

Abstract
Sphingosine kinase 1 (SK1) and its product sphingosine-1-phosphate (S1P) have been implicated in the regulation of many cellular processes including growth regulation, protection from apoptosis, stimulation of angiogenesis, and most recently as mediators of the TNF alpha inflammatory response. In this study we set out to examine the role of SK1/S1P in the RAW macrophage response to the potent inflammatory stimulus LPS. We show that LPS increases cellular levels of SK1 message and protein. This increase is at the transcriptional level and is accompanied by increased SK activity and generation of S1P. S1P is able to cause increases in COX-2 and PGE2 levels in RAW cells. Knock down of SK1 using siRNA is able to inhibit the TNF but not the LPS inflammatory response. Moreover, knock down of SK1 enhances both TNF and LPS-induced apoptosis. These data indicate that there is a dual and distinct role for SK1 and S1P in the TNF and the LPS inflammatory pathways.
INTRODUCTION
Sphingolipid molecules, once considered mainly structural components of cell membranes, are emerging as biologically important signaling molecules. Among the best studied in this class of lipids are ceramide, sphingosine, and sphingosine-1-phosphate (S1P). They serve as signaling molecules within or among cells, and thus play essential roles in the regulation of many different cellular events such as cell growth, differentiation, stress responses, and apoptosis (1,2).
One bioactive sphingolipid, S1P is important in signaling numerous prosurvival cellular responses. S1P is generated by the conversion of sphingosine to S1P by sphingosine kinase (SK) and acts as extracellular ligands for a family of G-protein coupled receptors called S1P receptors (3). It's also possible that S1P has yet unidentified intracellular targets (4). S1P has been implicated in several biological responses including protection from apoptosis, angiogenesis, cardiac and neuronal development, and immune cell trafficking (5).
There is growing research on the relationship between the SK/S1P pathway and early inflammatory activation. Many immune stimuli are capable of activating SK including proinflammatory cytokines such as TNFα and interleukin-1β, as well as inflammatory regulated growth factors such as platelet-derived growth factor (PDGF) (6,7). In platelets, S1P is found in granular stores and released upon activation of the coagulation cascade during acute inflammatory events (8). S1P stimulates the migration of mast cells following stimulation by IgE (9). In bronchial epithelial cells, S1P is involved in signaling the release of IL-8 which attracts neutrophils (10). S1P has also been shown to be involved in the activation, survival, and adherence of macrophages to the extra cellular matrix via integrins (11-14). In addition, S1P induces calcium release in monocytes as a marker for cell activation (11,15). Furthermore, knockdown of SK1 in macrophages inhibited TNF-α, IL-6, and IL-8 release induced by complement activation (16).
Recent work by Pettus et al showed that the SK1/S1P mediates the cyclooxygenase (COX-2) upregulation and subsequent prostaglandin E2 (PGE2) release following simulation by TNF-α in mouse fibroblasts (17). COX-2 is a common component in inflammatory reactions in response to cytokines and other inflammatory mediators such as lipopolysaccharide (LPS). Although Wu and co-workers implicated SK1 in protecting lipopolysaccharide-activated macrophages from apoptosis (18), the role of the SK1/S1P pathway on COX-2 stimulation during LPS signaling in innate immune response has not yet been investigated.
Macrophage stimulation by LPS is a well characterized model for studying innate immune response. LPS is a cell wall component of gram negative bacteria that activates macrophage antibacterial response and cytokine release. This well characterized inflammatory pathway involves the activation of the toll-like receptor TLR4 which activates IL-1 receptor resulting in the activation of NF-kB, and JNK pathways (19). Ultimately this will lead to changes in the cytoskeleton, production of cytokines, chemokines, and proinflammatory mediators such as PGE2 and nitric oxide species (20).
In this manuscript we set out to demonstrate whether the SK1/S1P pathway plays a role in the innate immune response of LPS. We demonstrate that LPS causes upregulation of message and protein levels of SK1 and an increase in S1P. However this activation of the SK1/S1P pathway did not appear to mediate LPS-induced COX-2 or PGE2 upregulation. Herein, we show that there are two independent signaling pathways resulting in induction of the COX-2 – PGE2 pathway one activated by TNF-α that is SK1 / S1P mediated and another activated by LPS and SK1 / S1P independent.
EXPERIMENTAL PROCEDURES
Cell lines and culture conditions
RAW 264.7 cells are maintained in RPMI containing 10% fetal calf serum at 37°C in a 5% CO2 incubator. For each experiment cells were treated at 60−80% confluency.
Measurement of sphingolipid production
10 × 106 cells were plated in 100 mm dishes 24 h prior to the experiment and treated with 500 ng/ml LPS (obtained from Calbiochem, E.Coli J5) for different times up to 24 h. After treatment, cells were collected and the lipids were extracted in 2 ml 70% isopropyl alcohol: ethyl acetate (2:3, v/v). The upper phase was collected and dried for S1P quantification using the positive mode electrospray ionization (ESI)/MS/MS analysis in the Lipidomics Core Facility at MUSC. The results were expressed as picomole S1P/mg of protein (21).
Real Time PCR
For the Real-time PCR analysis cells were collected and submitted to mRNA extraction using Qiagen RNeasy kit as described by manufacturer. 1 μg mRNA was converted to cDNA using Invitrogen SuperScript. Real-time RT-PCR was performed on a Biorad Icycler. The standard reaction volume was 25 ul and contained 12.5ul of QuantiTect SYBER Green, 1ul of 300ng cDNA template, 1 ul of the forward primer, 1ul of the reverse primer (both the forward and reverse primers at 3.125 uM concentration), and 9.5ul of water. Initial steps of RT-PCR were 3 min at 95 °C. Cycles (n = 40) consisted of a 10s melt at 95 °C, followed by a 45 second annealing/extension at 53 °C. The final step was a 95 °C incubation for 1 min and then 53°C for 1 min. All reactions were performed in triplicate. The threshold for cycle of threshold (Ct) analysis of all samples was set at 0.15 relative fluorescence units. Primer sequences were: SK1 (accession number NM_021972; forward, 5’ TGCCTTCTCATTGGACTGTGG -3’, reverse, 5’ GTAGCAGCACCAGCACCAG -3’), β-actin (accession number NM_001101; forward, 5’-ATTGGCAACGAGCGGTTCC -3’, reverse, 5’-TGTAGTTTCATGGATGCCACA -3’).
The β-actin gene was used as an internal reference control to normalize relative levels of gene expression. Results were analyzed using Q-Gene® software, which expresses data as mean normalized expression.
Western blot Analysis
Cells were scraped and lysed in a buffer containing 50 mM HEPES, pH 7.5, 0.5 M NaCl, 1% Triton X-100, 0.5% Tween 20, 1mM PMSF, and 0.1% protease inhibitor mixture (Sigma). The lysates were homogenized with a 23 gauge needle and centrifuged at 45000 × g for 45 min. Protein concentration of the supernatant was determined using Pierce BCA Protein Assay. Forty μg of protein was diluted in Tris-Gycine SDS sample buffer (Invitrogen) and applied to an Invitrogen precast 4−20% polyacrylamide gel under reducing conditions. Proteins were then transferred to a nitrocellulose membrane. For the COX-2 protein detection, the membrane was blocked with TBS containing 5% milk and 0.1% Tween (blocking buffer) for 3 hours at room temperature. The membrane was then probed with the COX-2 primary antibody (Santa Cruz C-20) in blocking buffer overnight at 4°C in, and probed with secondary antibody HRP-conjugated donkey anti-goat (Santa Cruz) for 1 hour. For SK1 protein detection, the membrane was incubated in blocking buffer overnight at 4°C. The membrane was probed with the SK1 primary antibody (22) for 3 hours in blocking buffer, and probed with secondary antibody HRP-conjugated goat anti-rabbit (Santa Cruz) for 1 hour. The chemiluminescence signal was detected using Amersham Biosciences ECL plus.
Generation of hSK1 promoter construct
The hSK1 promoter was cloned from human genomic DNA through PCR amplification of a 3 KB DNA segment upstream of the SK1 5’UTR as described in (23). This fragment was gel purified and cloned into TOPO-XL vector (Invitrogen) and verified by sequencing. The putative SK1 promoter was excised using KpnI and NheI and ligated into the PGL3 Basic luciferase vector.
Transient transfection and measurement of promoter activity
5 × 106 RAW 264.7 cells were transiently co-transfected by electoporation using Nucleofactor with 5ug SK1 promoter construct and 1ug β-galactosidase plasmid and seeded in a 6-wells plate. Sixteen hours after transfection cells were treated with either PBS or LPS (500ng/ml) for 3 hours. Cells were then lysed using Promega reporter lysis buffer. Luciferase and β-galactosidase measurements were made using Promega luciferase and B-galactosidase enzyme assay system kit (from Promega) according to manufacturer's instruction. Data are normalized with β-galactosidase activity in each sample.
Sphingosine kinase activity
Sphingosine kinase activity was determined as described previously with minor modifications (24). After stimulation with 500 ng/ml LPS for 24h, cells were washed with cold PBS and harvested in SK1 buffer (containing 20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.5 mM Deoxypyridoxine, 15 mM NaF, 1 mM β-mercaptoethanol, 1 mM Na-orthovanadate, 40 mM β-glycerophosphate, 0.4 mM PMSF, 10% glycerol, 0.5% Triton X-100, and EDTA-free protease inhibitor mixture (Roche)). After brief sonication and determination of protein concentration using Pierce BCA protein assay 60 μg of protein were incubated in 100 μl of reaction mixture containing sphingosine (50 μM, delivered in 4 mg/ml fatty acid-free bovine serum albumin), [γ-32P] ATP (5 μCi, 1 mM dissolved in 10 mM MgC12) and SK1 buffer for 30 min at 37°C. The reaction was terminated by the addition of 10 μl of 1N HC1 and 400 μl of chloroform/methanol/HC1 (100:200:1, v/v/v) and allowed to stand at room temperature for 10 min. Subsequently, 120 μl of chloroform and 120 μl of 2 M KCl were added, and samples were centrifuged at 3000 × g for 5 min. 200 μl of the organic phase was transferred to new glass tubes and dried. Samples were re-suspended in chloroform/methanol/HC1 (100:100:1, v/v/v). Lipid were then resolved on silica thin layer chromatography plates using 1-butanol/methanol/acetic acid/water (8:2:1:2, by volume) as solvent system and visualized by autoradiography. The radioactive spots corresponding to S1P were scraped from the plates and counted for radioactivity. Background values were determined in negative controls in which sphingosine was not added to the reaction mixture.
Measurement of PGE2 levels
RAW 264.7 cells were incubated for different times in the absence or presence of 500ng/ml LPS, 3μM-10μM S1P, or 3nM TNF-α. At the end of incubation, the conditioned medium was collected and analyzed for the presence of PGE2 using a PGE2 monoclonal enzyme immunoassay (EIA) kit from Cayman Chemical (cat # 514010, Ann Arbor, MI) according to the manufacturer's instructions. The results were calculated according to the PGE2 standard curve and expressed as pg/ml.
Measurement of TNF-α levels
RAW 264.7 cells were incubated for different times in the absence or presence of 500ng/ml LPS, 3μM-10μM S1P, or 3nM TNF-α. At the end of incubation, the conditioned medium was collected and analyzed for the presence of TNF-α using a TNF-α enzyme immunoassay (EIA) Kit from eBioscience (cat # 88−7324, San Diego, Ca) according to the manufacturer's instructions. The results were calculated according to the TNF-α standard curve and express as pg/ml.
Knockdown of mouse SK1
Knockdown of mouse SK1 was performed using siRNA consisting of 76 nucleotides from the SK1 start codon (GGGCAAGGCUCUGCAGCUCTdTd and GAGCUGCAGAGCCUUGCCCTdTd); a scrambled sequence (SCR) (CUGGUUACGAAGCGAAUCCTdTd and GGAUUCGCUUCGUAACCAGTdTd) was used as a control. RAW 264.7 cells were transfected with the 21-nucleotide duplexes using OligofectAMINE (Invitrogen, San Diego, CA, USA) as recommended by the manufacturer. After 48 or 72 hours the cells were lysed, and RNA extracted for Real-time PCR analysis to confirm SK1 knockdown (17).
Measurement of Cell Death
Apoptotic cell death was measured by Annexin V staining. RAW 264.7 cells were treated with 100 nM siRNA SK1 or siRNA scrambled for 48 hrs and then stimulated with LPS (500 ng/ml) or TNF-α (3nM) for 24 hrs. Cells were then fixed and stained using Annexin V apoptosis detection assay kit (Molecular Probes cat # A13203). Stained cells were counted and expressed as a fraction of the total cells.
RESULTS
LPS upregulates S1P and SK1 in RAW 264.7 macrophages
To determine the role for the sphingosine kinase and S1P pathway in the LPS inflammatory response, experiments were geared at whether LPS regulates this pathway and at the kinetics of this regulation in RAW 264.7 macrophage cells. To this end RAW 264.7 macrophage cells were initially treated with 500 ng/ml LPS and a time course determination of S1P levels was performed. Figure 1A demonstrates that LPS did not increase cellular S1P levels at 15 or 30 minutes and up to 6 hours of LPS treatment. However, S1P levels were significantly increased to approximately 2−3 fold by 16 hours of LPS treatment and this increase was sustained at 24 hours of treatment. Interestingly no significant increase was detected in secreted S1P in response to LPS (0.28 ± 0.26 pmol/mg cellular protein from control media vs. 0.33 ± 0.35 pmol/mg from media obtained after LPS treatment).
Figure 1. LPS up-regulates S1P and SK1 in RAW 264.7 macrophages.

A) RAW 264.7 cells were treated for the indicated times with 500 ng/ml LPS. At the end of incubation cells were collected and the relative levels of total cellular S1P were determined by MS/MS. Results are representative of three independent experiments performed in duplicate. Error bars represent standard error (SE). B) RAW 264.7 cells were stimulated with 500ng/ml of LPS or PBS for 0 to 32 hours. The data are expressed as mean normalized expression which is directly proportional to the amount of RNA of the target gene, SK1, relative to the amount of RNA of the reference gene, β-actin. Results are representative of three independent experiments performed in duplicate. Error bars represent SE. C) RAW 264.7 cells were transfected with 5 μg SK1 promoter luciferase construct and 1ug β-galactosidase plasmid. After 48h cells were treated with LPS (500 ng/ml) or PBS for the indicated times. Luciferase activity and β-galactosidase activity were measured using the luciferase assay system (Promega). Data were normalized with β-Gal activity (Promega) in each with sample. Results are representative of three independent experiments performed in duplicate. Error bars represent SE. D) Time course of SK1 protein upregulation. RAW 264.7 cells were treated with LPS (500ng/ml) or PBS for 0 to 24 hours. Cells were then harvested for immunoblot analysis of SK1 protein levels. Shown in the blot to the left is the PBS treated cells at 3, 6 and 24 hours followed by the LPS samples for the 3, 6, 16, and 24 hours. Results are representative of three independent experiments.
Next it was important to determine whether the long-term increases in S1P levels in response to LPS were due to up-regulation of SK levels in cells. To this end, cells were treated with LPS and their RNA was extracted at different time points. Figure 1B demonstrates real time PCR results for SK1 in response to LPS treatment, whereby SK1 message increased as early as two hours after LPS treatment and continued to increase peaking at 16 hours of LPS treatment. This increase in message was unique to SK1 as the SK2 message level did not change with LPS treatment (data not shown).
Next to evaluate if the increase in SK1 was at the transcriptional level, a promoter construct with 3124 bp upstream of the SK1 transcription start site (as described in (23)) was sub-cloned in front of the luciferase gene. Transient transfection of this construct into RAW 264.7 cells demonstrates a significant increase in luciferase activity by LPS within 3 hours of treatment, thus indicating that the SK1 response to LPS is at least in part due to transcriptional up-regulation of the SK1 message.
To determine if the increase in SK1 message levels was mirrored by an increase in SK1 protein levels, cells were treated with LPS and subjected to western blot analysis for SK1 protein. Figure 1D demonstrates a significant increase in SK1 protein levels after 6 hours of LPS treatment and this increase is sustained for up to 24 hours of treatment. In addition, measurement of SK activity demonstrated that SK activity in control RAW cells was 0.96±0.38 pmol/min/mg protein, and upon treatment with LPS (300ng/ml) for 24 hours, SK activity increased to 3.53±0.77 pmol/min/mg protein thus indicating that SK activity parallels the increase of SK1 protein and message levels.
S1P activates a pro-inflammatory pathway in RAW 264.7 macrophages
Having previously established a role for S1P and SK1 in activation of the COX-2/PGE2 pro-inflammatory pathway in L929 fibroblasts (17), we set out to determine if S1P was able to activate COX-2 and PGE2 in RAW 264.7 macrophages. To this end RAW 264.7 cells were treated with vehicle control or S1P for the indicated times and doses (Figure 2A top panel and lower panel respectively). Western blot analysis of COX-2 protein levels demonstrates a modest increase in COX-2 levels by S1P after 2 hours of treatment and a significant increase in COX-2 levels at 6 hours of S1P treatment (Figure 2A top panel). Moreover S1P concentrations of 1 μM caused a detectable increase in COX-2 protein levels that continued to increase significantly in response to 5 and 10 μM concentrations of S1P (Figure 2 A bottom panel).
Figure 2. S1P activates a pro-inflammatory pathway in RAW 264.7 macrophages.
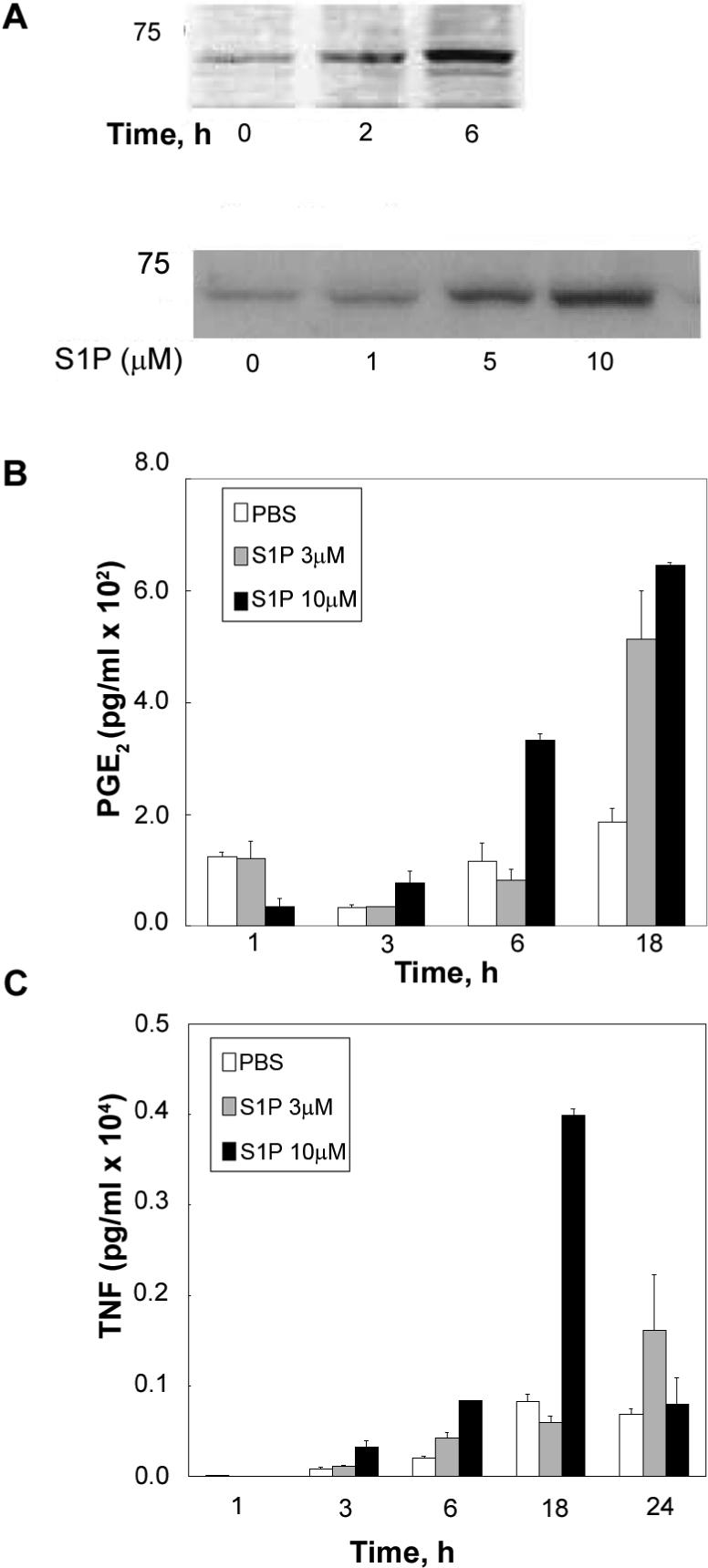
A) Upper panel: RAW 264.7 were treated for 0 to 6h with 1 μM S1P and then the cells were collected and analyzed by western for COX-2 protein levels. Lower panel: RAW 264.7 were incubated for 6.h in the presence or absence of different concentrations of S1P (0−10 μM) and submitted to western blot analysis as described in Experimental Procedures. Results are representative of three independent experiments. B) RAW 264.7 were incubated for up to 18h in the absence or presence of 3 μM or 10 μM S1P. At the end of incubation medium was collected and analyzed for PGE2 levels as described in Experimental Procedures. Results are representative of three independent experiments performed in duplicate. Error bars represent SE. C) RAW 264.7 were incubated for up to 24h in the absence or presence of 3 μM or 10 μM S1P. At the end of incubation medium was collected and analyzed for TNF-α levels as described in Experimental Procedures. Results are representative of three independent experiments performed in duplicate. Error bars represent SE.
The increase in COX-2 protein levels by S1P was mirrored by an increase in PGE2 levels (Figure 2B) such that 3 μM S1P increased PGE2 levels after 18 hours of treatment and 10 μM of S1P caused an increase in PGE2 levels at 6 hours after treatment which was sustained for up to 18 hours of S1P treatment.
Another indicator of the inflammatory response of macrophages is secretion of the pro-inflammatory cytokine TNF-α; therefore, the effect of S1P on TNF-α secretion in RAW 264.7 cells was next determined. Figure 2C demonstrates that TNF-α levels in the media were significantly increased in response to S1P treatment such that 10μM S1P caused a detectable increase in TNF-α secretion as early as 3 hours and a greater than 4 fold increase by 18 hours of S1P treatment.
SK1 is not necessary for the LPS inflammatory response in RAW 264.7 macrophages
Having established that LPS up-regulates SK1 and S1P levels and that S1P can induce a pro-inflammatory response in RAW 264.7 macrophage cells, it was important next to demonstrate if the SK1/S1P pathway was necessary to mediate the LPS inflammatory response. To this end RAW 264.7 cells were transfected with SK1 siRNA to knock down their SK1 levels. Figure 3A demonstrates that SK1 RNAi was effective at knocking down SK1 such that 100 nM of siRNA against SK1 significantly knocked down SK1 message levels by 48 hours of treatment. Next cells were transfected with scrambled or SK1 RNAi for 48 hours; cells were then treated with LPS for the indicated time. Figure 3B demonstrates that as expected LPS induced COX-2 protein levels in Raw 264.7 cells (that were transfected with scrambled RNA) as early as 3 hours and highly significantly by 18 hours of treatment. Interestingly SK1 knockdown did not at all inhibit the COX-2 increase by LPS. Moreover, SK1 knockdown had no effect on the significant PGE2 increases seen with LPS (figure 3C) nor did SK1 knockdown have any affect on TNF-α secretion in response to LPS (figure 3D). These data demonstrate that as expected LPS is a potent activator of the inflammatory response in RAW cells, and that SK1/S1P does not appear to mediate the release of TNF-α and PGE2 that occur in response to LPS.
Figure 3. SK1 is not necessary for the LPS induced-TNF-α or PGE2 release.

A) RAW 264.7 cells were treated with different concentration of scrambled (sc) siRNA or SK1 siRNA (25, 50, 100, 200 nM) for 72 h. At the end of incubation total RNA was extracted and SK1 mRNA levels were measured by Real-time PCR. Mean Normalized Expression is directly proportional to the amount of SK1 mRNA relative to the amount of β-actin mRNA. B) sc siRNA and SK1 siRNA-treated RAW 264.7 (100nM, 48 h) were stimulated with LPS (500 ng/ml) for the indicated time. At the end of incubation cells were collected and analyzed by western blot for COX-2 protein levels. Results are representative of two independent experiments. C) sc siRNA and SK1 siRNA-treated RAW 264.7 (100nM, 48 h) were incubated in the absence or presence of 500ng/ml LPS for different times (3, 6, 16, 24 and 48 h). At the end of the incubation the medium was collected and analyzed for the presence of PGE2 as described in Experimental Procedures. Results are representative of two independent experiments performed in duplicate. Error bars represent SE. D) sc siRNA and SK1 siRNA-treated RAW 264.7 (100nM, 48 h) were incubated in the absence or presence of 500ng/ml LPS for different times (3, 6, 16, 24 and 48 h). At the end of the incubation the medium was collected and analyzed for the presence of TNF-α as described in Experimental Procedures. Results are representative of two independent experiments performed in duplicate. Error bars represent SE.
SK1 is necessary for the TNF-α-induced inflammatory response in RAW 264.7 macrophages
Previous work by Pettus et al., established that SK1 was necessary for TNF-α induction of COX-2 and PGE2 in L929 fibroblasts and A549 lung cancer cells (17). It was therefore important to determine whether SK1 was also necessary for PGE2 induction by TNF-α in macrophages. To this end, RAW 264.7 cells were again transfected with scrambled or SK1 siRNA. Cells were then treated with TNF-α or vehicle control and PGE2 levels were assayed. Figure 4 demonstrates that TNF-α induces significant release of PGE2 as early as three hours, and PGE2 levels continue to accumulate for up to 24 hours. Interestingly, SK1 knockdown almost completely inhibited PGE2 release thus indicating that SK1 indeed mediates the TNF-α inflammatory response in macrophage cells.
Figure 4. SK1 is necessary for TNF-α inflammatory response.
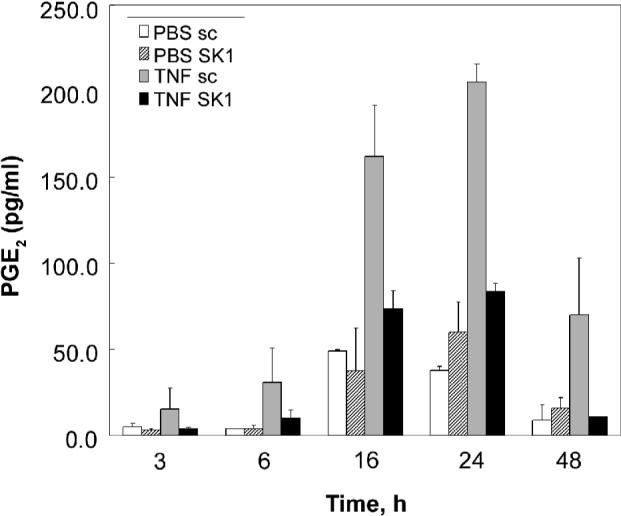
Sc siRNA and SK1 siRNA-treated RAW 264.7 (100nM, 48 h) were incubated in the absence or presence of 3nM TNF-α for different times (3, 6, 16, 24 and 48 h). At the end of the incubation the medium was collected and analyzed for the presence of PGE2 as described in Experimental Procedures. Results are representative of three independent experiments performed in duplicate. Error bars represent SE.
SK1 protects macrophages from LPS- and TNF-induced apoptosis in RAW 264.7 macrophages
To understand the physiologic significance of LPS-induced versus TNF-α- induced SK1 activation in macrophages, the role of SK1 in LPS- and TNF-α-induced apoptosis was evaluated. RAW 264.7 macrophages were transfected with scrambled or SK1 siRNA, and cells were then treated with LPS or TNF-α. Both LPS and TNF-α induced a modest amount of apoptosis by 24 hours in scrambled RNA transfected cells. When SK1 was knocked down, LPS and TNF-α induced a significant increase in apoptosis of RAW264.7 cells (Fig. 5). These data indicate that loss of SK1 significantly sensitizes RAW cells to both LPS and TNF-α-induced apoptosis. These data also imply that SK1 upregulation by LPS is likely protective from apoptosis in response to LPS thus possibly prolonging the LPS inflammatory response in a distinct manner from its role in the TNF-α inflammatory response.
Figure 5. SK1 protects RAW 264.7 macrophages from LPS- and TNF-induced apoptosis.
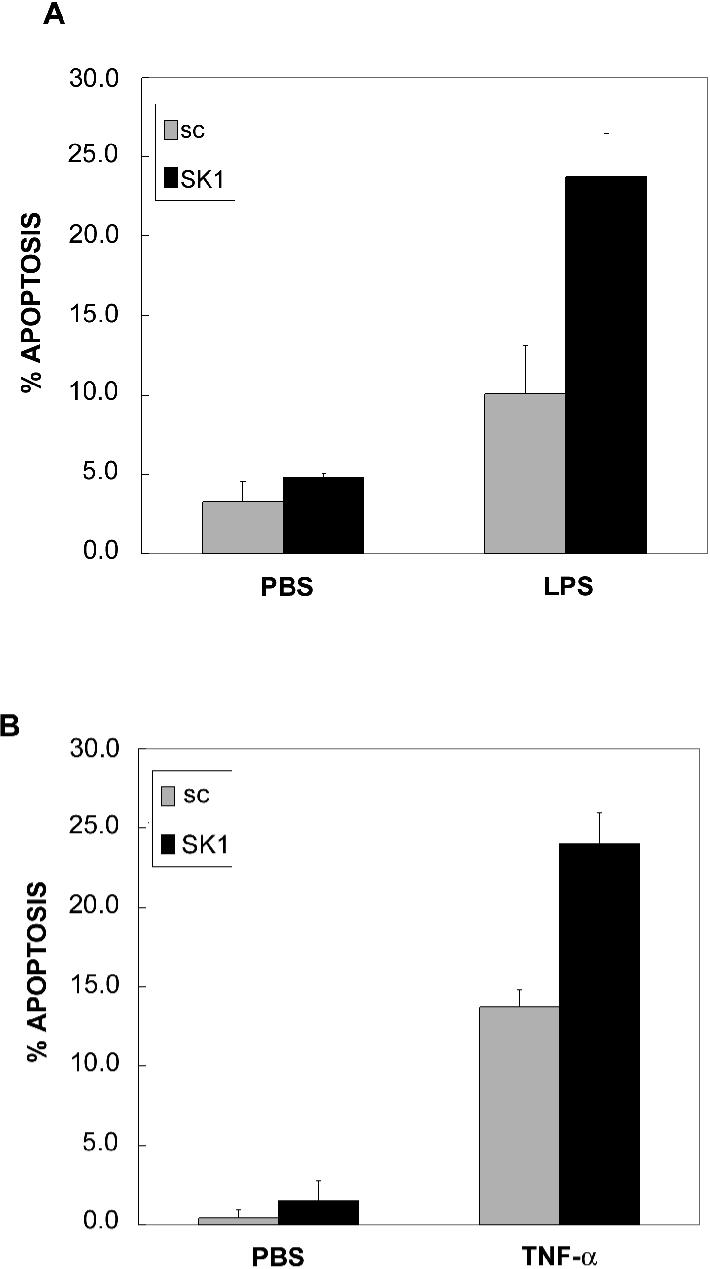
RAW 264.7 cells were treated with 100 nM sc siRNA or SK1 siRNA for 48 hrs and then stimulated with 500 ng/ml LPS (5A) or 3 nM TNFα (5B) for 24 hrs. Apoptotic cell death was measured by Annexin V staining as described in Experimental Procedures. Stained cells were counted and expressed as a fraction of the total cells. Results are representative of two independent experiments performed in duplicate. Error bars represent SE.
DISCUSSION
In this study, the role of S1P as a mediator of innate immune response was investigated using the classical model of LPS stimulation to a mouse macrophage cell line, RAW 264.7. The results showed that LPS activated the SK1/S1P pathway. This was seen first through the upregulation of SK1 message and protein and was followed by a rise in S1P. Next, we showed that S1P alone was sufficient to stimulate the pro-inflammatory pathway, such that, exogenous treatment of S1P resulted in an inflammatory response through the upregulation of COX-2 and the release of TNF-α and PGE2. Interestingly, when cells were treated with LPS following SK1 knockdown there was no change in COX-2/ PGE2 production. However using TNF-α as another model of inflammatory response, SK1 was shown to be necessary for induction of its inflammatory activity. In addition, the SK1/S1P pathway was shown to play a role in protection from apoptosis in both pathways suggesting multiple roles in innate immune signaling.
There are several observations in the literature that suggest a role for sphingolipids in LPS signaling. In 1995, it was suggested that LPS might activate cells via its direct stimulation of the sphingomyelin pathway due to a close structural similarity of the lipid A portion of the LPS molecule and ceramide (25). Furthermore, mice injected with LPS showed an increase in ceramide and sphingomylinase release from the liver and into the blood (26). Another study showed that preventing ceramide formation in monocytes by inhibiting neutral sphingomylinase with C11AG, a selective inhibitor, led to blocking the translocation of NF-kB and the induction of iNOS (27). Since the SK1/S1P pathway has been implicated in NF-kB activation (28) and iNOS induction (29) these studies suggest that previous findings attributed to sphingosine and ceramide on innate stimulation are possibly due to their intracellular metabolism to S1P.
In our studies we showed that LPS upregulates SK1 which leads to cellular S1P generation. Interestingly we did not detect a significant increase in secreted S1P in response to LPS. It is possible that S1P was rapidly metabolized by ectophosphatases. Alternatively, it is possible that intracellular rather than secreted S1P may be mediating LPS responses. Our data suggest that SK1/S1P upregulation provides macrophages with a prosurvival response, whereby, cells treated with LPS following SK1 knockdown showed an increase in apoptosis. This is in agreement with previous work (18) showing that inhibition of SK1 using N,N-dimethylsphingosine or dominant-negative SPK1 sensitized RAW 264.7 cells to LPS-induced apoptosis. It is possible that this LPS-induced upregulation of SK1 is a mechanism to prolong macrophage cell survival during an inflammatory response. This fits with the notion of a typical inflammatory response in vivo, whereby early proinflammatory genes (such as COX-2 and TNF-α) are induced as the macrophage enters the inflammatory site, and late genes (such as IL-10, protease inhibitors, TIMPs and many other products) that are directed toward cleaning up and cell survival (30). SK1 appears to belong to these later genes that enhance macrophage cell survival by LPS.
The cytokine TNF-α is known to be a strong mediator of both apoptotic and inflammatory responses. It is released as a response to stimulation of the innate immune system by LPS. This acts on blood vessels to increase blood flow and vascular permeability and to increase endothelial adhesiveness for leukocytes and platelets thus allowing for an influx of cells into the infected site. TNF-α also regulates the induction of COX-2 and PGE-2. Recent studies by Pettus et al. have implicated the SK1/S1P pathway in this regulation (17). In that study, it was shown that in fibroblasts TNF-α-induced COX-2 activation was inhibited by the down regulation of SK1. This drop in COX-2 production could then be rescued by the addition of S1P. Moreover, S1P levels increased three fold following ten minutes of stimulation by TNFα, and the levels returned to baseline following forty minutes of treatment. Further proof that S1P was important for COX-2 upregulation by TNF-α was demonstrated when RNAi to the S1P lyase and S1P phosphatase showed an increase in S1P followed by an increase in COX-2 (17). In our studies using the macrophage model, when SK1 was knocked down using RNAi, PGE2 levels in response to TNF-α were drastically reduced suggesting that this signaling pathway is also active in macrophage cell lines as well as fibroblasts.
COX-2 and PGE2 signaling in response to LPS and TNF-α are regulated through NF-k β signaling (19). However several studies have suggested other factors such as ERK, p38, AP-1, and PKC may be required (31,32). S1P has been implicated in the activation of NF-kβ, AP-1, and ERK (33,34). There is a biphasic model of NF-kB signaling in the LPS model involving two downstream signaling pathways (35). The first involves the initial upregulation of cytokines including TNF-α. This upregulation is necessary for the subsequent MYD88 dependent NF-kB response necessary for the perpetuation of the signal (35). It is possible that SK1 is not involved in the initial NF-kB response but instead is involved in the later NF-kB perpetuation of cell survival pathways.
In TNF-α signaling, TRAF2 has been shown to be necessary for the activation of SK1 through a TRAF2-specific binding motif (36). TLR4 also recruits a TRAF family member, TRAF6 (37). It is possible that SK1 might be activated by this pathway to activate alternate responses. In this model, later activation by TNF-α could potentially play a role in the perpetuation of inflammation. Thus multiple mechanisms of regulation of SK1 may be involved in perpetuation of an inflammatory response. In this study we have uncovered a novel mechanism of sustained SK1 upregulation by LPS namely at the transcriptional level.
In conclusion, this study shows that the SK1/S1P pathway plays an important role in the innate immune response. Treatment with S1P alone is sufficient to signal the upregulation of the COX-2/PGE2 pathway, a major inflammatory response. This study also suggests that different inflammatory stimuli may be differentially mediated by the SK1/S1P pathway, such that this pathway is necessary for the TNF-α but not the LPS inflammatory response. In addition the SK1/S1P pathway mediates survival of macrophages that are subjected to proinflammatory stimuli suggesting that the SK1/ S1P pathway maybe necessary for the perpetuation of the inflammatory signal. Continued investigation of S1P signaling pathways is necessary to determine the difference in activation between the LPS and TNF-α signaling pathways.
Acknowledgments
We thank Dr. Yusuf Hannun for helpful discussion and Ms. Kathy Wiita Fisk for administrative assistance. We also thank the Lipidomics Core at Medical University of South Carolina. This work was supported in part by the NIH/NIGMS R01 GM062887, NIH/NCI P01 CA097132, and a MERIT Award to LMO by the Office of Research and Development, Department of Veterans Affairs, Ralph H. Johnson VA Medical Center, Charleston, South Carolina.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hannun YA, Obeid LM. Biochemical Society transactions. 1997;25(4):1171–1175. doi: 10.1042/bst0251171. [DOI] [PubMed] [Google Scholar]
- 2.Spiegel S, Merrill AH., Jr. Faseb J. 1996;10(12):1388–1397. doi: 10.1096/fasebj.10.12.8903509. [DOI] [PubMed] [Google Scholar]
- 3.Hla T. Prostaglandins & other lipid mediators. 2001;64(1−4):135–142. doi: 10.1016/s0090-6980(01)00109-5. [DOI] [PubMed] [Google Scholar]
- 4.Pettus BJ, Chalfant CE, Hannun YA. Current molecular medicine. 2004;4(4):405–418. doi: 10.2174/1566524043360573. [DOI] [PubMed] [Google Scholar]
- 5.Spiegel S, Milstien S. Nature reviews. 2003;4(5):397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 6.Billich A, Bornancin F, Mechtcheriakova D, Natt F, Huesken D, Baumruker T. Cellular signalling. 2005;17(10):1203–1217. doi: 10.1016/j.cellsig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Olivera A, Edsall L, Poulton S, Kazlauskas A, Spiegel S. Faseb J. 1999;13(12):1593–1600. doi: 10.1096/fasebj.13.12.1593. [DOI] [PubMed] [Google Scholar]
- 8.Yatomi Y, Igarashi Y, Yang L, Hisano N, Qi R, Asazuma N, Satoh K, Ozaki Y, Kume S. Journal of biochemistry. 1997;121(5):969–973. doi: 10.1093/oxfordjournals.jbchem.a021681. [DOI] [PubMed] [Google Scholar]
- 9.Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. The Journal of experimental medicine. 2004;199(7):959–970. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Cummings R, Usatyuk P, Morris A, Irani K, Natarajan V. The Biochemical journal. 2002;367(Pt 3):751–760. doi: 10.1042/BJ20020586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fueller M, Wang DA, Tigyi G, Siess W. Cellular signalling. 2003;15(4):367–375. doi: 10.1016/s0898-6568(02)00117-1. [DOI] [PubMed] [Google Scholar]
- 12.Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, Brune B. Blood. 2006;108(5):1635–1642. doi: 10.1182/blood-2006-04-014852. [DOI] [PubMed] [Google Scholar]
- 13.Hammad SM, Taha TA, Nareika A, Johnson KR, Lopes-Virella MF, Obeid LM. Prostaglandins & other lipid mediators. 2006;79(1−2):126–140. doi: 10.1016/j.prostaglandins.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Hamada K, Nakamura H, Oda T, Hirano T, Shimizu N, Utiyama H. Biochemical and biophysical research communications. 1998;244(3):745–750. doi: 10.1006/bbrc.1998.8328. [DOI] [PubMed] [Google Scholar]
- 15.Melendez A, Floto RA, Gillooly DJ, Harnett MM, Allen JM. The Journal of biological chemistry. 1998;273(16):9393–9402. doi: 10.1074/jbc.273.16.9393. [DOI] [PubMed] [Google Scholar]
- 16.Melendez AJ, Ibrahim FB. J Immunol. 2004;173(3):1596–1603. doi: 10.4049/jimmunol.173.3.1596. [DOI] [PubMed] [Google Scholar]
- 17.Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, Chalfant CE, Obeid LM, Hannun YA. Faseb J. 2003;17(11):1411–1421. doi: 10.1096/fj.02-1038com. [DOI] [PubMed] [Google Scholar]
- 18.Wu W, Mosteller RD, Broek D. Molecular and cellular biology. 2004;24(17):7359–7369. doi: 10.1128/MCB.24.17.7359-7369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Neill LA. Current opinion in immunology. 2006;18(1):3–9. doi: 10.1016/j.coi.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Hwang D. Faseb J. 2001;15(14):2556–2564. doi: 10.1096/fj.01-0432com. [DOI] [PubMed] [Google Scholar]
- 21.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Methods (San Diego, Calif. 2006;39(2):82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Johnson KR, Becker KP, Facchinetti MM, Hannun YA, Obeid LM. The Journal of biological chemistry. 2002;277(38):35257–35262. doi: 10.1074/jbc.M203033200. [DOI] [PubMed] [Google Scholar]
- 23.Nakade Y, Banno Y, K TK, Hagiwara K, Sobue S, Koda M, Suzuki M, Kojima T, Takagi A, Asano H, Nozawa Y, Murate T. Biochimica et biophysica acta. 2003;1635(2−3):104–116. doi: 10.1016/j.bbalip.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Olivera A, Barlow KD, Spiegel S. Methods in enzymology. 2000;311:215–223. doi: 10.1016/s0076-6879(00)11084-5. [DOI] [PubMed] [Google Scholar]
- 25.Wright SD, Kolesnick RN. Immunology today. 1995;16(6):297–302. doi: 10.1016/0167-5699(95)80185-5. [DOI] [PubMed] [Google Scholar]
- 26.Lightle S, Tosheva R, Lee A, Queen-Baker J, Boyanovsky B, Shedlofsky S, Nikolova-Karakashian M. Archives of biochemistry and biophysics. 2003;419(2):120–128. doi: 10.1016/j.abb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 27.Amtmann E, Baader W, Zoller M. Drugs under experimental and clinical research. 2003;29(1):5–13. [PubMed] [Google Scholar]
- 28.Siehler S, Wang Y, Fan X, Windh RT, Manning DR. The Journal of biological chemistry. 2001;276(52):48733–48739. doi: 10.1074/jbc.M011072200. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Riley RT, Sharma RP. Toxicology. 2007;229(1−2):42–53. doi: 10.1016/j.tox.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni RG, Achaiah G, Sastry GN. Current pharmaceutical design. 2006;12(19):2437–2454. doi: 10.2174/138161206777698945. [DOI] [PubMed] [Google Scholar]
- 31.Sweet MJ, Hume DA. Journal of leukocyte biology. 1996;60(1):8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 32.Cuschieri J, Umanskiy K, Solomkin J. The Journal of surgical research. 2004;121(1):76–83. doi: 10.1016/j.jss.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Hsieh HL, Wu CB, Sun CC, Liao CH, Lau YT, Yang CM. Journal of cellular physiology. 2006;207(3):757–766. doi: 10.1002/jcp.20621. [DOI] [PubMed] [Google Scholar]
- 34.An S, Zheng Y, Bleu T. The Journal of biological chemistry. 2000;275(1):288–296. doi: 10.1074/jbc.275.1.288. [DOI] [PubMed] [Google Scholar]
- 35.Covert MW, Leung TH, Gaston JE, Baltimore D. Science (New York, N.Y. 2005;309(5742):1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 36.Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM, D'Andrea RJ, Gamble JR, Vadas MA. The Journal of biological chemistry. 2002;277(10):7996–8003. doi: 10.1074/jbc.M111423200. [DOI] [PubMed] [Google Scholar]
- 37.Takeda K, Akira S. Seminars in immunology. 2004;16(1):3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]