in muscle atrophy have been equivocal, and the resulting conclusions have been inconsistent. Generally, three approaches to assessing calpain involvement in muscle wasting have been employed, but none truly addresses the contributions of calpain to muscle proteolysis in vivo. Many investigations have assessed changes in the concentration of calpain or its mRNA in pathological, injured, or experimentally perturbed muscle (e.g. Medina et al. 1995). However, the concentration of calpain does not necessarily reflect its in vivo activity because activity varies with cytosolic calcium concentrations, and is further regulated by interactions with calpastatin or phospholipids (Croall & DeMartino, 1991). An alternative approach for assessing calpain involvement in muscle disease or injury processes has been to partially purify calpain from muscle samples, and then assess proteolysis of exogenous substrates in the presence of high calcium concentrations (Belcastro, 1993; Arthur et al. 1999; Reid & Belcastro, 2000). Although this assay will reflect calpain concentration in the tissue, it does not reflect in vivo activity where calcium concentrations would be lower and endogenous inhibitors would be present. A final approach has employed measuring calpain autolytic fragments, because autolysis indicates that calpain activation has occurred (Spencer et al. 1995, 1997). Although this assay reflects actual calpain activation in vivo, it is limited in that calpain can be rapidly inactivated by binding calpastatin. Thus, measurement of calpain autolytic fragments provides an index of calpain that has undergone activation in vivo, but does not necessarily indicate the actual levels of active calpain within the cells. In addition, each of these approaches to assessing calpain's possible contribution to proteolysis uses in vivo samples at a single point in time, which may not be sufficient to assess calpain's role over a prolonged period of muscle wasting.
In the present investigation, we test the role of the calpain-calpastatin system in muscle atrophy by generating transgenic (Tg) mice in which there is a muscle-specific overexpression of calpastatin (CS). An important advantage of this Tg approach is that it provides the tissue-specific, calpain-specific inhibition of the protease for evaluation of perturbations of calpain activity in vivo. We use the CS Tg mice to address the question of whether calpain-mediated proteolysis contributes to the response of muscle to unloading in vivo. Reduction in muscle cell size and changes in myofibrillar myosin content are assessed as indices of muscle adaptation to unloading. Our findings show that expression of CS transgenes in skeletal muscle greatly reduces calpain activity in muscle, and produces an increase in fibre number and a reduction of fibre size in ambulatory control animals. Expression of the CS transgene also causes significant reduction in muscle fibre wasting during muscle unloading. Finally, we find that there is no detectable change in the proportion of muscle fibres expressing slow or fast type isoforms of myosin heavy chain (MHC) during unloading of CS-overexpressing muscles, which indicates a key role for calpain-mediated proteolysis in fibre type transformations.
Methods
Production of calpastatin transgenic mice
The human skeletal muscle actin (HSA) promoter (provided by Dr Jeffrey Chamberlain) was used to drive muscle-specific expression of the full length, human calpastatin cDNA (kindly donated by Dr Masatoshi Maki) (Fig. 1). This promoter was first characterized in a Tg mouse by Dr Hardeman's group (Brennan & Hardeman, 1993), who showed skeletal muscle-specific expression of the transgene, although cardiac muscle was also found to express the transgene in some Tg mouse lines. This promoter contains -2139 to +239 of the HSA promoter and has been modified from Dr Hardeman's construct by the addition of the SV40 VP1 intron (isolated by HindIII/Xba I) between the promoter and the cDNA (Crawford et al. 2000) to provide a splice acceptor. This construct has been used previously to generate Tg mice with muscle-specific expression (Crawford et al. 2000; Wehling et al. 2001; Spencer et al. 2002). Tg mice were generated at the University of California, Irvine Tg Mouse Facility by microinjection of purified plasmid into zygotes from C57Bl/6J × Balb c parents. F1 mice were crossed with C57/BL10. All comparisons were made with age-matched littermates that were not Tg. PCR of tail-chop DNA was used to identify Tg mice (Spencer et al. 1997) using upstream primers in the HSA promoter (5′ CCC GAG CCG AGA GTA GCA GT 3′) and downstream primers in the vp1 intron (5′ CCC TTC CCT GTT GGC TAC T 3′).
Figure 1. Diagram of calpastatin cDNA construct used to generate calpastatin TG mice.

The CS cDNA is a 2.3 kb region that begins at the start codon at the 5′ end of exon 2, and excludes the 5′ and 3′ untranslated region. The construct encodes the L-domain and the four-repeat inhibitory domains. The vp1 intron was isolated from pSVL and contains a splice aceceptor. The SV40 polyadenylation signal was isolated from pCMVβ.
Zymogram analysis of calpain activity
Zymograms were performed according to the protocol of Croall et al. (2002). Whole muscle samples were homogenized in six to ten volumes of Mops buffer (50 mm Mops, 10 mm ethylene glycol-bis (β-aminoethyl ether) N,N,N’,N’-tetraacetic acid, 10 mm ethylene diamine tetraacetic acid, 5 mm β-mercaptoethanol, 50 μl ml−1 phenylmethyl-sulfonyl fluoride, 75 μl ml−1 benzamidine) and centrifuged at 12 000 g before loading to the gel. All buffers were made immediately before use. Each lane was loaded with 250 μg protein of total muscle extract. A pre-run of the gel was performed before loading. Gels were then run overnight at 7 °C, and over-run for 1 h at 125 V to enhance separation of the calpain isoforms. Gels were developed overnight in 5 mm CaCl2 with three changes of buffer.
Muscle unloading
All experimental protocols and use of animals were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the UCLA Institutional Animal Care and Use Committee. Two lines of CS Tg mice, designated as CS 69.1 and CS 381, were chosen for muscle unloading because they expressed relatively high levels of the transgene, as assessed by Western analysis. Mice from both lines (3-4 months of age; either sex) were subjected to hindlimb unloading for 10 days (Morey-Holton & Globus, 2002) which causes substantial loss of soleus muscle mass and a shift in MHC expression from slow to fast isoforms. In this procedure, the tail of each mouse is placed in a harness, which is used to elevate the pelvis so that the feet of the hindlimbs do not contact the cage floor. Other animals were subjected to 10 days of hindlimb unloading followed by reloading for 2 days by allowing the animals to return to normal ambulation. The effect of the CS transgene on muscle reloading was assessed because previous work has shown that muscle reloading after unloading produced an increase in calpain concentration and autolysis (Spencer et al. 1997). For each group, there were age-matched, ambulatory controls. Other controls included littermates of the Tg animals that did not express the transgene, but were subjected to identical hindlimb unloading or hindlimb unloading followed by reloading. Thus, the groups analysed were: ambulatory CS 69.1 Tg (10 mice), ambulatory CS 69.1 non-Tg (13 mice), unloaded CS 69.1 Tg (6 mice), unloaded CS 69.1 non-Tg (6 mice), reloaded CS 69.1 Tg (6 mice), reloaded CS 69.1 non-Tg (7 mice), ambulatory CS 381 Tg (6 mice), ambulatory CS 381 non-Tg (8 mice), unloaded CS 381 Tg (8 mice), unloaded CS 381 non-Tg (9 mice), reloaded CS 381 Tg (6 mice), and reloaded CS 381 non-Tg (6 mice).
Muscle preparation and analysis
At the end of experimental treatments, soleus and tibialis anterior muscles were dissected from each mouse and weighed. One soleus and a tibialis anterior muscle from each animal were frozen in isopentane cooled with liquid nitrogen and used for frozen sectioning and then morphological and immunohistochemical analysis. The second soleus and tibialis anterior muscles from each animal were frozen in liquid nitrogen and then used for Western analysis. Muscle samples that were used for Western analysis were homogenized in a Dounce homogenizer in reducing sample buffer (80 mm Tris, pH 6.8, containing 0.1 m dithiothreitol and 70 mm SDS), and then boiled for 1 min and centrifuged to remove particulate matter. The protein concentration of the supernatant fraction was measured by the method of Minamide & Bamburg (1990) and then 30 μg of each sample was loaded on 10 % polyacrylamide gels prepared according to Laemmli (1970). Gels were then electrophoretically transferred to nitrocellulose (Burnette, 1981). Protein blots were incubated with a mouse monoclonal antibody to domain 1 of muscle-type calpastatin (clone CSF1-2) (Takara Bio. Inc., Shiga, Japan) diluted 1 : 200 in 50 mm Tris, pH 7.6, containing 150 mmNaCl, 0.1 % NaN3, 0.05 % Tween 20, and 3 % BSA. After extensive buffer washing of the blots, they were incubated with a second antibody conjugated to horseradish peroxidase and the bound antibody was detected by enhanced chemiluminescence (Amersham). Levels of calpastatin expression in Tg muscles relative to non-Tg control muscles were compared by densitometry of immunoreactive bands in Western blots (Alpha Innotec, USA).
Muscles frozen in isopentane were cross-sectioned mid-belly and sections were stained either with haematoxylin or with a mouse monoclonal antibody to myosin fast type heavy chain (MHCf) that binds both MHC IIa and MHC IIb in mammalian muscle (clone WB-MHCf) (Novacastra Labs, Newcastle-upon-Tyne, UK). All fibres in each haematoxylin-stained section were counted, and the cross-sectional areas of all fibres were measured using a digital imaging system (Bioquant, Nashville, TN, USA). Sections used for immunohistochemistry were fixed with acetone and then immunolabelled with anti-MHCf. Binding to endogenous mouse IgG was blocked with an MOM kit (mouse-on-mouse; Vector). After primary antibody staining and buffer rinses, sections were incubated in biotin-conjugated anti-mouse IgG, followed by avidin-conjugated, horseradish peroxidase. Colour development was done with an AEC kit (3-amino-9-ethylcarbazole; Vector). The total numbers of MHCf-expressing fibres in the cross-section of each muscle were counted microscopically.
Statistical analysis
Experimental data presented in Fig. 5 were analysed by Student's t test to compare mean ± s.d. values between transgenic and non-transgenic mice. Data (means ± s.d.) in Fig. 6 and Fig. 9 were compared between groups by one-way analysis of variance, using the Bonferroni test for post hoc comparisons. The level of statistical significance was set at P = 0.05 for each analysis.
Figure 5. Total number of fibres in a mid-belly cross-section of soleus muscles.
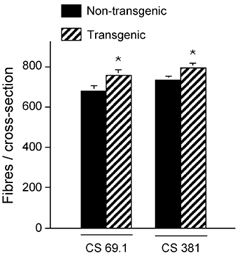
Muscles were collected from Tg mice and non-Tg littermates from the 69.1 and 381 lines. * Values differ significantly from non-Tg animals (P < 0.05).
Figure 6. Muscle fibre atrophy as indicated by percentage change in mean cross-sectional area of muscle fibres, relative to ambulatory controls.
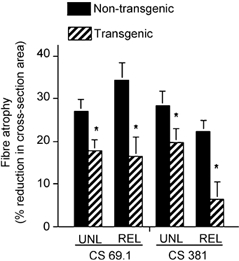
Mid-belly cross-sections of soleus muscle were analysed. Atrophy was assessed in muscles from mice experiencing 10 days of muscle unloading (UNL) or 10 days unloading followed by 2 days of reloading (REL). In every case, atrophy in CS Tg mice was significantly less (*P < 0.05) than in non-Tg littermates.
Figure 9. Proportion of total fibres in cross-section of soleus muscle that contain myofibrillar MHCf.
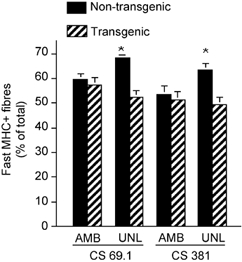
Data from ambulatory control mice (AMB) and from mice experiencing 10 days of muscle unloading (UNL) in lines 69.1 and 381 are shown. * Groups that differ significantly from ambulatory animals in the same line (P < 0.05).
Results
Expression of the CS transgene greatly reduces muscle calpain activity
Western blots were used to confirm expression of the transgene in skeletal muscles of all transgenic animals that were analysed in this investigation. Calpastatin expression was elevated approximately 50-fold (CS 69.1) or 30-fold (CS 381) in CS Tg mice compared to controls (Fig. 2). CS 381 mice also showed elevated CS expression in cardiac muscle, although the CS concentration in cardiac muscle of CS 69.1 mice did not differ from non-transgenic controls. Zymogram analysis of calpain activity in muscle homogenates showed obvious calpain 1 and calpain 2 activity in extracts of muscle from non-Tg animals; however, no detectable activity of either calpain 1 or 2 was apparent in extracts of CS Tg animals (Fig. 3). Furthermore, hindlimb unloading resulted in no significant decrease in the concentration of CS in the soleus muscles of Tg or control animals (Fig. 4).
Figure 2. Western analysis for CS in muscle samples.
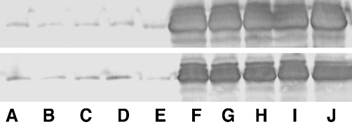
Upper panel shows relative CS levels in muscle from line 69.1; lower panel shows samples from line 381. Lanes A-E show samples from non-Tg littermates; lanes F -J show samples from CS Tg mice. CS mass in each sample was 105 kDa.
Figure 3. Zymogram analysis for calpain activity in muscle extracts.
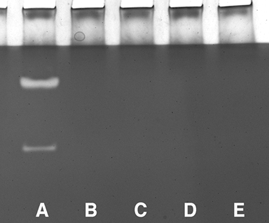
Lane A, extract from non-Tg mouse muscle. Lanes B-E, extracts from two Tg mice from line 69.1 (B and C) and from line 381(D and E). Upper band in lane A shows casein proteolysis by calpain 1; lower band shows casein proteolysis by calpain 2. No substrate proteolysis was detectable in muscle extracts from CS Tg mice.
Figure 4. Western analysis for CS in soleus muscle samples.
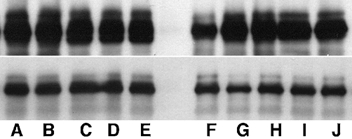
Upper panel shows relative CS levels in muscle from line 69.1 Tg mice; lower panel shows samples from line 381 Tg mice. Lanes A-E show extracts from ambulatory Tg mice; lanes F- J show extracts from Tg mice that had experienced hindlimb unloading for 10 days. CS mass in each sample was 105 kDa.
Features of CS Tg mice and muscle phenotype
CS Tg mice appeared healthy and no gross physiological, morphological or behavioural defects were observed. Age-matched Tg mice and non-Tg littermates did not differ significantly in body mass. Comparisons of the masses of muscles between age-matched Tg and non-Tg littermates showed that expression of the transgene did not significantly affect the mass of fast (tibialis anterior) or slow (soleus) muscles. We also used fibre cross-sectional area as an assessment of muscle size. Comparisons of soleus fibre cross-sectional area showed that muscle fibres in CS 381 Tg mice were 15 % smaller than in non-Tg littermate controls (CS 381 Tg = 2258 ± 177 μm2; non-Tg = 2637 ± 383 μm2; P < 0.05) and soleus fibre cross-sectional areas in CS 69.1 Tg mice were 17 % smaller than control littermates (CS 69.1 Tg = 2106 ± 341 μm2; non-Tg = 2561 ± 323 μm2; P < 0.05). Previous investigations have shown that the rate of reduction in fibre cross-sectional area in rat soleus muscles is independent of the initial cross-sectional area during muscle unloading by hindlimb suspension (Hauschka et al. 1987) or spaceflight (Miu et al. 1990).
Both lines showed a significant increase in the number of soleus muscle fibres in a mid-belly cross-section (Fig. 5). Expression of the CS Tg in CS 381 was associated with an 8.4 % increase in fibre number (CS 381 Tg = 798 ± 83.3; CS 381 non-Tg = 736 ± 89.6; P < 0.05), and a 13 % increase in fibre number in the soleus muscles of CS 69.1 (CS 69.1 Tg = 768 ± 104.3; CS 69.1 non-Tg = 680 ± 119.8; P < 0.05).
Expression of the CS transgene slows muscle atrophy during muscle unloading
Soleus muscle atrophy during hindlimb unloading was assessed by reduction in muscle fibre cross-sectional area in mid-belly cross-sections. Reduction in cross-sectional area was used rather than change in muscle mass, because differences in dissection technique could overwhelm treatment effects in the small soleus muscles, which are less than 8 mg in mass. Data were expressed as the percentage difference between the mean cross-sectional area of the muscle fibres at the end of the experimental treatment relative to the cross-sectional area of fibres in ambulatory control littermates.
Tg mice showed less muscle atrophy during muscle unloading, than non-Tg littermates (Figs 6-8). CS 381 Tg soleus muscles showed 30 % less loss of muscle cross-sectional area than non-Tg controls. Similarly, CS 69.1 Tg muscles showed 33 % less atrophy than non-Tg controls. Although the expression of the transgene had nearly identical effects on the extent of atrophy during unloading in both mouse lines, the increase in fibre diameter that occurred during 2 days of reloading differed distinctly between the two lines. CS 69.1 Tg mice showed no increase in fibre diameter during this reloading period, while fibre cross-sections of CS 381 Tg mice returned nearly to ambulatory control values after 2 days of reloading. Although previous investigations have suggested that calpain-mediated proteolysis could play a role in muscle adaptation to reloading following unloading (Spencer et al. 1997), the current findings do not provide further insights into that possibility.
Figure 8. Cross-sections of soleus muscles from Tg and non-Tg littermates from line 381.
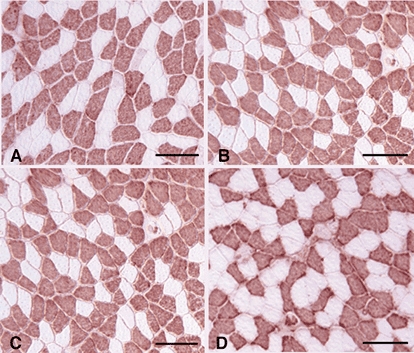
Sections were immunolabelled for MHCf (red reaction product). A, muscle from ambulatory non-Tg mouse. B, muscle from ambulatory Tg mouse. C, muscle from non-Tg mouse subjected to 10 days of muscle unloading. D, muscle from a Tg mouse subjected to 10 days of muscle unloading. Bars = 100 μm.
Expression of the CS transgene obviates changes in myosin isoform expression during muscle unloading
Unloading of skeletal muscle that expresses slow isoforms of MHC produces a frequently described replacement of slow type isoforms of MHC by fast isoforms (reviewed by Talmadge, 2000). Although the switch in myofibrillar myosin content is a prominent feature of the response of muscle to reduced neuromuscular activity, the process by which myosin turnover occurs is unknown. We measured the percentage of total muscle fibres expressing MHCf in entire mid-belly cross-sections of soleus muscles from ambulatory controls and unloaded muscles of CS 69.1 and CS 381 Tg mice and non-Tg littermates, to test whether the change in myosin isoform expression was influenced by levels of calpastatin. Expression of the transgene did not affect the proportion of fast fibres present in the soleus muscles of mice in either line (CS 381 Tg 51.4 ± 8.5 % fast fibres; CS 381 non-Tg 53.5 ± 9.6 %; CS 69.1 Tg 57.5 ± 9.1 %; CS 69.1 non-Tg 59.7 ± 7.5 %). However, 10 days of muscle unloading produced no increase in the proportion of fast fibres in the soleus muscles of Tg mice in either line (CS 381 = 49.4 ± 8.2 % fast fibres; CS 69.1 = 52.2 ± 6.7 %), although non-Tg littermates showed significant increases in fast fibres (CS 381 non-Tg = 63.5 ± 8.3 % fast fibres; CS 69.1 non-Tg = 68.2 ± 2.3 %; Fig. 9). Previous investigations have shown that approximately 30 % (Carlson et al. 1999) or 42 % (Burkholder et al. 1994) of muscle fibres in soleus muscle of adult, ambulatory mice express MHCf, with the differences in the proportion of MHCf fibres being attributable to differences in the mouse strain that was studied.
Immunohistochemical observations indicate that the lack of increase in the proportion of fast fibres in the soleus muscles of Tg mice resulted from the lack of myosin isoform conversion, rather than the occurrence of a compensatory switch from fast to slow isoforms of MHC expression to yield an unchanged ratio. Antibody staining for MHCf showed fibres that were apparently undergoing a switch to MHCf expression in some non-Tg animals, but no similar transitional fibres were observed in muscles from Tg animals (Fig. 7).
Figure 7. Cross-sections of soleus muscles from Tg and non-Tg littermates from line 69.1.
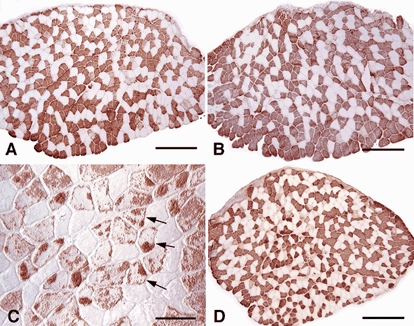
All sections were immunolabelled for fast myosin (red reaction product). A, muscle from ambulatory non-Tg mouse. B, muscle from ambulatory Tg mouse. C, muscle from non-Tg mouse subjected to 10 days of muscle unloading. Arrows indicate fibres that appear to be in the process of switching to MHCf expression, which is suggested by the clusters of MHCf-containing myofibrils that occupy a portion of the total cross-section of the individual fibres. D, muscle from a Tg mouse subjected to 10 days of muscle unloading. In A, B and D, scale bar = 240 μm. In C, scale bar = 50 μm.
Discussion
Previous investigations have suggested that inhibition of calpain activity could have therapeutic value in slowing muscle wasting (Spencer et al. 1995; Huang & Forsberg, 1998). However, this possibility has been difficult to address because of an inability to specifically perturb calpain activity in vivo so that its role could be assessed in a physiological context. The findings of the present investigation show that the calpain-calpastatin system plays a significant role in regulating the loss of muscle mass that occurs during reduced muscle use. Our data show that at least 30 % of the atrophy that occurs during the first 10 days of soleus muscle unloading results from calpain-mediated processes. It is likely that we did not achieve complete inhibition of muscle calpains, so that the actual contribution of the calpain-calpastatin system to atrophy could be greater than 30 %.
Although our data show a significant role of the calpain-calpastatin system in muscle atrophy during unloading, current knowledge of the proteolytic function of calpain indicates that calpains provide just one of the proteolytic systems that may function in series to degrade muscle proteins during atrophy. Calpains typically cleave proteins at only one or a few specific proteolytic sites, which is generally interpreted as indicating a regulatory function, rather than a degradative role for calpains (Croall & DeMartino, 1991). However, the limited proteolytic modification of select substrates by calpains could target substrates for further degradation by other proteolytic systems, especially the ubiquitin-proteasome system. Experimental observations have indicated that proteolysis of skeletal muscle proteins by the ubiquitin-proteasome system can be catalysed by modifications in the amino termini of the substrates (Solomon et al. 1998a,b), and it is feasible that limited proteolytic cleavage of substrate by calpains could provide this modification in the amino termini. Previous investigators have shown that there is an increase in expression of several components of the ubiquitin-proteasome system during muscle unloading, which may reflect an increase in proteolysis through that system (Riley et al. 1992; Taillandier et al. 1996; Bodine et al. 2001). In addition, muscle atrophy during unloading is associated with increases in protein ubiquination and increases in cathepsin L expression (Ikemoto et al. 2001), suggesting that both of these systems may further contribute to proteolysis during muscle unloading.
Our findings also support the expectation that the calpain-calpastatin system plays a significant role in muscle development. We observed that muscle fibres in CS Tg mice were smaller, but more numerous, so that muscle mass remained unchanged. The presence of more numerous, smaller fibres may reflect impaired muscle cell fusion into multinucleated fibres during development, which is consistent with in vitro observations concerning the role of calpains in muscle cell fusion. Previous investigators have shown that the fusion of myocytes to form multinucleated myotubes in vitro can be increased by calcium ionophore and is associated with relocation of calpain to the muscle cell membrane, where the calpains are expected to promote membrane fusion (Schollmeyer, 1986). Experimental findings have shown that the microinjection of CS into myoblasts in vitro reduces fusion to form myotubes in vitro, which further implicates the calpain-calpastatin system in this process (Temm-Grove et al. 1999). Endogenous regulation of calpain and CS in myocyte fusion apparently occurs by the regulation of CS expression. CS expression declines at the time of myocyte fusion which leads to an increase in the relative concentration of calpain : calpastatin, thereby promoting fusion (Barnoy et al. 1996, 2000).
The role of the calpain-calpastatin system in myofibrillar myosin isoform shifting was an unexpected finding. Many investigations have shown that reductions in neuromuscular activity are associated with a shift of myofibrillar myosin from slow to fast isoforms of MHC, although little is known of the mechanisms that regulate this shift. Spinal cord injuries (Dupont-Versteegden et al. 1998), limb immobilization (Jankala et al. 1998), prolonged bedrest (Widrick et al. 1997), spaceflight (Caiozzo et al. 1995) and reductions in neuromuscular activity by tetrodotoxin (Michel et al. 1996) can each produce shifts in myofibrillar MHC from slow to fast isoforms (e.g. review by Talmadge, 2000), which suggests that there may be common, underlying regulatory mechanisms that influence the response to these perturbations. Recent, important findings have shown that fibre type determination in skeletal muscle can be regulated by the transcriptional co-activator PGC-1α (Lin et al. 2002) and members of the MEF2 family of transcription factors (reviewed by Olson & Williams, 2000), which may be components of a common regulatory mechanism. The calpain-calpastatin system could feasibly contribute to the shift in myofibrillar myosin content through any of three general processes. First, it is possible that calpain could directly or indirectly modify the activity of factors that regulate the expression of specific isoforms of myosin. For example, if calpain-mediated cleavage were required for activation of a transcription factor that promotes MHCf expression or inhibits slow MHC expression, CS overexpression could prevent the shift to MHCf in atrophying muscles in Tg mice. Previous studies have shown that calpains cleave and modify the activity of transcription factors (Pariat et al. 2000; Oda et al. 2002). However, findings in the present investigation suggest that CS overexpression does not affect transcription of specific myosin isoforms because no difference was observed in the relative proportions of fibres that expressed MHCf in the soleus muscles of ambulatory CS Tg mice and non-Tg mice. A second possibility is that CS overexpression inhibits calpain-mediated cleavage of MHC, and thereby inhibits turnover. Several investigations have shown that skeletal muscle MHC is a substrate of calpains (Ishiura et al. 1979; Hara et al. 1983; Pemrick & Grebenau, 1984). However, incubation of myofibrils with calpain does not produce a reduction of MHC concentration or mass that is detectable by SDS-PAGE, which indicates that calpain-mediated cleavage of MHC is not extensive (Goll et al. 1991).
A final possible way in which overexpression of CS can prevent the shift of myofibrillar MHC content from slow to fast isoforms during muscle atrophy is that CS may inhibit the calpain-mediated release of myosin thick filaments from myofibrils, which are subsequently proteolysed through the ubiquitin-proteasome system. Strong evidence has shown that the ubiquitin-proteasome system plays a significant role in proteolysis during muscle unloading (Bodine et al. 2001) and in other models of muscle wasting (Solomon et al. 1998b; Mitch et al. 1999; Bailey & Mitch, 2000). In addition, MHC can be degraded by the ubiquitin-proteasome system, although myosin thick filaments are not ubiquinated and proteolysed when they are associated with thin filaments (Solomon & Goldberg, 1996). Thus, myosin thick filaments must be released from sarcomeres before degradation by the ubiquitin-proteasome system can occur. Several proteins in addition to myosin are calpain substrates that are important in maintaining the architecture of the sarcomere. In particular, titin plays a central role in structuring the sarcomere, it binds myosin and it is readily cleaved by calpain (Kim et al. 1995; Suzuki et al. 1996; Galvagni et al. 1998). Collectively, these observations suggest that the role of calpains in regulating the shift in myofibrillar myosin content may be to release thick filaments from the sarcomere, after which they are degraded by the ubiquitin-proteasome system, and replaced by other myosin thick filaments.
The results of the present investigation provide the first information concerning how perturbation of a specific proteolytic system affects muscle wasting in vivo. The significant reduction of muscle atrophy that occurs in CS overexpressing muscle during muscle unloading indicates that therapeutic approaches that target the calpain-calpastatin system may be useful in slowing muscle atrophy. Continuing studies are directed toward testing whether the calpain-calpastatin system contributes similarly to muscle wasting in other models of disease and injury.
Acknowledgments
This work was supported by the National Institutes of Health (AR40343 and AR48177) and the National Aeronautics and Space Administration. Katherine Wen and Baiyuan Cai provided expert technical assistance.
References
- Arthur GD, Booker TS, Belcastro AN. Exercise promotes a subcellular redistribution of calcium-stimulated protease activity in striated muscle. Canadian Journal of Physiology and Pharmacology. 1999;77:42–47. doi: 10.1139/cjpp-77-1-42. [DOI] [PubMed] [Google Scholar]
- Bailey JL, Mitch WE. Mechanisms of protein degradation: what do the rat studies tell us? Journal of Nephrology. 2000;13:89–95. [PubMed] [Google Scholar]
- Baracos VE, Greenberg RE, Goldberg AL. Influence of calcium and other divalent cations on protein turnover in rat skeletal muscle. American Journal of Physiology. 1986;250:E702–710. doi: 10.1152/ajpendo.1986.250.6.E702. [DOI] [PubMed] [Google Scholar]
- Barnoy S, Glaser T, Kosower NS. The role of calpastatin (the specific calpain inhibitor) in myoblast differentiation and fusion. Biochemical and Biophysical Research Communications. 1996;220:933–938. doi: 10.1006/bbrc.1996.0509. [DOI] [PubMed] [Google Scholar]
- Barnoy S, Supino-Rosin L, Kosower NS. Regulation of calpain and calpastatin in differentiating myoblasts: mRNA levels, protein synthesis and stability. Biochemical Journal. 2000;351:413–420. [PMC free article] [PubMed] [Google Scholar]
- Belcastro AN. Skeletal muscle calcium activated neutral protease (calpain) with exercise. Journal of Applied Physiology. 1993;74:1381–1386. doi: 10.1152/jappl.1993.74.3.1381. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. Journal of Biological Chemistry. 1993;268:719–725. [PubMed] [Google Scholar]
- Burkholder TJ, Fingado B, Baron S, Lieber RL. Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. Journal of Morphology. 1994;221:177–190. doi: 10.1002/jmor.1052210207. [DOI] [PubMed] [Google Scholar]
- Burnette WN. Western blotting’: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Analytical Biochemistry. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Caiozzo VJ, Haddad F, Baker MJ, Baldwin KM. Functional and cellular adaptations of rodent skeletal muscle to weightlessness. Journal of Gravitational Physiology. 1995;2:39–42. [PubMed] [Google Scholar]
- Carlson CJ, Booth FW, Gordon SE. Skeletal muscle myostatin mRNA expression is fiber-type specific and increases during hindlimb unloading. American Journal of Physiology. 1999;277:R601–606. doi: 10.1152/ajpregu.1999.277.2.r601. [DOI] [PubMed] [Google Scholar]
- Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. Journal of Cell Biology. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croall DE, DeMartino GN. Calcium-activated neutral protease (calpain) system: structure, function, and regulation. Physiological Reviews. 1991;71:813–847. doi: 10.1152/physrev.1991.71.3.813. [DOI] [PubMed] [Google Scholar]
- Croall DE, Moffett K, Hatch H. Casein zymography of calpains using a 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid-imidazole buffer. Analytical Biochemistry. 2002;304:129–132. doi: 10.1006/abio.2001.5606. [DOI] [PubMed] [Google Scholar]
- Dupont-Versteegden EE, Houle JD, Gurley CM, Peterson CA. Early changes in muscle fiber size and gene expression in response to spinal cord transection and exercise. American Journal of Physiology. 1998;275:C1124–1133. doi: 10.1152/ajpcell.1998.275.4.C1124. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Furuno K, Goldberg AL. The activation of protein degradation in muscle by calcium or muscle injury does not involve a lysosomal mechanism. Biochemical Journal. 1986;237:859–864. doi: 10.1042/bj2370859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno K, Goodman MN, Goldberg AL. Role of different proteolytic systems in the degradation of muscle proteins during denervation atrophy. Journal of Biological Chemistry. 1990;265:8550–8557. [PubMed] [Google Scholar]
- Galvagni F, Cartocci E, Oliviero S. The dystrophin promoter is negatively regulated by YY1 in undifferentiated muscle cells. Journal of Biological Chemistry. 1998;273:33708–33713. doi: 10.1074/jbc.273.50.33708. [DOI] [PubMed] [Google Scholar]
- Goll DE, Dayton WR, Singh I, Robson RM. Studies of the α-actinin/actin interaction in the Z-disk by using calpain. Journal of Biological Chemistry. 1991;266:8501–8510. [PubMed] [Google Scholar]
- Hara K, Ichihara Y, Takahashi K. Purification and characterization of a calcium-activated neutral protease from monkey cardiac muscle. Journal of Biochemistry. 1983;93:1435–1445. doi: 10.1093/oxfordjournals.jbchem.a134279. [DOI] [PubMed] [Google Scholar]
- Hauschka EO, Roy RR, Edgerton VR. Size and metabolic properties of single muscle fibers in rat soleus after hindlimb suspension. Journal of Applied Physiology. 1987;62:2338–2347. doi: 10.1152/jappl.1987.62.6.2338. [DOI] [PubMed] [Google Scholar]
- Huang J, Forsberg NE. Role of calpain in skeletal-muscle protein degradation. Proceedings of the National Academy of Sciences of the USA. 1998;95:12100–12105. doi: 10.1073/pnas.95.21.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto M, Nikawa T, Takeda S, Watanabe C, Kitano T, Baldwin KM, Izumi R, Nonaka I, Towatari T, Teshima S, Rokutan K, Kishi K. Space shuttle flight (STS-90) enhances degradation of rat myosin heavy chanin in association with activation of ubiquitin-proteasome pathway. FASEB Journal. 2001;10(1096/fj):00–0629fje. doi: 10.1096/fj.00-0629fje. [DOI] [PubMed] [Google Scholar]
- Imajoh S, Kawasaki H, Suzuki K. Limited autolysis of calcium-activated neutral protease (CANP): reduction of the Ca2+-requirement is due to the NH2-terminal processing of the large subunit. Journal of Biochemistry. 1986;99:1281–1284. doi: 10.1093/oxfordjournals.jbchem.a121755. [DOI] [PubMed] [Google Scholar]
- Ishiura S, Sugita H, Suzuki K, Imahori K. Studies of a calcium-activated neutral protease from chicken skeletal muscle. II. Substrate specificity. Journal of Biochemistry. 1979;86:579–581. doi: 10.1093/oxfordjournals.jbchem.a132558. [DOI] [PubMed] [Google Scholar]
- Jankala H, Harjola V-P, Petersen NE, Harkonen M. Myosin heavy chain mRNA transforms to faster isoforms in immobilized skeletal muscle: a quantitative PCR study. Journal of Applied Physiology. 1998;82:977–982. doi: 10.1152/jappl.1997.82.3.977. [DOI] [PubMed] [Google Scholar]
- Kim K, Homma Y, Ikeuchi Y, Suzuki A. Cleavage of connectin by calpain and cathepsin D. Bioscience, Biotechnology and Biochemistry. 1995;59:896–899. doi: 10.1271/bbb.59.896. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:497–601. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Medina R, Wing SS, Goldberg AL. Increase in levels of polyubiquitin and proteasome mRNA in skeletal muscle during starvation and denervation atrophy. Biochemical Journal. 1995;307:631–637. doi: 10.1042/bj3070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel RN, Parry DJ, Dunn SE. Regulation of myosin heavy chain expression in adult rat hindlimb muscles during short-term paralysis comparison of denervation and tetrodotoxin-induced neural inactivation. FEBS Letters. 1996;391:39–44. doi: 10.1016/0014-5793(96)00618-7. [DOI] [PubMed] [Google Scholar]
- Minamide LS, Bamburg JR. A filter paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Analytical Biochemistry. 1990;190:66–70. doi: 10.1016/0003-2697(90)90134-u. [DOI] [PubMed] [Google Scholar]
- Mitch WE, Bailey JL, Wang X, Jurkovitz C, Newby D, Price SR. Evaluation of signals activating ubiquitin-proteasome proteolysis in a model of muscle wasting. American Journal of Physiology. 1999;276:C1132–1138. doi: 10.1152/ajpcell.1999.276.5.C1132. [DOI] [PubMed] [Google Scholar]
- Miu B, Martin TP, Roy RR, Oganov V, Ilyina-Kakueva E, Marini JF, Leger JJ, Bodine-Fowler SC, Edgerton VR. Metabolic and morphologic properties of single muscle fibers in the rat after spaceflight, Cosmos 1887. FASEB Journal. 1990;4:64–72. doi: 10.1096/fasebj.4.1.2136839. [DOI] [PubMed] [Google Scholar]
- Morey-Holton ER, Globus RK. Hindlimb unlaoding rodent model: technical aspects. Journal of Applied Physiology. 2002;92:1367–1377. doi: 10.1152/japplphysiol.00969.2001. [DOI] [PubMed] [Google Scholar]
- Oda A, Wakao H, Fujita H. Calpain is a signal transducer and activator of transcription (STAT) 3 and STAT 5 protease. Blood. 2002;99:1850–1852. doi: 10.1182/blood.v99.5.1850. [DOI] [PubMed] [Google Scholar]
- Olson EN, Williams RS. Remodeling muscles with calcineurin. BioEssays. 2000;22:510–519. doi: 10.1002/1521-1878(200011)22:11<1049::AID-BIES14>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Pariat M, Salvat C, Bebien M, Brockly F, Altieri E, Carillo S, Jariel-Encontre I, Piechaczyk M. The sensitivity of c-Jun and c-Fos proteins to calpains depends on conformational determinants of the monomers and not on formation of dimers. Biochemical Journal. 2000;345:129–138. [PMC free article] [PubMed] [Google Scholar]
- Pemrick SM, Grebenau RC. Qualitative analysis of skeletal myosin as substrate of calcium-activated neutral protease: comparison of filamentous and soluble, native, and L2-deficient myosin. Journal of Cell Biology. 1984;99:2297–2308. doi: 10.1083/jcb.99.6.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid WD, Belcastro AN. Time course of diaphragm injury and calpain activity during resistive loading. American Journal of Respiratory and Critical Care Medicine. 2000;162:1801–1806. doi: 10.1164/ajrccm.162.5.9906033. [DOI] [PubMed] [Google Scholar]
- Riley DA, Ellis S, Giometti CS, Hoh JF, Ilyina-Kakueva EI, Oganov VS, Slocum GR, Bain JL, Sedlak FR. Muscle sarcomere lesions and thrombosis after spaceflight and suspension unloading. Journal of Applied Physiology. 1992;73:S33–43. doi: 10.1152/jappl.1992.73.2.S33. [DOI] [PubMed] [Google Scholar]
- Rodemann HP, Waxman L, Goldberg AL. The stimulation of protein degradation in muscle by Ca2+ is mediated by prostaglandin E2 and does not require the calcium-activated protease. Journal of Biological Chemistry. 1982;257:8716–8723. [PubMed] [Google Scholar]
- Ruff RL, Secrist D. Inhibitors of prostaglandin synthesis or cathepsin B prevent muscle wasting due to sepsis in the rat. Journal of Clinical Investigation. 1984;73:1483–1486. doi: 10.1172/JCI111352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schollmeyer JE. Role of calcium and calcium-activated-protease in myoblast fusion. Experimental Cell Research. 1986;162:411–422. doi: 10.1016/0014-4827(86)90346-0. [DOI] [PubMed] [Google Scholar]
- Solomon V, Baracos V, Sarraf P, Goldberg AF. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proceedings of the National Academy of Sciences of the USA. 1998b;95:12602–12607. doi: 10.1073/pnas.95.21.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon V, Goldberg AL. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. Journal of Biological Chemistry. 1996;271:26690–26697. doi: 10.1074/jbc.271.43.26690. [DOI] [PubMed] [Google Scholar]
- Solomon V, Lecker SH, Goldberg AL. The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. Journal of Biological Chemistry. 1998a;273:25216–25222. doi: 10.1074/jbc.273.39.25216. [DOI] [PubMed] [Google Scholar]
- Sorimachi H, Suzuki K. The structure of calpain. Journal of Biochemistry. 2001;129:653–664. doi: 10.1093/oxfordjournals.jbchem.a002903. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Croall DE, Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. Journal of Biological Chemistry. 1995;270:10909–10914. doi: 10.1074/jbc.270.18.10909. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Guyon JR, Sorimachi H, Potts A, Richard I, Herasse M, Chamberlain J, Dalkilic I, Kunkel LM, Beckmann JS. Stable expression of calpain 3 from a muscle transgene in vivo: immature muscle in transgenic mice suggests a role for calpain 3 in muscle maturation. Proceedings of the National Academy of Sciences of the USA. 2002;99:8874–8879. doi: 10.1073/pnas.132269299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer MJ, Lu B, Tidball JG. Calpain II expression is increased by changes in mechanical loading of muscle in vivo. Journal of Cellular Biochemistry. 1997;64:55–66. doi: 10.1002/(sici)1097-4644(199701)64:1<55::aid-jcb9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kim K, Ikeuchi Y. Proteolytic cleavage of connectin/titin. Advances in Biophysics. 1996;33:53–64. doi: 10.1016/s0065-227x(96)90022-2. [DOI] [PubMed] [Google Scholar]
- Taillandier D, Aurousseau E, Meynial-Denis D, Bechet D, Ferrara M, Cottins P, Ducastaing A, Bigard X, Guezennec C, Schmid H, Attaix D. Coordinate activation of lysosomal, calcium activated and ATP-ubiquitin-dependent proteinases in the unweighted rat soleus muscle. Biochemical Journal. 1996;316:65–72. doi: 10.1042/bj3160065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge RJ. Myosin heavy chain isoform expression following reduced neuromuscular activity: potential regulatory mechanisms. Muscle and Nerve. 2000;23:661–679. doi: 10.1002/(sici)1097-4598(200005)23:5<661::aid-mus3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Temm-Grove CJ, Wert D, Thompson VF, Allen RE, Goll DE. Microinjection of calpastatin inhibits fusion in myoblasts. Experimental Cell Research. 1999;246:293–303. doi: 10.1006/excr.1998.4362. [DOI] [PubMed] [Google Scholar]
- Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. Journal of Applied Physiology. 1990;68:1–12. doi: 10.1152/jappl.1990.68.1.1. [DOI] [PubMed] [Google Scholar]
- Tischler ME, Rosenberg S, Satarug S, Henriksen EJ, Kirby CR, Tome M, Chase P. Different mechanisms of increased proteolysis in atrophy induced by denervation or unweighting of rat soleus muscle. Metabolism. 1990;39:756–763. doi: 10.1016/0026-0495(90)90113-q. [DOI] [PubMed] [Google Scholar]
- Voisin L, Breuille D, Combaret L, Poutey C, Taillandier D, Aurousseau E, Obled C, Attaix D. Muscle wasting in a rat model of long-lasting sepsis results from the activation of lysosomal, calcium-activated, and ubiquitin-proteasome proteolytic pathways. Journal of Clinical Investigation. 1996;97:1610–1617. doi: 10.1172/JCI118586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. Journal of Cell Biology. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widrick JJ, Romatowski JG, Bain JL, Trappe SW, Thompson JL, Costill DL, Riley DA, Fitts RH. Effect of 17 days of bed rest on peak isometric force and unloaded shortening velocity of human soleus fibers. American Journal of Physiology. 1997;273:C1690–1699. doi: 10.1152/ajpcell.1997.273.5.c1690. [DOI] [PubMed] [Google Scholar]
- Zeman RJ, Kameyama T, Matsumoto K, Bernstein P, Etlinger JD. Regulation of protein degradation in muscle by calcium. Journal of Biological Chemistry. 1985;260:13619–13624. [PubMed] [Google Scholar]