

Abstract
The signal recognition particle (SRP)–dependent targeting pathway facilitates rapid, efficient delivery of the ribosome–nascent chain complex (RNC) to the protein translocation channel. We test whether the SRP receptor (SR) locates a vacant protein translocation channel by interacting with the yeast Sec61 and Ssh1 translocons. Surprisingly, the slow growth and cotranslational translocation defects caused by deletion of the transmembrane (TM) span of yeast SRβ (SRβ-ΔTM) are exaggerated when the SSH1 gene is disrupted. Disruption of the SBH2 gene, which encodes the β subunit of the Ssh1p complex, likewise causes a growth defect when combined with SRβ-ΔTM. Cotranslational translocation defects in the ssh1ΔSRβ-ΔTM mutant are explained by slow and inefficient in vivo gating of translocons by RNCs. A critical function for translocation channel β subunits in the SR–channel interaction is supported by the observation that simultaneous deletion of Sbh1p and Sbh2p causes a defect in the cotranslational targeting pathway that is similar to the translocation defect caused by deletion of either subunit of the SR.
Introduction
The signal recognition particle (SRP)–dependent targeting pathway allows rapid and efficient delivery of the ribosome–nascent chain complex (RNC) to the protein translocation channel. Formation of the SRP–RNC complex occurs when the signal sequence of the nascent polypeptide emerges from the polypeptide exit site on the large ribosomal subunit and is bound by SRP54 (for review see Walter and Johnson, 1994). Targeting of the SRP–RNC to the SRP receptor (SR) initiates the SRP54-SRα GTPase cycle that results in dissociation of SRP54 from the signal sequence and transfer of the RNC to the Sec61 heterotrimer (Rapiejko and Gilmore, 1997). Dissociation of SRP54 from the signal sequence and GTP hydrolysis by SRP54 and SRα are both blocked by the inactivation or absence of the Sec61 complex (Song et al., 2000), suggesting that the SR may locate an unoccupied translocon by interacting with the Sec61 heterotrimer. Dissociation of SRP from the RNC is a prerequisite for RNC attachment to the translocon because the SRP54-binding site on the large ribosomal subunit overlaps the Sec61-binding site (Halic et al., 2004). With the exception of the hypothetical SR–Sec61 heterotrimer interaction, each of these complexes (SRP–RNC, SRP–SR, SRP–SR–RNC, and RNC–translocon) has been characterized at the biochemical and structural levels (Beckmann et al., 2001; Morgan et al., 2002; Mandon et al., 2003; Egea et al., 2004; Focia et al., 2004; Halic et al., 2006).
The yeast Saccharomyces cerevisiae has distinct cotranslational and posttranslational protein translocation channels. The Sec61 heterotrimer (Sec61p, Sbh1p, and Sss1p) is a cotranslational protein translocation channel that also assembles with the Sec62–Sec63 complex to form the Sec complex (Deshaies et al., 1991; Panzner et al., 1995). The Sec complex lacks ribosome-binding activity and serves as the posttranslational translocation channel (Panzner et al., 1995). The nonessential Ssh1p heterotrimer (Ssh1p, Sbh2p, and Sss1p) does not assemble with the Sec62–Sec63 complex (Finke et al., 1996) but is instead a dedicated cotranslational translocation channel (Prinz et al., 2000; Wittke et al., 2002).
Unlike the eukaryotic SRα–SRβ heterodimer that is anchored to the membrane by the N-terminal transmembrane (TM) span of SRβ, the Escherichia coli SRα homologue (FtsY) is present in both membrane-bound and soluble forms in vivo (de Leeuw et al., 1997). Membrane-bound FtsY interacts with phospholipids and can also bind to the E. coli translocation channel (SecYEG; Angelini et al., 2005) to form a carbonate-resistant complex that is stabilized by blocking the GTPase activity of FtsY (Angelini et al., 2006). A different mode of interaction has been observed between the GTPase domain of yeast SRβ and the yeast translocon β subunits (Sbh1p or Sbh2p). The cytosolic domains of Sbh1p or Sbh2p stimulate the dissociation of GDP from yeast SRβ (Helmers et al., 2003). As GTP binding to SRβ is required for SRα–SRβ heterodimerization (Legate et al., 2000; Schwartz and Blobel, 2003), the proposed guanine nucleotide exchange factor (GEF) activity of the translocon β subunits would occur before the hypothesized interaction between the SR and a protein translocation channel.
In yeast, both SRα and SRβ are necessary for the cotranslational targeting pathway (Ogg et al., 1992, 1998). The observation that the GTPase domain but not the TM span of yeast SRβ is required for SR function (Ogg et al., 1998) is explained by the GTP-dependent interaction between SRα and SRβ (Legate et al., 2000; Schwartz and Blobel, 2003) and the presence of an uncharacterized binding site on the ER membrane for the soluble SR (SRα + SRβ-ΔTM). Our analysis of yeast strains that express the soluble SR indicates that the Ssh1 heterotrimer is critical for the SRP-targeting pathway, presumably as a result of a higher affinity between the Ssh1 complex and the soluble SR. Analysis of yeast strains that display synthetic slow growth phenotypes indicate that translocon β subunits are essential for the rapid and efficient gating of the translocon by RNCs that are targeted to the membrane by the SRP-dependent pathway.
Results
Genetic interactions between the soluble SR and the protein translocation channels
A haploid yeast strain (YJY101) was constructed to test the hypothesis that an interaction between the SR and the translocation channel is important for the cotranslational translocation pathway. In YJY101, chromosomal disruptions of the SEC61 and SRP102 genes are covered by a URA3-marked plasmid encoding both Sec61p and SRβ (encoded by SRP102). Disruption of the SSH1 gene in YJY101 yielded an additional starting strain (YJY102) that lacks the Ssh1p translocon. A plasmid shuffle procedure was used to replace the URA3-marked SEC61-SRP102 plasmid with pairs of plasmids encoding intact or soluble (SRβ-ΔTM) forms of SRβ and wild-type or mutant alleles of Sec61p and Ssh1p.
Yeast strains that lack the Ssh1 translocon have a minor growth defect on rich (YPD) media (Fig. 1 A; Cheng et al., 2005). Deletion of the TM domain of SRβ causes a growth rate defect at 30°C (Ogg et al., 1998), which is accentuated at 37°C (Fig. 1 A). Combining the ssh1Δ and SRβ-ΔTM mutations caused a severe synthetic growth defect at 30 and 37°C. SEC61 is an essential gene, so we tested whether point mutations in Sec61p (Cheng et al., 2005) cause a synthetic growth defect in the SRβ-ΔTM strain. The sec61R275E R406E mutant (sec61EE) was chosen because these two charge-reversal substitutions in cytoplasmic loops 6 and 8 selectively interfere with the cotranslational translocation pathway. Expression of Ssh1p suppresses the slow growth and defective translocation phenotypes of the sec61EE mutant (Cheng et al., 2005). The growth rate defect caused by the SRβ-ΔTM mutation is not accentuated by the sec61EE mutation but is instead partially suppressed (Fig. 1 A). Sequence conservation between Sec61p and Ssh1p allowed construction of the corresponding ssh1EE mutant (ssh1R278E R411E). Although the ssh1EE mutant grows at a wild-type rate when intact SRβ is present (not depicted), the ssh1EE SRβ-ΔTM mutant grows very slowly at 30°C and is inviable at 37°C (Fig. 1 A). Synthetic genetic phenotypes can be explained by inactivating lesions in parallel pathways or by partial impairment of interacting components within a linear or branching pathway (Huffaker et al., 1987). The synthetic interactions observed here are examples of the latter case in which a partially defective SR interacts weakly with the Sec61 heterotrimer and interacts unproductively with the defective ssh1EE heterotrimer.
Figure 1.

Genetic interactions between the SR and the translocons. (A) Haploid yeast strains expressing wild-type or soluble (ΔTM) forms of SRβ and wild-type or mutant alleles of Sec61 (sec61EE) or Ssh1 (ssh1EE) from plasmids as the sole source of each protein were grown to mid-log phase in SEG media at 30°C. Growth rates were compared by serial dilution analysis on YPD plates at both 30 and 37°C as described in Materials and methods. Plates were photographed after 2 (30°C) or 3 d (37°C) of growth. (B) Yeast strains (SSH1 or ssh1Δ) expressing wild-type Sec61p and SRβ-ΔTM from low-copy plasmids were transformed with high-copy plasmids (2μ) encoding SRβ-ΔTM, soluble SR (SRα + SRβ-ΔTM), or the Ssh1p complex (SSH1*; Ssh1p + Sbh2p + Sss1p). Growth rates of yeast strains on YPD media were compared as described in A. (C) Growth rates of selected yeast strains in liquid YPD media at 30°C. Plotted values are derived from a single experiment. Error bars represent SD.
If the soluble SR interacts with the Ssh1 translocon, increased expression of either the Ssh1 heterotrimer or the soluble SR (SRα + SRβ-ΔTM) should suppress the growth defect of the SRβ-ΔTM mutant by favoring the more rapid formation of SR–Ssh1 complexes. Because the Ssh1 heterotrimer shares one subunit (Sss1p) with the Sec61 complex, we expressed all three subunits of the Ssh1p complex (Ssh1p, Sbh2p, and Sss1p) from a single high-copy plasmid (Fig. 1 B, 2μ[SSH1*]). Overexpression of the Ssh1 complex reduces the growth defect of the SRβ-ΔTM mutant at 30°C and suppresses the temperature-sensitive phenotype. The 6-h cell division time for the SRβ-ΔTM mutant in liquid media at 30°C is reduced to 4 h when the Ssh1 complex is overexpressed (Fig. 1 C). Increasing the cellular content of the soluble SR (SRα + SRβ-ΔTM) also alleviates the slow growth phenotype (Fig. 1 B) and reduces the cell division time to 3.5 h in liquid media (Fig. 1 C). Overexpression of the soluble SR is ineffective in the ssh1Δ mutant, supporting the hypothesis that the soluble SR preferentially interacts with the Ssh1p translocon. Overexpression of SRβ-ΔTM alone (Fig. 1 B) did not increase the growth rate, indicating that the soluble SR, not just SRβ, is the growth rate–limiting factor. Instead, the overexpression of SRβ-ΔTM appears to be toxic in the absence but not in the presence of Ssh1p.
The slow cell division cycle of the ssh1Δ SRβ-ΔTM mutant in liquid media (>8 h/generation) is comparable with the growth rates of yeast strains that lack SRα, SRβ, or SRP54 (Hann and Walter, 1991; Ogg et al., 1992, 1998). The uniform pink to red colony color of the ssh1Δ and SRβ-ΔTM strains (Fig. 1, A and B) demonstrates that the single mutants are not petite (ρ−), unlike the SRα, SRβ, or Srp54 deletion mutants. However, the ssh1Δ SRβ-ΔTM mutant loses respiration competence with a 10-fold increased frequency (∼3%/generation; unpublished data) relative to a wild-type strain when grown on synthetic defined media containing dextrose as a carbon source (SD media). To prevent the accumulation of ρ− mutants, all yeast strains were maintained on synthetic minimal media containing ethanol and glycerol (SEG media).
Translocation defects of strains that express the soluble SR
Yeast strains were shifted from SEG media into SD media and grown for 4 or 24 h before being pulse labeled for 7 min with 35S amino acids. The vacuolar membrane protein dipeptidylaminopeptidase B (DPAPB) was used as a reporter because DPAPB RNCs are targeted to the translocon by the SRP-dependent pathway (Ng et al., 1996). Integration of DPAPB into the ER was detected by the reduced gel mobility caused by N-linked glycosylation. DPAPB integration was very efficient in wild-type and ssh1Δ cells after 4 h (Fig. 2 A) or 24 h (Fig. 2 B) of growth in SD media. Two important differences were observed when DPAPB integration was analyzed in cells that express the soluble SR. Less than 50% of the DPAPB products are integrated into the membrane in the SRβ-ΔTM mutant (Fig. 2 A). The translocation defect of the SRβ-ΔTM strain was enhanced in cells that lack the Ssh1 complex. We also observed a three- to fivefold reduced incorporation of radiolabel into DPAPB precursor (pDPAPB) and DPAPB after 4 h of growth in SD media (Fig. 2 A). The reduced incorporation of radiolabel is not specific for DPAPB or SRP-dependent translocation substrates but is instead indicative of a global reduction in the protein synthesis rate (unpublished data).
Figure 2.

Cotranslational translocation defects of yeast that express SRβ-ΔTM. (A–D) Wild-type (wt) and mutant yeast cells were grown to mid-log phase at 30°C in SEG media. The cultures were diluted into SD media and allowed to grow for 4 h (A, C, and D) or 24 h (B) at 30°C. Wild-type or mutant cells (4 A600) were collected and pulse labeled for 7 min. DPAPB immunoprecipitates was resolved by SDS-PAGE. Glycosylated (DPAPB) and nonglycosylated (pDPAPB) forms of DPAPB were quantified with a molecular imager to calculate the percent precursor. Plasmids encoding the soluble SR (SRα + SRβ-ΔTM) and the Ssh1 complex are designated as SR-ΔTM and SSH1*, respectively. (A and B) Total incorporation of Tran-35S-label (pDPAPB + DPAPB) in each strain is expressed relative to the wild-type strain.
Translocation defects were not observed previously in the SRβ-ΔTM mutant (Ogg et al., 1998). This discrepancy is primarily explained by an adaptation process that occurs upon continued growth of the SRβ-ΔTM strain in SD or YPD media. After 24 h, incorporation of 35S amino acids into DPAPB was usually proportional to the growth rate of the strain in liquid media, and pDPAPB accumulation had decreased several fold (Fig. 2 B). Adaptation of yeast to lesions in the SRP-targeting pathway has been observed previously (Hann and Walter, 1991) and occurs by enhanced expression of cytoplasmic chaperones and by decreased expression of gene products required for protein synthesis (Mutka and Walter, 2001). Growth rate differences observed on YPD plates (Fig. 1) reflect the postadapted state, as these images were recorded after 2–3 d of culture.
Additional pulse-labeling experiments were conducted after 4 h of culture in SD media to investigate how increased expression of the Ssh1 complex (Fig. 2 C) or the soluble SR (Fig. 2 D) impacts the cotranslational translocation pathway. The nontranslocated precursor in the ssh1Δ SRβ-ΔTM mutant is reduced in a dosage-dependent manner by expression of the Ssh1 complex from low- and high-copy plasmids (Fig. 2 C). Increased expression of the soluble SR heterodimer but not SRβ-ΔTM alone increases DPAPB integration provided that cells express the Ssh1 complex (Fig. 2 D).
In vivo kinetics of DPAPB integration
The in vivo kinetics of DPAPB integration can be analyzed using the Dap2 series of ubiquitin (Ub) translocation assay (UTA) reporters (Cheng and Gilmore, 2006). The Dap2 reporters consist of the N-terminal cytosolic and TM domains of DPAPB followed by a variable length spacer segment (49–265 residues) derived from the lumenal domain of DPAPB, Ub, a cleavage site for a Ub-specific protease, and HA epitope–tagged Ura3p (Fig. 3 A). Rapid folding of the Ub domain in the cytosol allows cleavage by Ub-specific proteases and release of the cleaved (Ura3-HA) reporter segment (Fig. 3 B). However, if Dap2-RNCs gate the translocon before the entire Ub domain emerges from the large ribosomal subunit, the intact reporter will be integrated into the ER. Mutations in SRP54 (Johnsson and Varshavsky, 1994) or Sec61α (Cheng and Gilmore, 2006) enhance UTA reporter cleavage by retarding reaction steps that precede translocon gating (Fig. 3 B). For example, the SRP–SR-dependent targeting pathway can be acutely blocked by shifting the temperature-sensitive SRβ mutant (srp102 K51I; Ogg et al., 1998) from the permissive to restrictive temperature. More than 95% of the Dap2–265 reporter was cleaved at the restrictive temperature (Fig. 3 D, open diamond). UTA assays were conducted after 4 h of cell growth in SD media (Fig. 3 C). The intact glycosylated reporters (e.g., g49) as well as the cleaved Ura3-HA domain were recovered by immunoprecipitation with anti-HA monoclonal antibody. In wild-type cells, Dap2 cleavage decreases as the spacer length is increased (Fig. 3, C [left] and D [squares]) from 49 to 103 residues, indicating that most Dap2-RNCs gate the translocon after 170 but before 224 residues of the reporter emerge from the large ribosomal subunit (N-terminal 45 residues + 103-residue spacer + 76-residue Ub domain = 224 residues). Further increases in spacer length have little impact on Dap2 reporter cleavage in wild-type cells (Fig. 3 D, squares). The difference between the plateau value for Dap2 reporter cleavage and the pDPAPB detected in a pulse-labeling experiment (Fig. 2 A) indicates that roughly 20% of nascent DPAPB is integrated by a posttranslational pathway in a wild-type cell.
Figure 3.

Slow targeting of Dap2-RNCs in the ssh1Δ SRβ-ΔTM mutant. (A) Dap2 reporters consist of (1) the N-terminal cytoplasmic domain of Dap2p (green; Dap2p1–29) followed by the TM span (black; Dap2p30–45), (2) 49–265-residue spacer segments (cyan) derived from Dap2p, (3) a Ub domain (red), (4) a 42-residue linker (blue) with a processing site (arrowhead) for a Ub-specific protease, and (5) a Ura3 reporter domain followed by a triple-HA epitope tag (yellow). Sites for N-linked glycosylation (Y-shaped symbols) are indicated. (B) Cleavage of the Dap2 reporter defines the in vivo kinetics of translocon gating in the SRβ-ΔTM strain. Soluble (dark blue) and membrane-bound (light blue) pools of the SR might interact with the SRP–RNC or the translocon. Any slow step (e.g., SRP binding, SRP–RNC targeting, and RNC docking or signal insertion) that precedes translocon gating allows folding of Ub in the cytosol and cleavage of the reporter. The color code for Dap2 segments is as defined in A. (C) In vivo cleavage of the Dap2 reporter in wild-type, SRβ-ΔTM, or ssh1Δ SRβ-ΔTM strains after 4 h of growth in SD media. Labels designate the intact glycosylated (e.g., g49) and cleaved (Ura3-HA) reporter domains. Downward-pointing arrowheads designate nonglycosylated intact reporters that are diagnostic of cytosolic Dap2 reporter aggregates (Cheng et al., 2005; Cheng and Gilmore, 2006). (D) Spacer length dependence of Dap2 reporter cleavage (percent cytosolic Ura3-HA) for the wild-type (squares), ssh1Δ (circles), SRβ-ΔTM (triangles), and ssh1Δ SRβ-ΔTM (closed diamonds) yeast strains after 4 h of growth in SD media. Dap2–265 cleavage was assayed 3 h after shifting the SRβ mutant (srp102K51I) to 37°C (open diamond). (E) In vivo cleavage of the Dap2 reporters in the absence or presence of cycloheximide (CH) after 24 h of growth in SD media. Molecular weight markers for C and E are identical. (F) Spacer length dependence of Dap2 reporter cleavage (percent cytosolic Ura3-HA) for the ssh1Δ (circles), SRβ-ΔTM (triangles), or ssh1Δ SRβ-ΔTM (closed diamonds, −CH; open diamonds, +CH) yeast strains after 24 h of growth in SD media. Data points in D and F are means of two or three experiments. Color-coded error bars in D and F designate individual data points for experiments that were performed twice or SDs for experiments performed three times.
Dap2 reporter cleavage was elevated in the SRβ-ΔTM strain after 4 h of growth in SD media (Fig. 3 C, middle). Quantification revealed a wider translocon gating window and an elevated plateau value (Fig. 3 D, triangles). Elimination of the Ssh1 translocon raised the plateau value for Dap2 cleavage as a result of a reduced membrane content of cotranslational translocons (Fig. 3 D, circles). Analysis of Dap2 reporter cleavage in the double mutant (ssh1Δ SRβ-ΔTM; Fig. 3 C) revealed an extremely slow and inefficient delivery of Dap2-RNCs to the Sec61 complex (Fig. 3 D, diamonds), which is diagnostic of a severe impairment of the cotranslational targeting pathway.
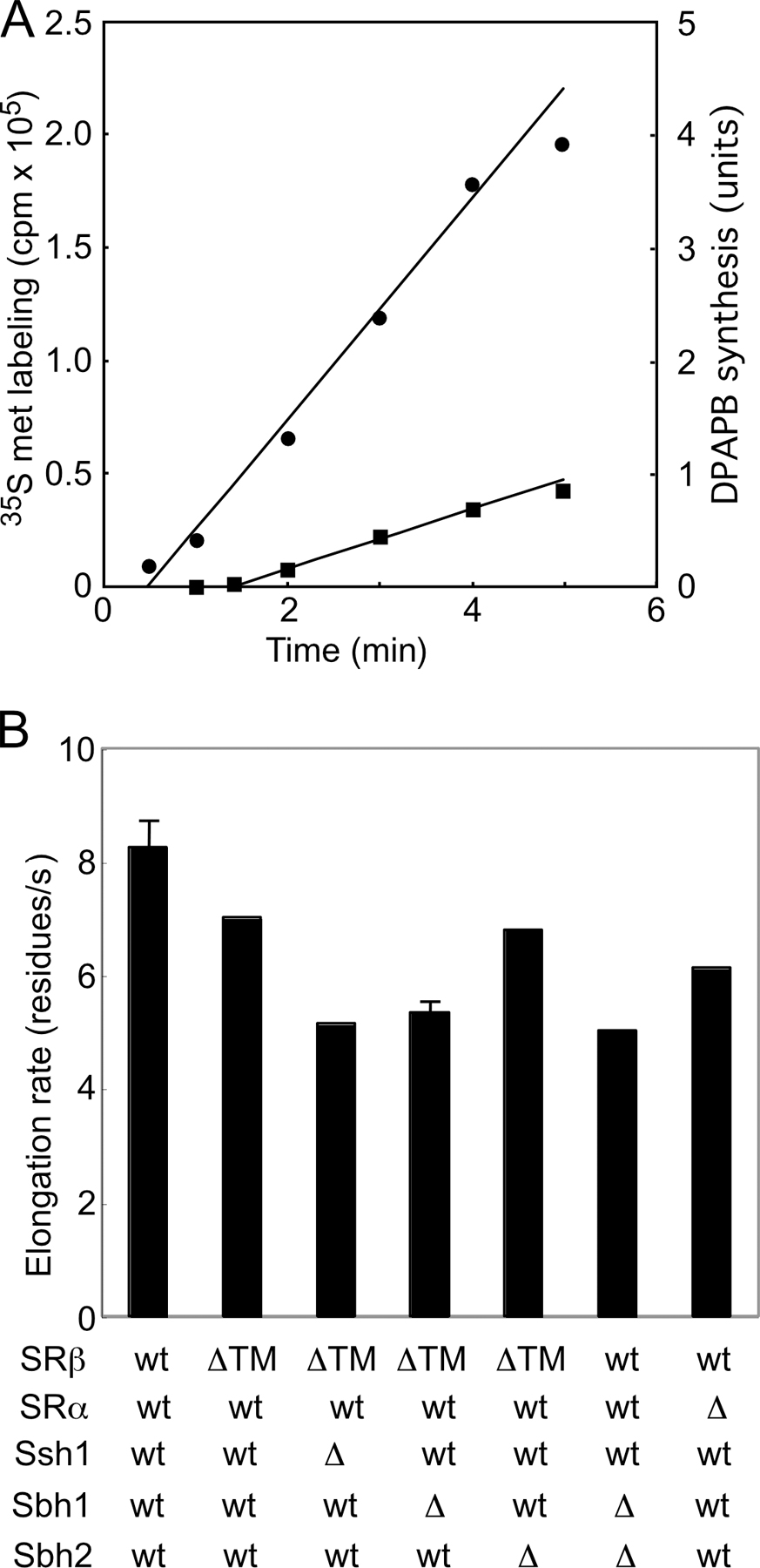
Dap2 reporter cleavage in wild-type (not depicted) and ssh1Δ cells (Fig. 3 F, circles) is very similar after 24 h of culture (Cheng and Gilmore, 2006) as a result of the increased expression of Sec61p in the ssh1Δ mutant (Fig. 4 B). Dap2 reporter cleavage was also reduced in the SRβ-ΔTM mutant after adaptation (Fig. 3 E, left). Quantification revealed a slight elevation in plateau value relative to a wild-type cell but apparently normal translocon gating kinetics (Fig. 3 F, triangles). More efficient targeting of the Dap2 reporter in the SRβ-ΔTM mutant could potentially be explained by a slower protein synthesis elongation rate. However, the elongation rate in the SRβ-ΔTM mutant (∼7 residues/s) is not markedly slower than in wild-type cells (∼8 residues/s; Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200707196/DC1). After adaptation, Dap2 reporter cleavage was also reduced in the ssh1Δ SRβ-ΔTM mutant (Fig. 3, E [middle] and F [closed diamonds]). Because protein synthesis elongation rates were slower in the ssh1Δ SRβ-ΔTM mutant (∼5 residues/s; Fig. S1), we next asked whether a further fourfold reduction in translation rate would allow normal gating kinetics by the ssh1Δ SRβ-ΔTM mutant. A low concentration of cycloheximide reduced cleavage considerably (Fig. 3, E [right] and F [open diamonds]); thus, we can conclude that additional time is required for the soluble SR to deliver an RNC to the Sec61 translocon.
Figure 4.

Genetic interactions between translocon β subunits and SRβ-ΔTM. (A) Growth rates of ssh1Δ, sbh1Δ, or sbh2Δ yeast strains expressing the soluble SR (SRα + SRβ-ΔTM) and wild-type Sec61p were determined by serial dilution analysis at 30 or 37°C as in Fig. 1. (B) Equal amounts of total protein (50 μg/lane) were resolved by PAGE in SDS for protein immunoblot analysis using a C-terminal–specific antibody to Sec61p. The blots were also probed with antisera to 3-phosphoglycerate kinase (PGK) as a loading control. (C) Diagrams of intact Sbh2p and N-terminal deletion mutants showing the location of the transmembrane (TM) domain and the proposed cytosolic GEF domain. All Sbh2 expression constructs retain the AUG initiation codon. The segment labeled Secβ structure is homologous to the segment of M. jannaschii Secβ (residues 21–52) that was resolved in the crystal structure of SecYEβ. (D) Growth rates of the SEC61sbh2Δ SRβ-ΔTM strain after transformation with a low-copy plasmid encoding intact or C-terminal fragments of Sbh2p.
Translocon β subunits are critical for the cotranslational translocation pathway
Additional strains were constructed to determine whether the lack of a translocon β subunit (either Sbh1p or Sbh2p) causes a synthetic growth defect in cells that express the soluble SR. Although lack of a single translocon β subunit does not cause a growth defect in a yeast strain that expresses wild-type SR (Finke et al., 1996), disruption of the SBH2 gene but not the SBH1 gene causes a growth defect in cells that express SRβ-ΔTM (Fig. 4 A). The synthetic growth defect was only slightly less severe than that caused by disruption of the SSH1 gene, demonstrating that Sbh2p is important for the interaction between the soluble SR and the Ssh1p complex.
Protein complexes can be destabilized by loss of a subunit. Although Sbh2p is unstable in the ssh1Δ mutant, the Ssh1p-Sss1p heterodimer is stable in the absence of Sbh2p (Finke et al., 1996), indicating that the synthetic growth defect of the sbh2Δ SRβ-ΔTM mutant cannot be explained by the absence of Ssh1 translocons. Protein extracts were prepared from selected mutants to determine whether Sec61p levels were altered upon disruption of the SSH1, SBH1, or SBH2 genes (Fig. 4 B). Interestingly, Sec61p levels increased in cells that lack Ssh1p regardless of whether SRβ was intact or lacked the TM span. Increased Sec61p levels in ssh1Δ cells might be explained by the lack of competition between Sec61p and Ssh1p for the shared pool of the translocon γ subunit (Sss1p). Lack of Sbh1p caused a slight reduction in Sec61p levels, whereas the absence of Sbh2p had little or no effect on Sec61 levels.
Eukaryotic translocon β subunits are C tail–anchored membrane proteins that expose a 55–60-residue N-terminal domain on the cytosolic side of the membrane. Methanococcus jannaschii Secβ has a smaller cytosolic segment than Sbh1p or Sbh2p, and most of this region of Secβ is disordered in the SecYEβ crystal structure (Van den Berg et al., 2004). Plasmids were constructed to express C-terminal fragments of Sbh2p (Fig. 4 C). An Sbh2 deletion mutant lacking as few as 10 N-terminal residues (sbh2Δ11) was unable to suppress the synthetic slow growth phenotype of the sbh2Δ SRβ-ΔTM mutant (Fig. 4 D). Increased expression of Sbh1p from a low- or high-copy plasmid did not suppress the growth defect of the sbh2Δ SRβ-ΔTM mutant at 30 or 37°C (unpublished data).
The synthetic growth defect of the sbh2Δ SRβ-ΔTM mutant suggests that the translocon β subunit is important for a functional interaction between the SR and the translocon. Single and double translocon β subunit deletion mutants were constructed to investigate the role of these proteins in the cotranslational translocation pathway. As reported previously (Finke et al., 1996), the double mutant grows at a wild-type rate at 30°C but is nearly inviable at 37°C (Fig. 5 E). Although pulse-labeling experiments conducted at 30°C indicate that the sbh1Δ mutant has a minor posttranslational translocation defect (Fig. 5 B), loss of a single translocon β subunit did not reduce the integration of DPAPB. Translocation of both cotranslational and posttranslational substrates was severely defective in the double mutant at the permissive temperature (Fig. 5, A and B). Finke et al. (1996) also observed a severe defect in the translocation of prepro-α-factor and Kar2p in the sbh1Δsbh2Δ mutant. The reduced electrophoretic mobility of pulse-labeled p1CPY and DPAPB synthesized by the double mutant (Fig. 5, A and B; asterisks) is caused by delayed trimming of glucose residues on the N-linked oligosaccharides (Feng et al., 2007).
Figure 5.

Translocation defects in sbh1Δsbh2Δ mutants. (A and B) The integration of DPAPB (A) and translocation of CPY (B) were evaluated by 7-min pulse labeling of wild-type (wt) and mutant yeast strains (sbh1Δ, sbh2Δ, and sbh1Δsbh2Δ) after 4 h of growth in SD media at 30°C. The glycosylated (DPAPB) and nonglycosylated (pDPAPB) forms of DPAPB and the ER (p1CPY) and precursor (ppCPY) forms of CPY are labeled. Asterisks designate incompletely glucose-trimmed forms of glycosylated DPAPB and p1CPY. (C) Protein immunoblot detection of DPAPB-HA or CPY in wild-type and sbh1Δsbh2Δ strains. Total cell extracts were prepared for SDS-PAGE with or without prior digestion by endoglycosidase H (Endo H). Precursor and mature forms of DPAPB-HA are labeled. Deglycosylated mature CPY (dgCPY) is resolved from vacuolar CPY (m), preproCPY (p), and a hypoglycosylated form of mCPY (−1). (D) Differential centrifugation of spheroplast lysates prepared from sbh1Δsbh2Δ, srp102(K51I), and wild-type yeast strains. Supernatant (S) and pellet (P) fractions were obtained after centrifugation at 100 Kg. SRα and SRβ were detected using antisera raised against yeast SR. (E–G) Plasmids encoding full-length or N-terminal deletion alleles of Sbh2 were transformed into the sbh1Δsbh2Δ strain. (E) Growth rates of wild-type and mutant strains were compared by serial dilution analysis as described in Fig. 1. (F) Integration of DPAPB and translocation of CPY were evaluated after 24 h of growth in SD media. (G) In vivo cleavage of Dap2 reporters after 24 h of growth in SD media. Labels designate the intact glycosylated (e.g., g265), intact nonglycosylated (arrowheads), and cleaved (Ura3-HA) reporter domains. Spacer length dependence of Dap2 reporter cleavage in wild type (squares), SRα null (triangles), sbh1Δsbh2Δ (closed circles), and the sbh1Δsbh2Δ mutant expressing sbh2Δ37 (open circles). Data points are means of two experiments, one of which is shown above the graph. Data for the wild-type strain is taken from Fig. 3 D. Color-coded error bars designate individual data points for experiments that were performed twice.
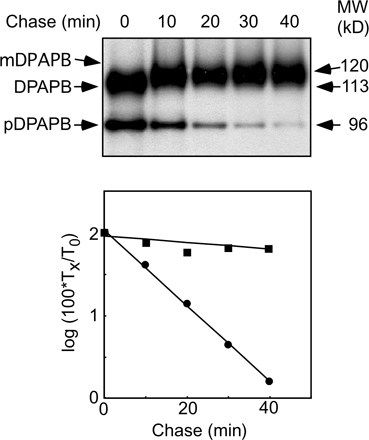
DPAPB-HA (Ng et al., 1996) was expressed under control of the 3-phosphoglycerate kinase promoter to allow facile protein immunoblot detection of pDPAPB. Nontranslocated precursor (pDPAPB-HA) was detected by protein immunoblotting in extracts prepared from the double mutant (Fig. 5 C), but the precursor was less abundant than mature DPAPB-HA. A pulse-chase experiment indicated that pDPAPB is rapidly degraded (t 1/2 = 7 min) by the double mutant (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200707196/DC1). Cell extracts were digested with endoglycosidase H to remove N-linked oligosaccharides from carboxypeptidase Y (CPY) before protein immunoblot analysis. The CPY precursor (pp-CPY), if present, would comigrate with vacuolar CPY (e.g., mCPY) before endoglycosidase H digestion. The untranslocated CPY precursor was not detected in extracts prepared from double mutant cells. Mature CPY synthesized by the double mutant migrates as a glycoform doublet because of incomplete N-glycosylation. The defects in transfer and processing of N-linked oligosaccharides are probably indirect consequences of the chronic translocation defect of the double mutant.
If the GEF activity of Sbh1p and Sbh2p is essential in vivo, the lack of SRβ GEF activity in the double mutant should favor the accumulation of SRβ-GDP and cause the cytosolic accumulation of SRα, as observed previously for a GTP-binding impaired SRβ mutant (Ogg et al., 1998). Cell extracts prepared from the double mutant and two control strains were fractionated into soluble and membrane-bound pools by differential centrifugation. Protein immunoblot experiments show that SRα is membrane bound in extracts prepared from wild-type or sbh1Δsbh2Δ cells but is roughly 50% soluble in extracts prepared from the SRβ (K51I) mutant (Fig. 5 D).
C-terminal fragments of Sbh2p (Fig. 4 C) were expressed in the sbh1Δsbh2Δ mutant to identify regions of the translocon β subunits that are important for function (Fig. 5 E). In contrast to what was observed in the sbh2Δ SRβ-ΔTM strain (Fig. 4 D), expression of the minimal Sbh2 fragment (sbh2Δ54) allowed partial suppression of the temperature-sensitive growth defect of the sbh1Δsbh2Δ mutant, whereas larger Sbh2p fragments (sbh2Δ11 and sbh2Δ37) strongly suppressed the growth defect (Fig. 5 E). Translocation of CPY and integration of DPAPB was analyzed in cells expressing the Sbh2p fragments (Fig. 5 F). Expression of sbh2Δ11 restored DPAPB integration and CPY translocation to a level that resembled the sbh1Δ mutant, whereas sbh2Δ37 allowed intermediate suppression of the sbh1Δsbh2Δ translocation defect. Expression of the Sbh2 deletion mutants in yeast was evaluated by appending an HA epitope tag to the C terminus of Sbh2p. As observed for the untagged derivatives of Sbh2p, the two larger Sbh2p fragments (sbh2Δ11-HA and sbh2Δ37-HA) retained the ability to compensate for the loss of intact Sbh1p and Sbh2p (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200707196/DC1). Protein immunoblot analysis of microsomes prepared from strains expressing HA-tagged derivatives of Sbh2p suggest that the incomplete suppression by sbh2Δ54-HA, and by analogy sbh2Δ54, is not explained by instability of the shortest deletion mutant (Fig. S3 B). Recently, C-terminal fragments of Sbh1p (e.g., sbh1Δ33 and sbh1Δ49) were also shown to suppress the growth defect of the sbh1Δsbh2Δ mutant and to assemble with Sec61p in vivo (Feng et al., 2007).
Cleavage of the Dap2 UTA reporters was evaluated in the double mutant to determine whether inefficient DPAPB integration is explained by slower translocon gating kinetics (e.g., a wide gating window) or is instead explained by a reduced number of functional translocons (e.g., an elevated plateau value). Remarkably, the translocon gating kinetics for Dap2 RNCs in the double mutant was very slow, with no apparent plateau value (Fig. 5 G, closed circles). Expression of the sbh2pΔ37 fragment in the double mutant increased the percentage of Dap2 RNCs that engage the cotranslational pathway (Fig. 5 G, open circles) but did not restore wild-type translocon gating kinetics.
Yeast strains that do not express SRP54 or either subunit of the SR adapt to loss of the SRP-dependent targeting pathway (Ogg et al., 1998). Analysis of Dap2 reporter cleavage in an SRβ deletion mutant (not depicted) or in an SRα deletion mutant (Fig. 5 F, triangles) yielded similar results. Yeast cells that adapt to loss of the SR translocate 35–40% of Dap2–265 by a cotranslational pathway. Remarkably, translocon gating by Dap2 RNCs was as slow in the sbh1Δsbh2Δ mutant as in a yeast strain that lacks the SR. Protein synthesis elongation rates in the sbh1Δsbh2Δ and SRα deletion mutant are similar (5–6 residues/s; Fig. S1).
Discussion
Interaction between SR and the Ssh1p complex
Cell fractionation and protein purification experiments indicate that the yeast ER contains three roughly equal-sized pools of Sec61p that correspond to the heptameric Sec complex as well as ribosome-bound and unbound Sec61 heterotrimers (Panzner et al., 1995). Ssh1 heterotrimers appear to be similar in abundance to Sec61 heterotrimers (Finke et al., 1996). In vivo proximity between SRβ and the yeast cotranslational translocons (Ssh1 and Sec61 heterotrimers) but not the heptameric Sec complex has been detected using the split-Ub assay system (Wittke et al., 2002). In wild-type cells, the majority of Dap2 RNCs gate the translocon within 30 s after initiation of translation (Cheng and Gilmore, 2006), as calculated from the protein synthesis elongation rate and the translocon gating window. When the SR is anchored to the membrane by the N-terminal TM span of SRβ, the search for a vacant translocon is restricted to the two-dimensional surface of the ER membrane and is not compromised by either the absence of a single translocon β subunit (Sbh1p or Sbh2p) or the Ssh1p translocon. Thus, membrane-bound SR can interact productively with both Ssh1 and Sec61 heterotrimers, but this interaction is severely compromised with β subunit–deficient translocons. The SR in SRβ-ΔTM cells is present in both membrane-bound and soluble pools (Ogg et al., 1998). Before adaptation, SRβ-ΔTM cells have a significant defect in the cotranslational targeting pathway that is explained by a delay in the rate and a reduction in the efficiency of translocon gating by RNCs (Fig. 3). This is consistent with the view that a three-dimensional search for a vacant translocon by the SRP–SR–RNC complex will require additional time. After adaptation, the apparently normal translocon gating kinetics of Dap2 RNCs in the SRβ-ΔTM strain is explained by a 15% decrease in the protein synthesis elongation rate and a global reduction in protein synthesis capacity that together accounts for the twofold reduction in the flux of nascent polypeptides through the cotranslational translocation pathway.
Synthetic slow growth defects observed in double mutant strains (e.g., ssh1Δ SRβ-ΔTM) indicate that the Ssh1p translocon is the preferred interaction site for the soluble SR. Genetic evidence for a direct interaction between the Ssh1p translocon and the soluble SR was provided by the demonstration that increased expression of either the Ssh1 heterotrimer or the soluble SR (SRα + SRβ-ΔTM), but not SRβ-ΔTM alone, largely alleviates the growth and translocation defects of the SRβ-ΔTM mutant. Analysis of Dap2 reporter cleavage in the ssh1Δ SRβ-ΔTM mutant before adaptation revealed a near complete block in the cotranslational translocation pathway that is comparable with that observed in the SRβ temperature-sensitive mutant (Fig. 3 D). In contrast to the single mutants (SRβ-ΔTM and ssh1Δ), the double mutant (ssh1Δ SRβ-ΔTM) did not display normal translocon gating kinetics after adaptation.
A critical role for the yeast Ssh1p translocon was first observed at the semipermissive temperature in the temperature-sensitive sec61-2 mutant (Finke et al., 1996). More interestingly, the expression of Ssh1p suppresses the growth and translocation defects caused by point mutations in the cytoplasmic loops (L6 and L8) of Sec61p (Cheng et al., 2005). Point mutations in L8 of Sec61p (e.g., R406E) inhibit ribosome binding; thus, these mutations block a step downstream from the targeting defect caused by deletion of the TM span of SRβ. Sequential defects in the cotranslational translocation pathway likely explain the dramatically different phenotypes of the ssh1EE SRβ-ΔTM and sec61EE SRβ-ΔTM mutants (Fig. 1 A). The soluble SR delivers the SRP–RNC to the defective ssh1EE translocon in preference to the wild-type Sec61 translocon.
A synthetic slow growth phenotype was also observed when the soluble form of SRβ was expressed in cells that lack Sbh2p, the β subunit of the Ssh1p translocon (Fig. 4). There were several remarkable aspects of the synthetic slow growth phenotype of the sbh2Δ SRβ-ΔTM stain. The lack of a similar phenotype for the sbh1Δ SRβ-ΔTM mutant supports the conclusion that the Ssh1p complex is the preferred interaction site for the soluble SR. The observation that loss of Sbh2p mimics loss of the Ssh1p translocon demonstrates that Sbh2p is critical for the function of the Ssh1p complex as a cotranslational translocon. Interestingly, the temperature-sensitive growth defect of the sbh2Δ SRβ-ΔTM mutant could not be suppressed by the expression of C-terminal Sbh2p fragments or by overexpression of intact Sbh1p. The larger C-terminal fragments of Sbh2p (e.g., sbh2pΔ11 or sbh2Δ37) are stable when expressed in yeast (Fig. S3) and are able to suppress the temperature-sensitive growth defect of a yeast strain that lacks intact translocon β subunits (Fig. 5). The TM span of Sbh2p provides the targeting information that directs integration of this tail-anchored membrane protein (Stefanovic and Hegde, 2007); thus, Sbh2 fragments likely assemble with Ssh1p.
Adaptation of yeast strains to loss of the SRP-targeting pathway
Several of the double mutant strains described here (e.g., ssh1Δ SRβ-ΔTM) share features with more conventional SRP pathway mutations, including an increased frequency of the petite (ρ−) phenotype and reductions in protein synthesis elongation rate and protein synthesis capacity. Yeast cells that lack the SRP or the SR GTPases undergo an adaptation process that allows growth in the absence of the SRP-targeting pathway. Adaptation has been most thoroughly characterized using cells that express a temperature-sensitive SRβ mutant (srp102(K51I)) or a dominant-negative allele of SRP54 (Mutka and Walter, 2001). Adaptation to loss of the SRP pathway involves a substantial, yet temporary, induction of cytosolic chaperones and long-term repression of gene products required for protein synthesis (Mutka and Walter, 2001). The reduction in protein synthesis capacity and protein elongation rate reduces the precursor load for the translocation channels, thereby allowing a bypass of the SRP-targeting pathway (Mutka and Walter, 2001). However, an open question was whether cells that lack the SRP-targeting pathway retain a vestige of a cotranslational translocation pathway or instead transport all substrates in a posttranslational mode through the heptameric Sec complex. Here, we addressed this question using the Dap2 series of UTA reporters. Interestingly, yeast strains that lack the SR transport roughly one third of Dap2 precursors by a cotranslational translocation pathway (Fig. 5 G). Cotranslational translocon gating in SRα-deficient cells is slower than in wild-type cells as revealed by a wider gating window. What mechanism could explain RNC targeting to the translocon when a core component of the targeting pathway is absent? An SRP-independent cotranslational pathway has been investigated using the mammalian cell-free system, where it has been shown that RNCs assembled by translation of a termination codon-deficient mRNA can engage translocons on ribosome-stripped membranes (Wiedmann et al., 1994; Jungnickel and Rapoport, 1995; Lauring et al., 1995; Raden and Gilmore, 1998). Ribosome-Sec61 and signal sequence–Sec61 interactions promote translocon gating by this SRP-independent targeting pathway. Alternatively, an 80S ribosome that was prebound to a translocation channel could initiate translation of an mRNA encoding a secretory protein (Potter and Nicchitta, 2000). These SRP-independent targeting mechanisms may allow cotranslational integration of hydrophobic membrane proteins that may not be optimal substrates for the posttranslational translocation pathway because of their potential for aggregation in the cytosol.
Diverse roles for translocon β subunits
Previous studies concerning the role of translocon β subunits have suggested diverse and somewhat contradictory roles for these subunits in the cotranslational and posttranslational translocation pathways. The initial evidence that translocon β subunits are not essential in yeast was provided by an analysis of single and double mutant strains (Finke et al., 1996). As observed here for CPY translocation, the sbh1Δ mutant analyzed by Finke et al. (1996) had a very mild in vivo defect in prepro-α-factor transport that was accentuated upon the deletion of Sbh2p. CPY and prepro-α-factor are translocated through the heptameric Sec complex, not through the Ssh1p complex (Wittke et al., 2002), so the severe posttranslational translocation defect caused by the deletion of both translocon β subunits is best explained by an indirect mechanism. When both translocon β subunits are missing, the reduced efficiency of translocon gating by the heterodimeric Sec61p and Ssh1p cotranslational translocons causes increased precursor flux through the posttranslational translocon (Sec complex) and competition between co- and posttranslational substrates for cytosolic chaperones. Although the expression of cytosolic chaperones is up-regulated by lesions in the cotranslational translocation pathway, expression of the heptameric Sec complex does not increase (Mutka and Walter, 2001). Although we favor this indirect explanation for the posttranslational translocation defect, a previous study indicates that microsomes isolated from the sbh1Δsbh2Δ mutant transport prepro-α-factor at a reduced rate in vitro (Finke et al., 1996). However, the in vitro defect in translocation of prepro-α-factor by microsomes isolated from the sbh1Δsbh2Δ mutant has not been confirmed by subsequent investigators (Feng et al., 2007). Drastically reduced stability of Ssh1p and Sec61p complexes in the sbh1Δsbh2Δ mutants can be eliminated as a potential explanation for the translocation defect, as β subunit deletions have relatively subtle effects on Sec61p and Ssh1p expression (Fig. 4 B; Finke et al., 1996).
Likewise, multiple roles have been proposed for the mammalian translocon β subunit (Sec61β). Proteoliposomes reconstituted with Sec61β-depleted canine Sec61 complexes are defective in cotranslational translocation unless the SRP–RNC-targeting step is performed at a reduced temperature (Kalies et al., 1998). Based on this result, Sec61β was proposed to mediate a posttargeting event in cotranslational translocation that might correspond to signal sequence insertion into the Sec61 pore. Although mammalian Sec61β is dispensable for the ribosome-binding activity of the Sec61 complex (Kalies et al., 1994), purified recombinant Sec61β binds ribosomes with an affinity comparable with the Sec61 heterotrimer (Levy et al., 2001). Canine Sec61β can be cross-linked to SPC25, a subunit of the signal peptidase complex, suggesting that the cytoplasmic domain of canine Sec61β recruits signal peptidase to the translocon when an RNC is bound to Sec61α (Kalies et al., 1998).
When assayed in vitro, the cytosolic domains of Sbh1p and Sbh2p act as GEFs for the GDP-bound form of yeast SRβ (Helmers et al., 2003). However, the limited sequence alignment between the yeast translocon β subunits and the Sec7 GEF domain (Helmers et al., 2003) does not appear to be well conserved in vertebrate, plant, or insect translocon β subunits. The SecYEβ crystal structure does not provide significant insight into the structure of the cytosolic domain of the eukaryotic translocon β subunit as a result of both the low conservation between archaebacterial Secβ and eukaryotic Sec61β (Sbh1p/Sbh2p) and the disordered structure of the M. jannaschii Secβ N termini (Van den Berg et al., 2004). Unassembled cytosolic SRα is not detected in the sbh1Δsh2Δ mutant (Fig. 5 D), indicating that SR heterodimer formation is not compromised in cells that lack translocon β subunits. Although our UTA assay experiments using the Dap2 reporters demonstrate that sbh1Δsbh2Δ cells have a severe cotranslational translocation defect, expression of a short segment of Sbh2p (e.g., sbh2pΔ37) that lacks the majority of the proposed GEF domain partially suppress the abnormal translocon gating kinetics. Collectively, these results demonstrate that the translocation defect in the sbh1Δsbh2Δ mutant is not caused by the lack of an SRβ GEF activity, but, instead, translocon β subunits mediate a critical step in the recognition of a vacant translocon by the SRP–SR–RNC complex. An interaction between the translocon β subunit and the SR heterodimer that facilitates recognition of the unoccupied translocon is an attractive explanation for the network of genetic interactions observed here between the soluble SR, translocon β subunits, and the Ssh1p complex.
Materials and methods
Plasmid and strain constructions
A hygromycin B resistance (HPH) gene flanked by 5′ (bp −512 to −1) and 3′ (bp 736–941) regions from the SRP102 gene encoding SRβ was used to transform the diploid yeast strain BWY100 (MATa/MATα, trp1-1/trp1-1, ade2/ade2, leu2-3,112/leu2-3,112, ura3/ura3, his3-11/his3-11, can1/can1, sec61∷HIS3/SEC61; provided by C. Stirling, University of Manchester, Manchester, UK). Hygromycin B–resistant colonies were selected, transformed with pJY01 (SEC61, SRP102-HA, and URA3), and induced to sporulate. Disruption of the SRP102 gene in the Hph+ His+ Ura+ haploid progeny (YJY101) was confirmed by PCR. The SSH1 gene in YJY101 was disrupted as described previously (Cheng et al., 2005) to obtain the strain YJY102.
The coding sequence of amino acids 3–24 of SRβ was deleted using recombinant PCR to construct plasmids containing SRβ-ΔTM under control of its own promoter. A SacI–EcoRI-digested fragment containing the 5′ (bp −440–6), coding, and 3′ (bp 73–1,945) segments of the SRP102 gene was inserted into SacI–EcoRI-digested pRS314 to obtain pJY209 (SRβ-ΔTM TRP1). A SacI–KpnI fragment encoding SRβ-ΔTM was obtained by digestion of pJY209 and ligated into SacI–KpnI-digested pRS424 (Christianson et al., 1992) to produce the high-copy plasmid pJY210. An EcoRI–BamHI-digested DNA fragment (bp −480–3,101) from the SRP101 gene encoding SRα was inserted into EcoRI–BamHI-digested pJY210 to obtain the high-copy plasmid (pJY211) encoding the soluble SR.
Yeast strain ZCY101 (MATα, trp1-1, ade2, leu2-3,112, ura3, his3-11, can1 s61∷HIS3[pBW11]) expressing full-length wild-type Sec61p was derived from BWY12. The SBH1 gene in ZCY101 was disrupted as described previously (Cheng et al., 2005) to obtain YJY103. A DNA fragment containing the HPH gene flanked by 5′ (bp −545 to −1) and 3′ (bp 268–758) regions from the SBH2 gene was generated and used to transform ZCY101 and YJY103, giving rise to the sbh2Δ strain (YJY104) and sbh1Δsbh2Δ strain (YJY105), respectively. The SBH1 and SBH2 genes were disrupted in YJY1209 to obtain YJY3209 (sbh1Δ SRβ-ΔTM) and YJY4209 (sbh2Δ SRβ-ΔTM), respectively, using the same method.
To construct plasmids expressing wild-type Sbh2p or Sbh2p C-terminal fragments, an XhoI–HindIII-digested PCR product corresponding to the 5′ untranslated region of SBH2 (bp −598 to −1), HindIII–XbaI-digested PCR products containing the coding sequence of Sbh2p or the Sbh2p truncations (Δ11, Δ37, and Δ54), and the 3′ untranslated region (bp 169–671) were ligated and inserted into XhoI–XbaI-digested pRS316 to obtain pJY300, pJY311, pJY337, and pJY354, respectively. All Sbh2p deletion constructs retained the AUG initiation codon.
A SacI–XhoI-digested PCR product corresponding to the SSH1 gene (bp −442–2,710) was inserted into SacI–XhoI-digested pRS315 to construct a plasmid encoding wild-type SSH1. Oligonucleotide primers containing point mutations (R278E or R411E) were used in recombinant PCR to produce the ssh1EE mutant. Mutations were confirmed by sequencing. An Apa1–Sac1 DNA fragment encoding the three subunits of the Ssh1 complex (Ssh1p, Sbh2p, and Sss1p) was produced using recombinant PCR with yeast DNA (for SSS1) or plasmid DNA (SSH1 and SBH2) as templates. The final PCR product (ApaI-SSH1 [bp −442–2,710]-XhoI-SBH2 [bp −600–810]-BamHI-SSS1 [bp −330–63]-SacI) was cloned into Apa1–Sac1-digested pRS425 to obtain pJY213.
A plasmid shuffle procedure (Sikorski and Boeke, 1991) was used to replace plasmid-born wild-type genes (e.g., SRP102 plus SEC61) with mutant alleles (e.g., SRβ-ΔTM) or overexpression plasmids (e.g., 2μ SRβ-ΔTM). For example, the plasmids pJY209 (SRβ-ΔTM) and pBW11 (SEC61 and LEU2; provided by C. Stirling) were cotransformed into YJY101 or YJY102; the Trp+ Leu+ Ura− prototrophs were selected on SD media containing 5-fluoroorotic acid, giving rise to YJY1209 and YJY2209, respectively. Plasmids expressing wild-type SSH1, ssh1EE, or the Ssh1 complex were cotransformed with pJY022 (SEC61, SRβ-ΔTM, and TRP1) into YJY102, and the Trp+ Leu+ Ura− prototrophs were selected on SD media containing 5-fluoroorotic acid.
Growth rate
For serial dilution experiments, yeast strains were grown at 30°C in SEG media to mid-log phase. After dilution of cells to 0.1 OD at 600 nm, 5-μl aliquots of 10-fold serial dilutions were spotted onto YPD plates that were incubated at 30 or 37°C for 2–3 d. For liquid growth rate experiments, yeast cells were first grown in SEG media to mid-log phase at 30°C. The cells were collected, diluted to 0.1 OD at 600 nm in fresh YPD media, and allowed to grow for 10 h at 30°C. Aliquots were taken every hour to measure A600.
Immunoprecipitation of radiolabeled proteins and protein immunoblots
After growth at 30°C in SEG media to mid-log phase (0.4–0.6 OD at 600 nm), yeast were collected by centrifugation, resuspended in SD media, and grown for an additional 4 or 24 h at 30°C. Yeast cells were collected by centrifugation, resuspended in fresh SD media at a density of 4 A600/ml, and pulse labeled for 7 min with 100 μCi/OD Tran-35S-label. Radiolabeling experiments were terminated by the addition of an equal volume of ice-cold 20 mM NaN3 followed by freezing in liquid nitrogen. Rapid lysis of cells with glass beads and immunoprecipitation of yeast proteins was performed as previously described (Rothblatt and Schekman, 1989). Immunoprecipitated proteins were resolved by SDS-PAGE and detected with a molecular imager (FX; Bio-Rad Laboratories).
Total protein extracts were prepared as described previously (Arnold and Wittrup, 1994) from cells after 24 h of growth at 30°C in SD media. Aliquots of the protein extracts were digested with endoglycosidase H (New England Biolabs, Inc.) before CPY immunoblots. The protein concentration in extracts was determined using the Dc protein assay kit (Bio-Rad Laboratories) so that identical protein amounts (50 μg/lane) were analyzed on Sec61 immunoblots. Protein immunoblots were performed as described previously (Cheng et al., 2005). Antibodies specific for yeast Sec61p and the yeast SR (SRα and SRβ) were provided by R. Schekman (University of California, Berkeley, Berkeley, CA) and T. Schwartz (Massachusetts Institute of Technology, Cambridge, MA), respectively.
UTA
The Dap2 series of UTA reporters (Dap249–265) have been described previously (Cheng and Gilmore, 2006). Cells expressing Dap2 reporters were radiolabeled with Tran-35S-label as described in the previous paragraph. The intact reporters and the Ura3-HA fragments were immunoprecipitated with anti-HA monoclonal antibodies. For experiments conducted in the presence of cycloheximide, the cells were incubated with SD media containing 0.2 μg/ml cycloheximide for 10 min at 30°C before radiolabeling. The distribution of methionine and cysteine residues in the intact UTA reporter and the Ura3-HA fragment was determined, and the cleavage percentage was calculated as described previously (Cheng and Gilmore, 2006).
Online supplemental material
Fig. S1 shows the in vivo protein synthesis elongation rate at 30°C for wild-type and seven yeast strains (e.g., ssh1Δ SRβ-ΔTM) used in this study. Fig. S2 shows that nontranslocated pDPAPB is rapidly degraded (t 1/2 = 7 min) in sbh1Δsbh2Δ cells. Fig. S3 shows that HA epitope–tagged Sbh2 fragments are expressed in the sbh1Δsbh2Δ mutant and are able to suppress the growth defect of the sbh1Δsbh2Δ mutant at 37°C. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200707196/DC1.
Supplemental Material
Acknowledgments
We thank Thomas Schwartz and Randy Schekman for providing antisera to the SR and Sec61p, respectively.
This work was supported by National Institutes of Health grant GM35687.
Z. Cheng's present address is Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, CA 94720.
Abbreviations used in this paper: CPY, carboxypeptidase Y; DPAPB, dipeptidylaminopeptidase B; GEF, guanine nucleotide exchange factor; pDPAPB, DPAPB precursor; RNC, ribosome–nascent chain complex; SR, SRP receptor; SRP, signal recognition particle; TM, transmembrane; Ub, ubiquitin; UTA, Ub translocation assay.
References
- Angelini, S., S. Deitermann, and H.G. Koch. 2005. FtsY, the bacterial signal-recognition particle receptor, interacts functionally and physically with the SecYEG translocon. EMBO Rep. 6:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini, S., D. Boy, E. Schiltz, and H.G. Koch. 2006. Membrane binding of the bacterial signal recognition particle receptor involves two distinct binding sites. J. Cell Biol. 174:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold, C.E., and K.D. Wittrup. 1994. The stress response to loss of signal recognition particle function in Saccharomyces cerevisiae. J. Biol. Chem. 269:30412–30418. [PubMed] [Google Scholar]
- Beckmann, R., C.M. Spahn, N. Eswar, J. Helmers, P.A. Penczek, A. Sali, J. Frank, and G. Blobel. 2001. Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell. 107:361–372. [DOI] [PubMed] [Google Scholar]
- Cheng, Z., and R. Gilmore. 2006. Slow translocon gating causes cytosolic exposure of transmembrane and lumenal domains during membrane protein integration. Nat. Struct. Mol. Biol. 13:930–936. [DOI] [PubMed] [Google Scholar]
- Cheng, Z., Y. Jiang, E.C. Mandon, and R. Gilmore. 2005. Identification of cytoplasmic residues of Sec61p involved in ribosome binding and cotranslational translocation. J. Cell Biol. 168:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson, T.W., R.S. Sikorski, M. Dante, J.H. Shero, and P. Hieter. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene. 110:119–122. [DOI] [PubMed] [Google Scholar]
- de Leeuw, E., D. Poland, O. Mol, I. Sinning, C.M. ten Hagen-Jongman, B. Oudega, and J. Luirink. 1997. Membrane association of FtsY, the E. coli SRP receptor. FEBS Lett. 416:225–229. [DOI] [PubMed] [Google Scholar]
- Deshaies, R.J., S.L. Sanders, D.A. Feldheim, and R. Schekman. 1991. Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature. 349:806–808. [DOI] [PubMed] [Google Scholar]
- Egea, P.F., S.O. Shan, J. Napetschnig, D.F. Savage, P. Walter, and R.M. Stroud. 2004. Substrate twinning activates the signal recognition particle and its receptor. Nature. 427:215–221. [DOI] [PubMed] [Google Scholar]
- Feng, D., X. Zhao, C. Soromani, J. Toikkanen, K. Romisch, S.S. Vembar, J.L. Brodsky, S. Keranen, and J. Jantti. 2007. The transmembrane domain is sufficient for Sbh1p function, its association with the Sec61 complex, and interaction with Rtn1p. J. Biol. Chem. 282:30618–30628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke, K., K. Plath, S. Panzer, S. Prehn, T.A. Rapoport, E. Hartmann, and T. Sommer. 1996. A second trimeric complex containing homologues of the Sec61p complex functions in protein transport across the ER membrane of S. cerevisiae. EMBO J. 15:1482–1494. [PMC free article] [PubMed] [Google Scholar]
- Focia, P.J., I.V. Shepotinovskaya, J.A. Seidler, and D.M. Freymann. 2004. Heterodimeric GTPase core of the SRP targeting complex. Science. 303:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halic, M., T. Becker, M.R. Pool, C.M. Spahn, R.A. Grassucci, J. Frank, and R. Beckmann. 2004. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 427:808–814. [DOI] [PubMed] [Google Scholar]
- Halic, M., M. Gartmann, O. Schlenker, T. Mielke, M.R. Pool, I. Sinning, and R. Beckmann. 2006. Signal recognition particle receptor exposes the ribosomal translocon binding site. Science. 312:745–747. [DOI] [PubMed] [Google Scholar]
- Hann, B.C., and P. Walter. 1991. The signal recognition particle in S. cerevisiae. Cell. 67:131–144. [DOI] [PubMed] [Google Scholar]
- Helmers, J., D. Schmidt, J.S. Glavy, G. Blobel, and T. Schwartz. 2003. The beta-subunit of the protein-conducting channel of the endoplasmic reticulum functions as the guanine nucleotide exchange factor for the beta-subunit of the signal recognition particle receptor. J. Biol. Chem. 278:23686–23690. [DOI] [PubMed] [Google Scholar]
- Huffaker, T.C., M.A. Hoyt, and D. Botstein. 1987. Genetic analysis of the yeast cytoskeleton. Annu. Rev. Genet. 21:259–284. [DOI] [PubMed] [Google Scholar]
- Johnsson, N., and A. Varshavsky. 1994. Ubiquitin-assisted dissection of protein transport across membranes. EMBO J. 13:2686–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungnickel, B., and T.A. Rapoport. 1995. A posttranslational signal sequence recognition event in the endoplasmic reticulum membrane. Cell. 82:261–270. [DOI] [PubMed] [Google Scholar]
- Kalies, K.-U., D. Görlich, and T.A. Rapoport. 1994. Binding of ribosomes to the rough endoplasmic reticulum is mediated by the Sec61p-complex. J. Cell Biol. 126:925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalies, K.-U., T.A. Rapoport, and E. Hartmann. 1998. The β-subunit of the Sec61p complex facilitates cotranslational protein transport and interacts with the signal sequence. J. Cell Biol. 141:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring, B., H. Sakai, G. Kreibich, and M. Wiedmann. 1995. Nascent polypeptide-associated complex protein prevents mistargeting of nascent chains to the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 92:5411–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate, K.R., D. Falcone, and D.W. Andrews. 2000. Nucleotide-dependent binding of the GTPase domain of the signal recognition particle receptor beta-subunit to the alpha-subunit. J. Biol. Chem. 275:27439–27446. [DOI] [PubMed] [Google Scholar]
- Levy, R., M. Wiedmann, and G. Kreibich. 2001. In vitro binding of ribosomes to the beta subunit of the Sec61p protein translocation complex. J. Biol. Chem. 276:2340–2346. [DOI] [PubMed] [Google Scholar]
- Mandon, E.C., Y. Jiang, and R. Gilmore. 2003. Dual recognition of the ribosome and the signal recognition particle by the SRP receptor during protein targeting to the endoplasmic reticulum. J. Cell Biol. 162:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, D.G., J.F. Menetret, A. Neuhof, T.A. Rapoport, and C.W. Akey. 2002. Structure of the mammalian ribosome-channel complex at 17A resolution. J. Mol. Biol. 324:871–886. [DOI] [PubMed] [Google Scholar]
- Mutka, S.C., and P. Walter. 2001. Multifaceted physiological response allows yeast to adapt to the loss of the signal recognition particle-dependent protein-targeting pathway. Mol. Biol. Cell. 12:577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, D.T.W., J.D. Brown, and P. Walter. 1996. Signal sequences specify the targeting route to the endoplasmic reticulum. J. Cell Biol. 134:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg, S.C., M.A. Poritz, and P. Walter. 1992. Signal recognition particle receptor is important for cell growth and protein secretion in Saccharomyces cerevisiae.Mol. Biol. Cell. 3:895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg, S.C., W.P. Barz, and P. Walter. 1998. A functional GTPase domain, but not its transmembrane domain, is required for function of the SRP receptor β-subunit. J. Cell Biol. 142:341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzner, S., L. Dreier, E. Hartmann, S. Kostka, and T.A. Rapoport. 1995. Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell. 81:561–570. [DOI] [PubMed] [Google Scholar]
- Potter, M.D., and C.V. Nicchitta. 2000. Regulation of ribosome detachment from the mammalian endoplasmic reticulum membrane. J. Biol. Chem. 275:33828–33835. [DOI] [PubMed] [Google Scholar]
- Prinz, A., E. Hartmann, and K.U. Kalies. 2000. Sec61p is the main ribosome receptor in the endoplasmic reticulum of Saccharomyces cerevisiae. Biol. Chem. 381:1025–1029. [DOI] [PubMed] [Google Scholar]
- Raden, D., and R. Gilmore. 1998. Signal recognition particle-dependent targeting of ribosomes to the rough endoplasmic reticulum in the absence and presence of the nascent polypeptide-associated complex. Mol. Biol. Cell. 9:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapiejko, P.J., and R. Gilmore. 1997. Empty site forms of the SRP54 and SRα GTPases mediate targeting of ribosome-nascent chain complexes to the endoplasmic reticulum. Cell. 89:703–713. [DOI] [PubMed] [Google Scholar]
- Rothblatt, J., and R. Schekman. 1989. A hitchhiker's guide to the analysis of the secretory pathway in yeast. Methods Cell Biol. 32:3–36. [DOI] [PubMed] [Google Scholar]
- Schwartz, T., and G. Blobel. 2003. Structural basis for the function of the beta subunit of the eukaryotic signal recognition particle receptor. Cell. 112:793–803. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and J.D. Boeke. 1991. In vitro mutagenesis and plasmid shuffling: from cloned genes to mutant yeast. Methods Enzymol. 194:302–318. [DOI] [PubMed] [Google Scholar]
- Song, W., D. Raden, E.C. Mandon, and R. Gilmore. 2000. Role of Sec61alpha in the regulated transfer of the ribosome-nascent chain complex from the signal recognition particle to the translocation channel. Cell. 100:333–343. [DOI] [PubMed] [Google Scholar]
- Stefanovic, S., and R.S. Hegde. 2007. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell. 128:1147–1159. [DOI] [PubMed] [Google Scholar]
- Van den Berg, B., W.M. Clemons Jr., I. Collinson, Y. Modis, E. Hartmann, S.C. Harrison, and T.A. Rapoport. 2004. X-ray structure of a protein-conducting channel. Nature. 427:36–44. [DOI] [PubMed] [Google Scholar]
- Walter, P., and A.E. Johnson. 1994. Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu. Rev. Cell Biol. 10:87–119. [DOI] [PubMed] [Google Scholar]
- Wiedmann, B., H. Sakai, T.A. Davis, and M. Wiedmann. 1994. A protein complex required for signal-sequence-specific sorting and translocation. Nature. 370:434–440. [DOI] [PubMed] [Google Scholar]
- Wittke, S., M. Dunnwald, M. Albertsen, and N. Johnsson. 2002. Recognition of a subset of signal sequences by Ssh1p, a Sec61p-related protein in the membrane of endoplasmic reticulum of yeast Saccharomyces cerevisiae. Mol. Biol. Cell. 13:2223–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}