

Abstract
The time to suppression of HIV-1 viremia to below the limit of detection of standard clinical assays is an important prognostic indicator for patients on highly active antiretroviral therapy (HAART). Recent clinical trials of the integrase inhibitor raltegravir have demonstrated more rapid viral decay than previously seen with reverse transcriptase (RT) or protease inhibitor-based regimens. Because of the therapeutic importance of drugs that target different steps in the virus life cycle, it is imperative to consider whether viral dynamics are affected by the stage of the viral life cycle at which an antiretroviral drug acts. We use a mathematical model to investigate the effects of various drug classes on the dynamics of HIV-1 decay and show that the stage at which a drug acts affects the dynamics of viral decay. We find that the drug class acting latest in the viral life cycle dictates the dynamics of HIV-1 decay. In general, we find that the later in the life cycle an inhibitor acts, the more rapid the decay in viremia, and we illustrate this by comparing the effect of RT and integrase inhibitors on viral dynamics. We conclude that the rapid decay observed in patients on integrase-inhibitor-containing regimens is not necessarily an indication of greater drug efficacy but rather an expected consequence of the fact that this drug acts later in the life cycle. We propose that clinically observed viral decay rates for HAART regimens should be evaluated in the context of the drug classes that are represented.
Keywords: highly active antiretroviral therapy, integrase inhibitor, viral decay, viral dynamics, raltegravir
Early studies of viral dynamics in patients on antiretroviral therapy revealed the decay characteristics of free virus and the turnover of virus-producing cells in HIV-1-infected individuals (1–4). As a result of these and subsequent clinical studies (5, 6), combination antiretroviral therapy quickly became the standard of care for treatment of HIV-1 infection. This therapy, termed highly active antiretroviral therapy (HAART), can reduce viremia to below the clinical limit of detection (<50 HIV-1 RNA copies/ml) in adherent patients (7).
Upon initiation of HAART, viremia decays with at least three phases (1–3, 7). The first phase reflects the decay of productively infected CD4+ T lymphoblasts, whereas the second phase results from the decay of chronically infected cells, possibly macrophages or CD4+ T cells in a lower state of activation (3, 8–11). An extremely slow third phase of decay occurs after the viral load drops below the limit of detection of clinical assays and is believed to be due to release of virus from latently infected resting CD4+ T cells that become activated (7, 9, 12–15) and from some other unidentified source (16).
Over the years, HAART has been refined based on clinical data (17). New classes of antiretroviral drugs, including those acting at different stages of the viral life cycle, are being introduced (18, 19), and it is important to understand the effects that these drugs will have on the decay of viral load. Raltegravir (MK-0518), a promising integrase inhibitor, provides an excellent example (18). Patients taking a raltegravir-based HAART regimen have a faster time to suppression of viremia than patients taking a HAART regimen based on the potent nonnucleoside reverse transcriptase (RT) inhibitor efavirenz (20, 45). Because the rate of viral-load decay is used as a measure of the effectiveness of antiretroviral drugs and drug regimens (21–23), this observation suggests that raltegravir may be an even more effective drug than efavirenz, a drug that is the mainstay of the recommended initial HAART regimen. However, an analysis of viral dynamics in the presence of the different drug classes is required before such a conclusion can be drawn.
Here, we describe a model of HIV-1 dynamics based on previous models (3, 8, 24–27), but which incorporates the effects of drugs acting at different stages in the viral life cycle on the first- and second-phase decay rates of plasma virus. Our model predicts that the stage in the life cycle at which different drugs act (e.g., RT vs. integrase inhibitors) will affect the observed decay rates of viremia independent of drug efficacy. Because viral decay rates have been used to assess the efficacy of HAART regimens (21–23), these results have wide implications.
Results
Development of a Mathematical Model of Viral Dynamics.
To examine whether inhibition at different stages of the viral life cycle results in different decay characteristics of HIV-1 viremia, we construct a mathematical model of host cell and viral dynamics based on well established models (3, 8, 24–27). Our model (Fig. 1) considers two populations of target cells, the CD4+ T lymphoblasts (T), which produce most of the plasma virus (28) and are responsible for first-phase decay, and the cells responsible for second-phase decay. The latter may be macrophages, CD4+ T cells in a lower state of activation or another cell type, but for convenience, they are designated here as M cells. Each of these cell types can be in an uninfected state (TU, MU), an early stage of infection (T1, M1), or a late stage of infection (T2, M2). We do not a priori specify a particular point in the virus life cycle that distinguishes early-stage-infected cells from late-stage-infected cells. Thus, our model and its solutions are general and may be used to compare the viral dynamics caused by any two drugs acting at different stages of the viral life cycle. Below, we first develop the general model, and then we compare different pairs of drugs. For each comparison, we define the boundary between the early and the late stages, such that the drug acting latest in the viral life cycle prevents transition from the early to the late stage. Consequently, we must make parameter choices about the duration of early and late stages in the context of the specific two drugs we are comparing.
Fig. 1.

General schematic of model reflecting uninfected CD4+ T lymphoblasts (TU), early-stage-infected CD4+ T lymphoblasts (T1), late-stage-infected CD4+ T lymphoblasts (T2), uninfected M cells (MU), early-stage-infected M cells (M1), and late-stage-infected M cells (M2). The death rates of these cell are represented by δTU, δT1, δT2, δMU, δM1, and δM2, respectively. kT and kM define the rates at which T1 transition to T2 and M1 transition to M2, respectively. Free virus (V) is cleared at a rate c and produced by T2 with a burst size of NT and by M2 with burst size NM. λT and λM represent CD4+ T lymphoblast and M cell production. βT and βM reflect virus infectivity with respect to CD4+ T lymphoblasts and M cells.
All production of CD4+ T lymphoblasts (e.g., by activation of resting CD4+ T cells and thymic or homeostatic production) is reflected by a zero-order rate constant, λT, and production of M cells is similarly reflected by zero-order constant λM. Free virus infects CD4+ T lymphoblasts to produce early-stage-infected lymphoblasts at a rate controlled by the constant βT, which represents the viral infectivity. Similarly, free virus infects M cells at a rate controlled by βM. Upon infection, target cells become early-stage-infected cells. Early-stage-infected CD4+ T lymphoblasts (T1) transition to late-stage-infected CD4+ T lymphoblasts (T2) at a rate kT. Early-stage-infected M cells (M1) transition to late-stage-infected M cells (M2) at a rate kM. Early- and late-stage-infected CD4+ T lymphoblasts die with rates δT1 and δT2, respectively. We do not make any assumptions about how these cells die [e.g., from cytotoxic T lymphocyte (CTL) response or the cytopathic effects of viral proteins]. We assume only that infected cells die at the specified constant death rates. Other models of HIV dynamics have included an explicit role for CTL killing (27). However, in our analysis, the mechanism of cell death is not critical; only the time scale on which infected cells die matters to our results. Uninfected CD4+ T lymphoblasts can be lost by conversion into nonpermissive resting T cells or by cell death. Resting T cells cannot be productively infected by HIV-1 and are therefore irrelevant for our model. We therefore incorporate all loss of uninfected CD4+ T lymphoblasts under the rate constant δTU. Similarly, we represent loss of second-phase target cells (M cells) with the rate constant δMU and the death rate constants of early- and late-stage-infected M cells as δM1 and δM2, respectively.
Even though we do not specify at this point what the two stages of infection are, we assume that only cells in the late stage (T2, M2) produce free virus (V) with production rates of NT and NM. We believe this assumption is reasonable, because virus production is typically the last stage of the viral life cycle. Nevertheless, the model may easily be changed to accommodate virus production by early-stage-infected cells as well.
Our model is defined by a system of seven ordinary differential equations:
Parameter values were chosen to be consistent with previously reported measurements and are discussed under “Parameter choices” in supporting information (SI) Text.
Decay of Viremia May Be Affected by Which Stages of the Viral Life Cycle Are Inhibited by HAART.
The drugs in a HAART regimen may inhibit viral replication by affecting different stages of the viral life cycle. In our model, defined by Eqs. 1–7, a drug can block either the early-stage infection of target cells or the transition from early- to late-stage infected cells. These two different modes of inhibition are reflected by a reduction in β or k, respectively. In the following, we analyze how the first- and second-phase decay rates of HIV-1 viremia will differ for drugs that reduce either β or k.
To obtain explicit solutions for the dynamics of infected cells and free virus after a drug is given (i.e., a complete block in at least one critical process of the viral life cycle), we make several simplifying assumptions. First, because first-phase decay is governed by decay of virus-producing infected CD4+ T lymphoblasts (T2 cells in our model) and insignificantly affected by chronically infected cells (8, 24), we ignore infected M cells for analysis of the first-phase decay rate. Because free virions have a high turnover rate (3, 24, 29) and are made by late-stage infected CD4+ T lymphoblasts (T2), their dynamics closely resemble those of T2 cells. Inclusion of the decay of free virions results in a slight “shoulder” in the decay curve for plasma virus, lasting only for ≈1/c or ≈1 h (3, 8, 24, 30). After that time, the decay of plasma virus parallels the decay of T2 cells. Therefore, we also remove the explicit representation of free virions from our model and instead represent them with T2 cells. Thus, the rate of infection is represented as a nonlinear term (βTUT2) proportional to both the target (uninfected) CD4+ T lymphoblasts and the late-stage-infected CD4+ T lymphoblasts (31). Second, we assume that TU changes slowly compared with T1 or T2 in the setting of HAART (8). Therefore, we can substitute the pretherapy steady-state value of target cells (T̄U) for TU in Eqs. 1 and 2 (substitution of the posttherapy value of T̄U does not change our calculations). These simplifications allow us to reduce our model to a system of two linear ordinary differential equations:
and
With the initial conditions that T1 and T2 are at their pretherapy steady-state levels (T̄1 and T̄2, respectively), and under the assumption that either kT or βT or both are zero, we find that
 |
where A = δT2 − δT1 − kT and . Eq. 8 describes the dynamics of the late-stage-infected CD4+ T cells at time t after one or more drugs have completely blocked at least one critical process of the viral life cycle (i.e., 100% drug efficacy at kT, βT, or both). The drug-specific solutions are obtained by setting kT, βT, or both equal to zero, depending on the drug or drugs that are given. Although both V and T2 are rapidly changing, the time scale on which T2 decays is much slower than the time scale on which V decays. Therefore, T2 is in a quasisteady state relative to V during the first-phase decay of viremia, and we can approximate that V(t) = NTT2(t)/c. Implicit in our calculations is the assumption that the death rate of infected cells is not affected by any variation in CTL number, which may occur after initiation of HAART (32). Whether CTL number varies significantly over the time scale that we are concerned with (i.e., 4 weeks) remains an open question (32).
To understand how inhibition of viral replication by drugs acting at kT vs. βT affects the decay of viremia, we assume complete suppression of viral replication by the drugs. By setting Eq. 3 equal to zero, we see that the pretherapy steady-state values of T1 and T2, T̄1 and T̄2 respectively, are related by the expression T̄1 =(δT2/kT)T̄2. We consider three cases for V(t) = NTT2(t)/c:
- Inhibition of viral replication by blocking the transition from T1 to T2. In this case, kT = 0. Thus, V(t) is reduced to
In this case, viremia decays according to a strict exponential decay with a rate of δT2.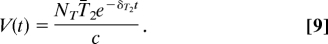
- Inhibition of viral replication by blocking new infection events. In this case, βT. Here, V(t) is reduced to:
In this case, viremia asymptotically decays as an exponential decay with a decay rate equal to the smaller of δT2 or δT1 after an initial shoulder phase that lasts for a period of 1 over the greater of δT2 or (δT1 + kT).
- Inhibition of viral replication by two drugs, one of which stops new infection events and the other of which blocks the transition of from T1 to T2. In this situation, βT = 0 and kT = 0. V(t) is reduced to Eq. 9:
Clearly, the equations for cases 1 and 2 are different. In case 1, which represents inhibition at an intermediate or late stage in the virus life cycle, the first-phase decay of viremia is governed entirely by the death rate of T2. This is a reasonable finding, because, without the further generation of virus-producing cells, the first-phase decay of the viremia should reflect the decay of the remaining virus-producing cells. In case 2, which represents inhibition of early stages of infection, the first-phase decay of viremia reflects the decay of virus-producing cells and the decay of early-stage-infected CD4+ T lymphoblasts and the time it takes for these cells to become virus-producing cells, as reflected by the rate constant kT. The precise behavior of the first-phase decay of viremia, as reflected by Eq. 8, depends on the relative magnitudes of δT1, δT2, and kT, although the decay rate asymptotically reaches the smaller of (δT1 + kT) or δT2. Based on the behavior described in cases 1 and 2, there are two possible scenarios that may occur depending on the relative magnitudes of δT1, δT2, and kT (Table 1). If the decay rate of viremia increases when inhibition occurs at kT compared with βT, then that suggests δT2 > (δT1 + kT). However, if the decay rate does not change but instead the decay curve is shifted left (i.e., because of elimination of the “shoulder” phase) when inhibition occurs at kT compared with βT, then that suggests δT2 < (δT1 + kT). Finally, case 3 demonstrates that the decay kinetics are determined solely by the latest stage in the viral life cycle that is inhibited during treatment with combinations of early- and late-stage inhibitors.

Table 1.
Effect of inhibition at k compared with β
| δ2 > δ1 + k | δ1 + k > δ2 | |
|---|---|---|
| Decay rate | Increases | No change |
| “Shoulder” length | Decreases | Decreases |
To study second-phase decay dynamics, we make similar assumptions. During the second phase, the majority of virus is produced by infected M cells. Therefore, we can ignore virus production by infected CD4+ T lymphoblasts. Furthermore, because free virions have a high turnover rate and are made by late-stage-infected M cells during the second-phase decay of viremia, their dynamics closely resemble those of late-stage infected M cells. Therefore, we again remove the explicit representation of free virions and instead represent them with M2 cells. Thus, the rate of infection is represented as a nonlinear term, βMMUM2. Finally, we assume that MU changes slowly compared with M1 or M2 in the setting of drug therapy, and so we can substitute the pretherapy steady-state value of target cells (M̄U) for MU in Eqs. 4 and 5 (substitution of the post-HAART value of M̄U does not change our calculations). These simplifying assumptions allow us to reduce our model to a two-equation system:
 |
from which we can show (similar to our analysis above) that the decay of free virus released by M cells may be expressed as
 |
for kM = 0 and βM = 0, respectively. Eqs. 11 and 12 have behaviors corresponding to cases 1–3 and Table 1 described above.
Effects of an Integrase Inhibitor on the First- and Second-Phase Decay.
Recent clinical trials of the integrase inhibitor raltegravir have demonstrated very promising results, with patients on a raltegravir-based HAART regimen exhibiting a decreased time to suppression of viremia to <50 copies/ml (20, 45). In patients on a raltegravir-based HAART regimen, preliminary reports suggest unique viral dynamics including a slightly faster first-phase decay rate and reduced contribution of the second-phase decay to viremia. We emphasize that the decay dynamics on raltegravir are not yet well understood. However, because the preliminary data from the raltegravir trials are so striking and might suggest a greater efficacy of this class of drugs, it is important to determine whether the unique decay dynamics and the decreased time to suppression of viremia in patients treated with an integrase inhibitor could be a consequence of the stage in the viral life cycle that is affected. The most recent clinical trials have compared raltegravir on the background of the nucleoside/nucleotide analogue RT inhibitors (NRTI) tenofovir and 3TC to efavirenz on the same background (20, 45). In the setting of complete suppression of viral replication by the integrase inhibitor raltegravir, the decay kinetics will be determined by raltegravir (the drug acting latest in the viral life cycle; case 3 above). By assuming complete suppression of viral replication by efavirenz alone or in combination with NRTIs (which act at the same stage in the viral life cycle), we use our model to directly compare HAART regimens where the final inhibited stage of the viral life cycle is at reverse transcription vs. integration of the HIV-1 genome.
Because integrase inhibitors act at a later point in the viral cycle than do RT inhibitors, we distinguish early- and late-stage-infected cells by integration of HIV-1 into the host genome. Therefore, for this comparison of RT inhibitors vs. integrase inhibitors, we assume that T1 represents infected CD4+ T lymphoblasts before integration, whereas T2 represents infected lymphoblasts after integration. In this context, kT represents the average rate at which infected lymphoblasts complete integration. Furthermore, δT1 represents the death rate of infected lymphoblasts before integration, and δT2 represents the death rate of infected lymphoblasts containing an integrated HIV-1 genome.
From Eqs. 9 and 10, we see that decay depends on the relative values of kT, δT1, and δT2. If we assume that raltegravir blocks viral replication by reducing kT to zero, then viremia will decay according to Eq. 9 as
Therefore, the model predicts that raltegravir would cause viremia to demonstrate a first phase of decay that reflects the death rate of infected CD4+ T lymphoblasts with an integrated HIV-1 genome. As discussed above, RT inhibitors act early in the life cycle, and for this analysis, we can consider they prevent the establishment of infection by reducing β to zero. In the presence of RT inhibitors, viremia will decay according to Eq. 10. However, the behavior of Eq. 10 depends on the relative magnitude of δT2 compared with kT + δT1 (Table 1). Examination of data from recent clinical trials suggests that the first-phase decay rate is slightly faster in the presence of raltegravir compared with efavirenz (45). This observation and our model therefore suggest that δT2 > δT1 + kT for in vivo-infected CD4+ T lymphoblasts (Table 1). Infected lymphoblasts that do not contain integrated HIV-1 DNA have limited ability to express viral genes and are less likely to be susceptible to CTL and viral cytopathic effects, whereas infected lymphoblasts with integrated HIV-1 DNA express viral genes are therefore more susceptible to CTL killing and the cytopathic effects of such viral proteins as gag, vpr, nef, and env, suggesting that δT2 > δT1. Although the process of integration is in general rapid in infected CD4+ T cell lymphoblasts, this process is not efficient in the sense that many integrated proviruses are not replication-competent (33–35). This inefficiency may be reflected in a low value for kT, where kT represents the rate of integration events leading to productively infected cells. Therefore, it is reasonable that δT2 > δT1 + kT, and indeed we can simulate first-phase decay dynamics of viremia that resemble those observed in raltegravir clinical trials using parameters meeting the criterion δT2 > δT1 + kT (Fig. 2A). Although our model also predicts a slight shoulder in the decay of viremia with the RT inhibitor, which is not present with an integrase inhibitor, the time scale of this shoulder (1/δT2) is very short and does not contribute to the observed viral dynamics as much as the actual first-phase exponential decay rate.
Fig. 2.

The decay of virus production upon initiation of HAART in the presence of an integrase inhibitor (red; kT = kM = 0) or RT inhibitor (blue; βT = βM = 0) from late-stage-infected CD4+ T lymphoblasts (A), late-stage-infected M cells (B), and both late-stage-infected CD4+ T lymphoblasts and late-stage-infected M cells (C). Unless otherwise noted, parameters used kT = 0.1 day−1, kM = 0.1 day−1, δTU = 0.02 day−1, δT1 = 0.50 day−1, δT2 = 1.00 day−1, δMU = 0.0495 day−1, δM1 = 0.0495 day−1, δM2 = 0.0495 day−1, λT = 2 × 109 cells per day, λM = 4.95 × 108 cells per day, c = 23 day−1, NT = 1,000 virions per cell per day, NM = 100 virions per cell per day, βT = 8 × 10−12 virions−1·day−1, βM = 8 × 10−15 virions−1·day−1.
The finding that the first-phase decay extends further with a raltegravir-based HAART regimen suggests that raltegravir decreases the contribution of infected M cells to viremia. However, the clinical data show that second-phase rate of decay is not different between a raltegravir-based compared with an efavirenz-based HAART regimen (45). We can use our model to gain some insight into the origin and dynamics of the second-phase decay of viremia.
If the rate of the second-phase decay does not differ when infection is blocked at the integration step vs. the RT step, then from Eqs. 11 and 12, we can conclude that δM1 + kM > δM2 in infected M cells (Table 1). Thus, in the long term, viremia decays exponentially in the presence of RT or integrase inhibitor with rate δM2, which is approximately equal to −ln(2)/14 days) = −0.0495 day−1. This is consistent with idea that the second phase of decay represents chronically infected cells that are less sensitive to the cytopathic effects of the virus (9, 36).
From raltegravir clinical trials and our model predictions, the cellular source of the second-phase decay of viremia must have the property that δM1 + kM > δM2. Several candidates for the cellular source of the second-phase decay have been proposed. The two primary candidates are infected macrophages and resting CD4+ T cells in a state of preintegration latency. In HIV-1-infected macrophages, which decay slowly and are resistant to the cytopathic effects of HIV-1 gene expression (i.e., small δM2), the time from infection to integration and subsequent HIV-1 gene expression (≈1/kM) is on the order of 3 days (37, 38), corresponding to kM = 1/3 day−1. Therefore, macrophages meet our inferred criteria that δM1 + kM > δM2 for the cellular source of the second-phase decay (Fig. 2B). In any event, the cellular source of the second-phase decay of viremia has a reduced contribution to viremia in the presence of an integrase inhibitor compared with an RT inhibitor, because no virus is ever produced by the subset of infected cells that are beyond reverse transcription but before integration (M1 cells). In the presence of an RT inhibitor, these cells would have produced virus and therefore delayed the decay of viremia on the order of 1/(δM1 + kM) before reaching an exponential decay. In contrast, infected resting CD4+ T cells with unintegrated HIV-1 DNA produce virus only after activation and then would be expected to have a rapid decay rate (39). From our previously reported work on the kinetic properties of these cells (39), it is clear that δM2 > δM2 + kM for resting CD4+ T cells in the state of preintegration latency. If the second-phase decay originated from resting CD4+ T cells in a state of preintegration latency, we would have expected that patients taking the integrase inhibitor raltegravir would exhibit a faster second-phase viral decay rate than patients on efavirenz (Table 1). Finally, although our analysis suggests that infected macrophages demonstrate kinetics of infection that are consistent with experimental observations of the second-phase decay, we emphasize that our analysis cannot be used to establish that macrophages produce the second-phase decay of viremia.
Combining our findings for the first- and second-phase decay dynamics with parameter choices that we inferred from previously observed viral dynamics, we find viral dynamics that resemble those observed in integrase inhibitor trials (Fig. 2C). From case 3 described above, these result extends to any HAART regimen composed of RT and/or protease inhibitors compared with one that is based on an integrase inhibitor. These simulations demonstrate that the clinically observed viral dynamics and decreased time to suppression of viremia to <50 copies/ml upon treatment with a raltegravir- vs. efavirenz-based HAART regimen may be explained by the faster average death rate of infected CD4+ T lymphoblasts that have progressed beyond HIV-1 integration compared with the death rate of infected CD4+ T lymphoblasts that have progressed beyond reverse transcription along with raltegravir's ability to eliminate the “shoulder” phase delay of infected M cells. More importantly, the results of our simulations demonstrate that antiretroviral drugs acting at different stages of the life cycle (e.g., RT inhibitor compared with integrase inhibitor) may cause different decay rates of viremia without any difference in ability to suppress viral replication.
Discussion
Mathematical modeling of HIV-1 viral dynamics has offered many insights into the pathogenesis and treatment of HIV-1 (1, 2, 8, 14, 24, 25, 27, 30, 40–43). In addition to providing a theoretical basis for clinical observations of steady-state viremia, these studies have also provided a framework for evaluation of the performance of HAART regimens. In particular, modeling has been useful in explaining the three observed phases of decay of viremia after the initiation of HAART. Because mathematical models suggest that the rates of decay may be faster in the presence of greater suppression of viral replication, drug regimens that maximize these decay rates are sought in clinical trials.
A number of new antiretroviral drugs target different stages of the viral life cycle. In addition to RT and protease inhibitors, entry inhibitors, fusions inhibitors, integrase inhibitors, and maturation inhibitors have been investigated in the last few years (44). As new drugs go through the clinical trial process, it is important to understand whether an observed difference in the decay dynamics between drugs acting at different stages of the viral life cycle is due to differences in drug efficacy. In this article, we present a mathematical model that reflects different stages of the viral life cycle in infected CD4+ T lymphoblasts and infected virus producing M cells. We explicitly model two stages in the viral life cycle that are not specified a priori but are determined by the drug comparison that will be performed. Previous work has demonstrated how the presence of multiple discrete stages in the viral life cycle within infected cells can lead to more complex viral dynamics than a simple exponential decay (25). Our results support these findings, in that our simulations also show how decay of infected cells may cause a shoulder phase in the decay of viremia. Our model incorporates only one early and one late stage of the viral life cycle in infected cells, although these stages could be further divided into arbitrarily more stages reflecting different processes in the viral life cycle. For short-lived infected CD4+ T cells, models incorporating additional stages of the viral life cycle, such as a virus-producing stage (27), can be reduced to a two-stage model because of the short time scales of the stages not immediately relevant to our analysis. In general, addition of more stages of the viral life cycle to the model complicates the predicted viral dynamics but does not change the results, namely that only stages in the viral life cycle after the latest-acting drug affect the decay of viremia, that the asymptotic exponential decay is determined by the slowest-decaying of these stages, and that the shape of the shoulder phase before the asymptotic decay is influenced by the number and individual decay rates of all these stages (SI Text). Therefore, our results would not qualitatively change with incorporation of more stages of the viral life cycle into the model. However, because our model includes only two stages of the viral life cycle, it is limited in its ability to predict the exact shape of the viral load decay curve, an issue that is common to every model of HIV-1 viral dynamics that does not incorporate multiple stages of the viral life cycle.
With our model, we are able to separately analyze the first- and second-phase decay dynamics of viremia to understand how drugs acting at different stages of the viral life cycle affect these decay dynamics. In doing so, we assume complete suppression of viral replication by the drugs, i.e., each drug has 100% efficacy, regardless of which stage of the viral life cycle is inhibited. Although we do not study the complex viral dynamics that may result from rapid expansion of T lymphoblasts, which compensates for the effects of drugs during incomplete suppression of viral replication (27), we do develop a clear understanding of how drugs acting at different stages of the viral life cycle may differentially affect viral dynamics. Interestingly, we find that the decay rates that characterize the first and second phases of decay of viremia are affected by where in the viral life cycle a drug acts. Our model predicts that the maximum attainable decay rates for the different phases of decay could come from drugs that block viral replication at late phases of the viral life cycle. Using previously reported information on the kinetics of the HIV-1 life cycle in infected CD4+ T lymphoblasts and other cell types such as macrophages, we have shown that the reported differences in the viral decay between raltegravir- and efavirenz-based regimens (20, 45) could easily be due to the stage in the viral life cycle at which each respective drug acts rather than differences in the degree of suppression of viral replication. Although the analysis and interpretation of raltegravir trial data are ongoing, our analysis of the current interpretations of the raltegravir data is a timely reminder that enhanced suppression of viral replication is not the only mechanism through which the time to suppression of viremia to <50 copies/ml may be decreased. In fact, we have explicitly described how the decay dynamics of the different phases of viral decay may change depending on the kinetic properties of infected CD4+ T lymphoblasts and infected M cells.
Based on the results presented above, we predict that future drugs acting at later stages of the HIV-1 life cycle may be capable of producing even faster decay rates than the most potent protease, RT, or integrase inhibitors. In particular, assembly and maturation inhibitors that prevent release of any viral particles from infected cells may cause viremia to decay with a rate equal to the clearance rate of free virus (although maturation inhibitors that allow release of noninfectious viral particles will exhibit the same viral dynamics as protease inhibitors). These decay dynamics will be a consequence of the stage in the viral life cycle at which these drugs act and not drug efficacy.
In the past, the time to suppression of viremia has been identified as an important prognostic indicator for patient performance on HAART (22, 23). Our results show that the stage in the HIV-1 life cycle at which a drug acts may affect the observed decay dynamics. Nonetheless, we do not discount the evidence that observed viral decay rates have prognostic value. Clearly, suppression will take longer in patients taking drugs that produce suboptimal suppression of viral replication. We do, however, propose that the observed viral decay rates must be viewed in the context of the drugs in the HAART regimen. Before the advent of integrase inhibitors, all available antiretroviral drugs acted at relatively early stages in the HIV-1 life cycle, so that viral decay dynamics were not significantly different. In this situation, comparing viral decay rates across all HAART regimens may have been appropriate as an indicator of drug efficacy. However, as drugs that act later in the HIV-1 life cycle are introduced, we predict that each drug class will have a maximum viral decay rate which occurs at 100% suppression of viral replication, and which will correspond to the decay rates of virus-producing cells beyond the block in replication produced by each drug class. Furthermore, the maximum potential viral decay rate of a HAART regimen will be dictated by the drug in the regimen that acts furthest in the HIV-1 life cycle. Therefore, we believe that the clinically observed viral decays produced by HAART must be evaluated in the context of the drugs in the regimen, especially as antiretroviral drugs become increasingly potent, thus likely reducing viral replication to inconsequential levels. These results are also applicable to chemotherapeutic treatment and subsequent decay of other pathogenic agents. Therefore, we believe that the principles proposed in this article should be considered in any clinical setting where the dynamics of a pathogenic agent are used as a marker for treatment efficacy.
Methods
Model Simulations.
All simulations and calculations were performed with Matlab Version 7.2.0.232 (The MathWorks). In particular, simulations were generated by solving Eqs. 1–7 with the standard Matlab ODE suite of programs.
Supplementary Material
Acknowledgments.
This work was supported by National Institutes of Health Grants AI43222 and AI51178, by a grant from the Doris Duke Charitable Foundation, and by the Howard Hughes Medical Institute. C.O.W. was supported by National Institutes of Health Grant AI 065960.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0711372105/DC1.
References
- 1.Ho DD, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 2.Wei X, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 3.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 4.Cavert W, et al. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science. 1997;276:960–964. doi: 10.1126/science.276.5314.960. [DOI] [PubMed] [Google Scholar]
- 5.Gulick RM, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 6.Hammer SM, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 7.Maldarelli F, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007;3:e46. doi: 10.1371/journal.ppat.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonhoeffer S, May RM, Shaw GM, Nowak MA. Virus dynamics and drug therapy. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevenson M. HIV-1 pathogenesis. Nat Med. 2003;9:853–860. doi: 10.1038/nm0703-853. [DOI] [PubMed] [Google Scholar]
- 10.Haase AT. Perils at mucosal front lines for HIV and SIV and their hosts. Nat Rev Immunol. 2005;5:783–792. doi: 10.1038/nri1706. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434:1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 12.Finzi D, Siliciano RF. Viral dynamics in HIV-1 infection. Cell. 1998;93:665–671. doi: 10.1016/s0092-8674(00)81427-0. [DOI] [PubMed] [Google Scholar]
- 13.Dornadula G, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. J Am Med Assoc. 1999;282:1627–1632. doi: 10.1001/jama.282.17.1627. [DOI] [PubMed] [Google Scholar]
- 14.Strain MC, et al. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc Natl Acad Sci USA. 2003;100:4819–4824. doi: 10.1073/pnas.0736332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nettles RE, et al. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. J Am Med Assoc. 2005;293:817–829. doi: 10.1001/jama.293.7.817. [DOI] [PubMed] [Google Scholar]
- 16.Bailey JR, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol. 2006;80:6441–6457. doi: 10.1128/JVI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammer SM, et al. Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society-USA panel. J Am Med Assoc. 2006;296:827–843. [PubMed] [Google Scholar]
- 18.Hazuda DJ, et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science. 2004;305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 19.Kilby JM, et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 20.Markowitz M, et al. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: Results of a 48-week controlled study. J Acquir Immune Defic Syndr. 2007;46:125–133. doi: 10.1097/QAI.0b013e318157131c. [DOI] [PubMed] [Google Scholar]
- 21.Kuritzkes DR, et al. Plasma HIV-1 RNA dynamics in antiretroviral-naive subjects receiving either triple-nucleoside or efavirenz-containing regimens: ACTG A5166s. J Infect Dis. 2007;195:1169–1176. doi: 10.1086/512619. [DOI] [PubMed] [Google Scholar]
- 22.Louie M, et al. Determining the relative efficacy of highly active antiretroviral therapy. J Infect Dis. 2003;187:896–900. doi: 10.1086/368164. [DOI] [PubMed] [Google Scholar]
- 23.Polis MA, et al. Correlation between reduction in plasma HIV-1 RNA concentration 1 week after start of antiretroviral treatment and longer-term efficacy. Lancet. 2001;358:1760–1765. doi: 10.1016/s0140-6736(01)06802-7. [DOI] [PubMed] [Google Scholar]
- 24.Perelson AS, et al. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 25.Lloyd AL. The dependence of viral parameter estimates on the assumed viral life cycle: limitations of studies of viral load data. Proc Biol Sci. 2001;268:847–854. doi: 10.1098/rspb.2000.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dixit NM, Perelson AS. Complex patterns of viral load decay under antiretroviral therapy: influence of pharmacokinetics and intracellular delay. J Theor Biol. 2004;226:95–109. doi: 10.1016/j.jtbi.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Rouzine IM, Sergeev RA, Glushtsov AI. Two types of cytotoxic lymphocyte regulation explain kinetics of immune response to human immunodeficiency virus. Proc Natl Acad Sci USA. 2006;103:666–671. doi: 10.1073/pnas.0510016103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286:1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 29.Mittler JE, Markowitz M, Ho DD, Perelson AS. Improved estimates for HIV-1 clearance rate and intracellular delay. AIDS. 1999;13:1415–1417. doi: 10.1097/00002030-199907300-00023. [DOI] [PubMed] [Google Scholar]
- 30.Wodarz D, Nowak MA. Mathematical models of HIV pathogenesis and treatment. BioEssays. 2002;24:1178–1187. doi: 10.1002/bies.10196. [DOI] [PubMed] [Google Scholar]
- 31.De Boer RJ, Perelson AS. Target cell limited and immune control models of HIV infection: a comparison. J Theor Biol. 1998;190:201–214. doi: 10.1006/jtbi.1997.0548. [DOI] [PubMed] [Google Scholar]
- 32.Ogg GS, et al. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J Virol. 1999;73:797–800. doi: 10.1128/jvi.73.1.797-800.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chun TW, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 34.Embretson J, et al. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- 35.Harper ME, Marselle LM, Gallo RC, Wong-Staal F. Detection of lymphocytes expressing human T-lymphotropic virus type III in lymph nodes and peripheral blood from infected individuals by in situ hybridization. Proc Natl Acad Sci USA. 1986;83:772–776. doi: 10.1073/pnas.83.3.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lum JJ, Badley AD. Resistance to apoptosis: mechanism for the development of HIV reservoirs. Curr HIV Res. 2003;1:261–274. doi: 10.2174/1570162033485203. [DOI] [PubMed] [Google Scholar]
- 37.Schrier RD, et al. T-cell-induced expression of human immunodeficiency virus in macrophages. J Virol. 1990;64:3280–3288. doi: 10.1128/jvi.64.7.3280-3288.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Brien WA. HIV-1 entry and reverse transcription in macrophages. J Leukocyte Biol. 1994;56:273–277. doi: 10.1002/jlb.56.3.273. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Y, Zhang H, Siliciano JD, Siliciano RF. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J Virol. 2005;79:2199–2210. doi: 10.1128/JVI.79.4.2199-2210.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 41.Bonhoeffer S, Nowak MA. Pre-existence and emergence of drug resistance in HIV-1 infection. Proc Biol Sci. 1997;264:631–637. doi: 10.1098/rspb.1997.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perelson AS. Modelling viral and immune system dynamics. Nat Rev Immunol. 2002;2:28–36. doi: 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- 43.Rouzine IM, Coffin JM. Evolution of human immunodeficiency virus under selection and weak recombination. Genetics. 2005;170:7–18. doi: 10.1534/genetics.104.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reeves JD, Piefer AJ. Emerging drug targets for antiretroviral therapy. Drugs. 2005;65:1747–1766. doi: 10.2165/00003495-200565130-00002. [DOI] [PubMed] [Google Scholar]
- 45.Murray JM, et al. Antiretroviral with the integrase inhibitor raltegravir alteus decay kinetics of HIV, significantly reducing the second phase. AIDS. 2007;21:2315–2321. doi: 10.1097/QAD.0b013e3282f12377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
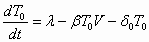{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}