

Abstract
The herpes simplex virus type 1 (HSV-1) latency associated transcript (LAT) gene has anti-apoptosis activity that directly or indirectly enhances the virus’s reactivation phenotype in small animal models. The first 1.5 kb of the primary 8.3 kb LAT is sufficient and some or all of it is necessary for LAT’s anti-apoptosis in transient transfection assays and for LAT’s ability to enhance the reactivation phenotype. Based on LAT’s genomic sequence, the first 1.5 kb contains eight potential open reading frames (ORFs) defined as an ATG followed by an in frame termination codon. In this study, point mutations were introduced into the ATGs of ORFs present in the 1.5 Kb fragment of LAT. Mutagenesis of all 8 ATGs in LAT ORFs consistently reduced the anti-apoptotic activity of LAT in transiently transfected mouse neuroblastoma cells regardless of whether apoptosis was induced by caspase 8 or caspase 9. Mutation of the 6 ATGs located in the stable intron sequences within the 1.5 Kb LAT had a dramatic effect on caspase 9, but not caspase 8, induced apoptosis. For both caspase 8 and caspase 9 induced apoptosis, mutating the two ATGs in the exon of the LAT 1.5 KB fragment reduced, but did not eliminate the anti-apoptotic activity of LAT. These studies suggest that altering the fine structure of regulatory RNA or expression of a putative LAT ORF regulates the anti-apoptosis activity of LAT. These studies also indicate that more than one function is present in the 1.5 Kb LAT fragment.
Keywords: Herpes simplex virus, LAT, latency, latency associated transcript, apoptosis
As many as 90% of adults in the US harbor latent herpes simplex virus type 1 or type 2 (HSV-1, HSV-2) infections [1]. HSV-1 causes cold sores in and around the mouth, genital herpes, encephalitis, and corneal disease. In the US, HSV-1 is the leading cause of corneal blindness due to an infectious agent [2]. HSV-1 induced corneal blindness is due to scarring of the cornea, mostly in response to recurrent rather than primary infection. Following a primary ocular HSV-1 infection the virus enters sensory nerves and travels to the nerve bodies located in the trigeminal ganglia (TG). Here life long viral latency is established. Sporadic viral reactivations can occur throughout the life of the infected individual, at which time virus returns to, is shed at the ocular surface, and can cause recurrent disease leading to loss of vision. Understanding the molecular mechanisms by which the HSV-1 latency--reactivation cycle is regulated is therefore important for the eventual control and elimination of recurrent herpetic disease.
During neuronal latency the latency associated transcript, or LAT gene, is the only abundantly transcribed viral gene [3, 4]. LAT plays an important role in the HSV latency-reactivation cycle since LAT null mutants have significantly reduced reactivation phenotypes in mice and rabbits while otherwise having wild type like replication [5–11]. LAT has anti-apoptosis activity [12–19]. Mutants in which LAT is replaced by an alternative anti-apoptosis gene have efficient, wild type-like reactivation phenotypes [20–22], indicating that LAT’s anti-apoptosis activity is sufficient to account for LAT’s ability to support the wild type reactivation phenotype in experimental animal models.
The mechanism by which LAT’s anti-apoptosis activity enhances the reactivation phenotype remains to be elucidated. In addition, how LAT’s anti-apoptosis activity is mediated also remains to be fully elucidated. However, the most critical LAT functions appear to be encoded within the first 1.5 kb of LAT, since the first 1.5 kb of the primary 8.3 kb primary LAT (approximately the first 18%) is sufficient for both supporting a wild type virus reactivation phenotype [23] and for efficiently blocking apoptosis in transient transfection assays [13, 16].
Based on sequence analysis of the LAT DNA, the first 1.5 kb of LAT contains 8 potential open reading frames (ORFs; an ATG followed by an in frame stop codon). However, the nucleotide sequence in this region is more highly conserved among functional LATs from 3 different HSV-1 strains (>95%) than are the predicted amino acid sequences (<80%). This suggests that there is more selective pressure to maintain the RNA sequence than protein coding sequences, suggesting that this region of LAT does not encode an important functional protein. We therefore proposed that LAT functions via its RNA rather than via a LAT protein. Consistent with this, it was recently reported that LAT encodes a miRNA that has anti-apoptosis activity [17]. However, this miRNA is unlikely to account for LAT’s full function as it is encoded from a location that is neither essential for [15, 24] nor sufficient for [13] LAT’s ability to block apoptosis or enhance the reactivation phenotype. Thus, this miRNA does not rule out the possibility of a LAT protein or additional regulatory RNAs.
The work presented here was initiated in the hope of demonstrating that none of the potential 8 LAT ORFs discussed above play a role in the ability of LAT to block apoptosis and by extension also not involved in LAT’s ability to enhance the viral reactivation phenotype. Surprisingly we report here that changing the relevant ATGs to TTGs to knock out expression of these putative proteins, had a significant impact on LAT’s anti-apoptosis activity.
We constructed three mutant LAT plasmids (Fig. 1), the parental plasmid of which (LAT3.3A) contains LAT nts −1801 to +1499 [13, 23]. The presence of the entire LAT promoter results in this plasmid expressing high levels of the first 1.5 kb of LAT following transfection of tissue culture cells. It should also be noted that LAT3.3A does not encode the entire stable 2 Kb LAT intron, but still maintains high levels of anti-apoptosis activity. In plasmid LAT3.3E the first two LAT ATGs were mutated to TTG, thereby knocking out both of the potential ORFs located within the exon (LAT nts 1–660). In plasmid LAT3.3I, all 6 potential ORFs within the region of the first 2kb LAT that is located within the first 1.5 kb of the LAT transcript (LAT nts 661–1499) were similarly knocked out. All 8 potential ORFs were similarly knocked out in LAT3.3U.
Figure 1.

LAT derived plasmids. A: LAT3.3A, the parental plasmid, contains LAT nts −1801 to +1499 relative to the start of LAT transcription. It therefore contains the entire LAT promoter and upstream control sequences and transcribes the first 1.5 kb of the primary 8.3 kb LAT. Nts +1 to 661 correspond to the first LAT exon. Nts 662 to 1499 correspond to the first 837 nts of the 2 kb stable LAT intron. The solid rectangles show the relative locations of all 8 potential ORFs (ATG followed by an in frame stop codon) within the first 1499 nts of the primary LAT transcript. The nt positions of the ORFs relative to the start of LAT transcription at LAT nt +1 are: ORF 1 = nts 217–376; ORF 2 = nts 486–669; ORF 3 = nts 916–1123; ORF 4 = 980–1079; ORF 5 = nts 1091–1298; ORF 6 = 1167–1242; ORF 7 = 1279–1360; ORF 8 = 1365–1497 [29]. All of the other plasmids are identical to LAT3.3A except for knock out of various ORFs by mutation of the relevant ATG to TTG by PCR site directed mutation. All changes were confirmed by complete sequencing of the final DNA fragments. B: LAT3.3E is knocked out for ORF1 and ORF2 in the exon but retains all 6 ORFs in the first 878 nts of the 2 kb intron. C: LAT3.3I is knocked out for all 6 ORFs in the intron but retains both ORFs in the exon. D: LAT3.3U is knocked out for all 8 ORFs.
In mammals, there are two major apoptotic pathways; the death receptor mediated pathway or the mitochondrial pathway [25–27]. The receptor mediated death pathway activates caspase 8, which induces a caspase cascade including caspase 3. Activation of the mitochondrial death pathway results in release of several important proapoptotic molecules, including cytochrome C and Smac/Diablo [27]. Released cytochrome C associates with Apaf-1 leading to caspase 9 activation, which culminates in activation of the effector caspases (including caspase 3). Thus, it was important to compare the results obtained with caspase 8 induced apoptosis to those obtained by inducing apoptosis with caspase 9.
Consequently, Neuro-2A cells were transfected with each of the LAT plasmids plus a plasmid that induces apoptosis by expressing caspase 8 (Fig. 2). Cell survival was determined 48 hours later as previously described [15, 16]. The number of cells in control cultures transfected with the same total amount of empty plasmid (no LAT; no caspase 8) was set to 100%. Approximately 38% of cells survived with the caspase 8 plasmid alone. As we previously reported, LAT3.3A significantly protected against caspase 8 induced death (90% survival; P<0.05 versus caspase 8, ANOVA two way analysis of variance). LAT3.3E, LAT3.3I, and LAT3.3U all had significantly less survival than the control and LAT3.3A (P<0.05). LAT3.3U did not appear to provide any protection against caspase 8 induced apoptosis (P>0.05) and was significantly different from all of the other plasmids (P<0.05). LAT3.3E and LAT3.3I both appeared to provide intermediate protection as they were each significantly different from the control, LAT3.3A, and caspase 8 (P<0.05) but were not different from each other (P>0.05). Thus, knocking out either ORFs 1 and 2 (LAT3.3E), ORFs 3–8 (LAT3.3I), or all 8 ORFs appeared to reduce the protective effect of LAT against caspase 8 induced death. Knocking out all 8 ORFs appeared to more effectively prevent LAT’s anti-apoptosis activity than knocking out just ORFs 1–2 or just ORFs 3–8.
Figure 2.
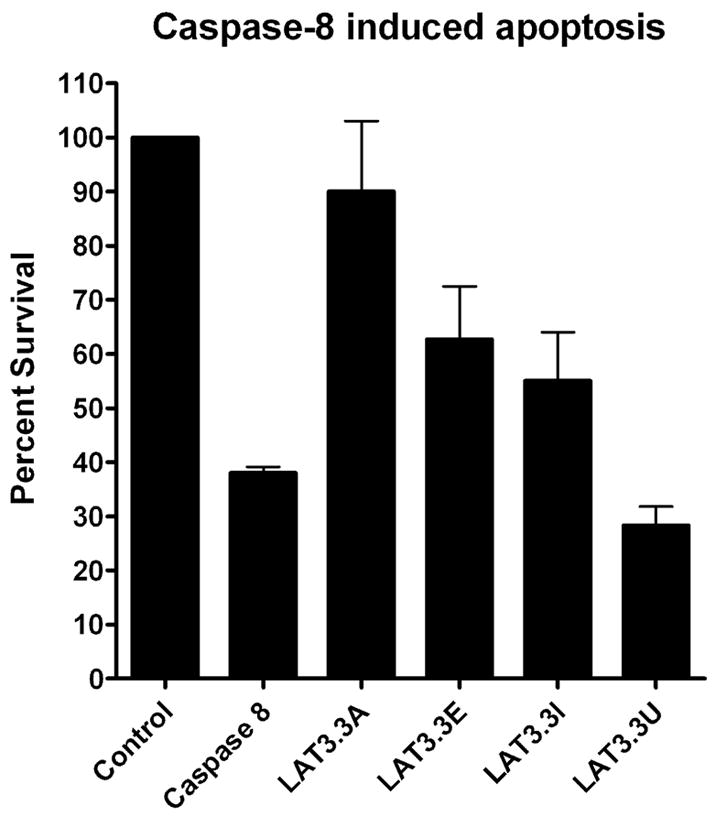
Inhibition of caspase 8 induced apoptosis by LAT constructs. Using our standard apoptosis blocking assay [15, 16] Neuro2A cells were co-transfected with a caspase 8 expressing plasmid to induce apoptosis and one of the LAT expressing plasmids (LAT3.3A, LAT3.3E, LAT3.3I, or LAT3.3U). Control cells were transfected with the same total amount of empty plasmid. Caspase 8 cells were co-transfected with the caspase 8 plasmid and the same amount of empty plasmid used in the various LAT samples. Each bar represents the average +/− SE of 3 independent experiments, each done at least in triplicate.
To determine the effects of these ORF knockouts on caspase 9 induced apoptosis, experiments performed as above were done using a caspase 9, rather than a caspase 8, expressing plasmid to induce apoptosis (Fig 3). Caspase 9 reduced cell survival to approximately 30% of the control. As with caspase 8 induced apoptosis, the intact LAT3.3A plasmid efficiently blocked caspase 9 induced cell death (P<0.05 versus caspase 9). As with caspase 8 induced apoptosis, the LAT3.3E plasmid, knocked out for the first 2 ORFs, provided intermediate protection. LAT3.3E provided significant protection against caspase 9 induced apoptosis (P<0.05), but the protection was significantly less than that provided by LAT3.3A (P<0.05). In contrast to the caspase 8 results, LAT3.3I was significantly different from LAT3.3E (P<0.05) and did not appear to provide any protection against caspase 9 induced apoptosis (P>0.05). On the other hand, as for caspase 8 induced apoptosis LAT3.3U did not appear to provide any protection against caspase 9 induced apoptosis. LAT3.3U was not significantly different from caspase 9 (P>0.05), but was significantly different from the control, LAT3.3A, or LAT3.3E (P<0.05). Thus, as with protection against caspase 8 induced apoptosis, knocking out either ORFs 1 and 2, ORFs 3–8, or all eight ORFs, appeared to reduce the ability of LAT to interfere with apoptosis. Knocking out all eight ORFs appeared to more effectively inhibit LAT’s anti-apoptosis activity compared to knocking out just the first 2 ORFs. Knocking out just ORFs 1 and 2 produced intermediate protection, while knocking out all 8 ORFs or just ORFs 3 to 8, appeared to completely block LAT’s ability to block caspase 9 induced apoptosis.
Figure 3.
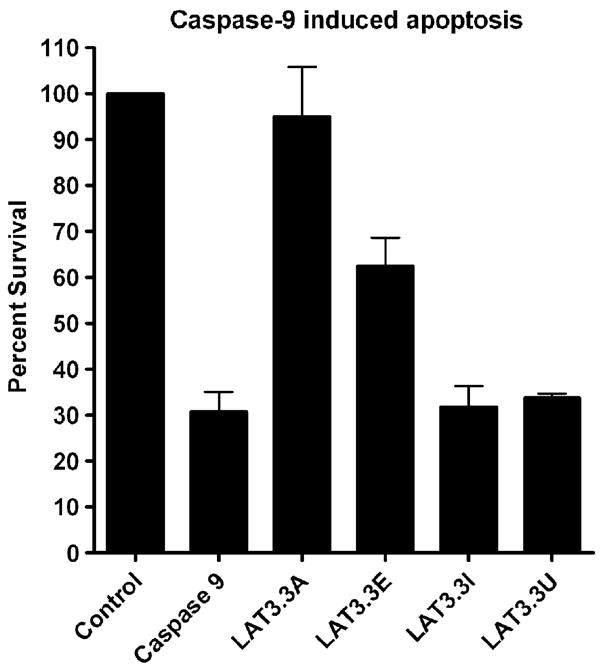
Inhibition of caspase 9 induced apoptosis by LAT constructs. Experiments were done as described for Fig. 2, except that a caspase 9 expressing plasmid was used instead of a caspase 8 expressing plasmid. Each bar represents the average +/− SE of 4 independent experiments, each done at least in triplicate.
The above results show that preventing or knocking out potential protein synthesis from the 8 potential LAT ORFs located within the functional first 1.5 kb of LAT can reduce that ability of a LAT plasmid to block apoptosis induced by either caspase 8 or caspase 9. However, whether these results indicate that there is one of more LAT protein involved in LAT’s anti-apoptosis activity remains unclear. These experiments were undertaken with the expectation that knocking out the LAT ORFs by introducing single nucleotide changes (ATG to TTG) would have little or no impact on LAT’s anti-apoptosis activity thus confirming that none of the eight potential LAT proteins was critical for LAT’s anti-apoptosis activity and hence LAT’s ability to enhance the reactivation phenotype.
If interpretation of the results are artificially restricted only to effects on translation of potential LAT proteins, one would have to conclude that LAT encodes at least two proteins, one from the exon (ORFs 1 or 2) and one from the beginning of the 2 kb intron (ORFs 3, 4, 5, 6, 7, or 8). However, other explanations are at least as likely. The single nucleotide changes may have affected the overall structure of the LAT RNA thus reducing an anti-apoptosis activity that is mediated by a large portion of the first 1.5 kb of LAT. The changes may have affected stability of the transcript thus decreasing the steady state amount of functional LAT product, regardless of its nature. However, this is unlikely because of the differential effect of LAT3.3I on caspase 8 versus caspase 9 induced apoptosis (see below). Alternatively, LAT may encode numerous miRNAs or other small RNAs that have a cumulative affect on suppressing apoptosis. Although none of the mutations introduced overlap with the first LAT miRNA reported [17], we have detected 2 additional small LAT derived RNAs both of which would be affected by the introduced mutations (unpublished). It is also possible that one or more as yet undetected small splices could fuse some of the ORFs together to generate a larger functional protein.
Finally, the ability to block apoptosis induced by caspase 8 versus caspase 9 appeared similar for each of the plasmids except LAT3.3I. This plasmid which is knocked out for ORFs 3, 4, 5, 6, 7, and 8 provided partial protection against caspase 8 induced apoptosis that was similar to the level of protection provided by LAT3.3E, which is knocked out for ORFs 1 and 2. In contrast, LAT3.3I provided no apparent protection against caspase 9 induced apoptosis. This suggests that within the first 1.5 kb of LAT, different LAT regions interfere with apoptosis by influencing different parts of the apoptotic pathways
Inhibiting apoptosis appears to be the most important LAT function involved in enhancing the virus’ reactivation phenotype since we have shown that two different anti-apoptosis genes can restore the wild type reactivation phenotype to a LAT negative virus [20–22, 28]. Determining if LAT interferes (directly or indirectly) with apoptosis via its RNA or via a LAT protein is important to deciphering the molecular mechanism(s) by which LAT inhibits apoptosis, thereby enhancing the viral reactivation phenotype. This in turn will be important for developing a highly efficacious clinical intervention against HSV-1 reactivation and recurrent disease. Regardless of the exact mechanism, the results reported here indicate that very minor changes to the LAT sequence can result in significant changes to LAT’s anti-apoptosis activity.
Acknowledgments
This work was supported by Public Health Service grants EY13191, EY12823, EY16663, EY14900; P20RR15635, USDA grants 2005-01554, 2006-01627, The Discovery Eye Foundation, The Henry L. Guenther Foundation, and Research to Prevent Blindness. Dr. Wechsler is an RPB Senior Scientific Investigator. Dr. BenMohamed is an RPB Special Award Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–8. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 2.Nesburn AB, editor. Report of the corneal disease panel: Vision research: A national plan 1983–1987. C.V. Mosby Co; St. Louis: 1983. [Google Scholar]
- 3.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, et al. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol. 1987;61:3820–6. doi: 10.1128/jvi.61.12.3820-3826.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science. 1987;235:1056–9. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 5.Block TM, Deshmane S, Masonis J, Maggioncalda J, Valyi-Nagi T, Fraser NW. An HSV LAT null mutant reactivates slowly from latent infection and makes small plaques on CV-1 monolayers. Virology. 1993;192:618–30. doi: 10.1006/viro.1993.1078. [DOI] [PubMed] [Google Scholar]
- 6.Bloom DC, Devi-Rao GB, Hill JM, Stevens JG, Wagner EK. Molecular analysis of herpes simplex virus type 1 during epinephrine- induced reactivation of latently infected rabbits in vivo. J Virol. 1994;68:1283–92. doi: 10.1128/jvi.68.3.1283-1292.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J Virol. 1994;68:1271–82. doi: 10.1128/jvi.68.3.1271-1282.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, et al. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol. 1989;63:2893–900. doi: 10.1128/jvi.63.7.2893-2900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, et al. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol. 1994;68:8045–55. doi: 10.1128/jvi.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. The effect of latency-associated transcript on the herpes simplex virus type 1 latency-reactivation phenotype is mouse strain-dependent. J Gen Virol. 2001;82:1117–22. doi: 10.1099/0022-1317-82-5-1117. [DOI] [PubMed] [Google Scholar]
- 11.Trousdale MD, Steiner I, Spivack JG, Deshmane SL, Brown SM, MacLean AR, et al. In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-associated transcript variant in a rabbit eye model. J Virol. 1991;65:6989–93. doi: 10.1128/jvi.65.12.6989-6993.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perng G, Jones C, Ciacci-Zanella H, Henderson G, Yukht A, Slanina S, et al. Virus induced neuronal apoptosis blocked by the herpes simplex virus latency associated transcript (LAT) Science. 2000;287:1500–3. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 13.Inman M, Perng G, Henderson G, Ghiasi H, Nesburn A, Wechsler S, et al. Region of Herpes Simplex Virus type 1 latency-associated transcript sufficient for wild type spontaneous reactivation promotes cell survival in tissue culture. J Virol. 2001;75:3636–46. doi: 10.1128/JVI.75.8.3636-3646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson G, Peng W, Jin L, Perng GC, Nesburn AB, Wechsler SL, et al. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J Neurovirol. 2002;8:103–11. doi: 10.1080/13550280290101085. [DOI] [PubMed] [Google Scholar]
- 15.Jin L, Peng W, Perng GC, Brick DJ, Nesburn AB, Jones C, et al. Identification of Herpes Simplex Virus Type 1 Latency-Associated Transcript Sequences That both Inhibit Apoptosis and Enhance the Spontaneous Reactivation Phenotype. J Virol. 2003;77:6556–61. doi: 10.1128/JVI.77.11.6556-6561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng W, Jin L, Henderson G, Perng GC, Brick DJ, Nesburn AB, et al. Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J Neurovirol. 2004;10:260–5. doi: 10.1080/13550280490468690. [DOI] [PubMed] [Google Scholar]
- 17.Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature. 2006;442:82–5. doi: 10.1038/nature04836. [DOI] [PubMed] [Google Scholar]
- 18.Branco FJ, Fraser NW. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol. 2005;79:9019–25. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed M, Lock M, Miller CG, Fraser NW. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol. 2002;76:717–29. doi: 10.1128/JVI.76.2.717-729.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin L, Perng GC, Carpenter D, Mott KR, Osorio N, Naito J, et al. Reactivation phenotype in rabbits of a herpes simplex virus type 1 mutant containing an unrelated antiapoptosis gene in place of latency-associated transcript. J Neurovirol. 2007;13:78–84. doi: 10.1080/13550280601164333. [DOI] [PubMed] [Google Scholar]
- 21.Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, et al. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol. 2005;79:12286–95. doi: 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perng GC, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, et al. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol. 2002;76:1224–35. doi: 10.1128/JVI.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3- kilobase primary transcript. J Virol. 1996;70:976–84. doi: 10.1128/jvi.70.2.976-984.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. A 371-nucleotide region between the herpes simplex virus type 1 (HSV-1) LAT promoter and the 2-kilobase LAT is not essential for efficient spontaneous reactivation of latent HSV-1. J Virol. 1996;70:2014–8. doi: 10.1128/jvi.70.3.2014-2018.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247–54. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitz I, Kirchhoff S, Krammer PH. Regulation of death receptor-mediated apoptosis pathways. Int J Biochem Cell Biol. 2000;32:1123–36. doi: 10.1016/s1357-2725(00)00048-0. [DOI] [PubMed] [Google Scholar]
- 27.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–33. [PubMed] [Google Scholar]
- 28.Mott KR, Osorio N, Jin L, Brick DJ, Naito J, Cooper J, et al. The bovine herpesvirus-1 LR ORF2 is critical for this gene’s ability to restore the high wild-type reactivation phenotype to a herpes simplex virus-1 LAT null mutant. J Gen Virol. 2003;84:2975–85. doi: 10.1099/vir.0.19421-0. [DOI] [PubMed] [Google Scholar]
- 29.Drolet BS, Perng GC, Cohen J, Slanina SM, Yukht A, Nesburn AB, et al. The region of the herpes simplex virus type 1 LAT gene involved in spontaneous reactivation does not encode a functional protein. Virology. 1998;242:221–32. doi: 10.1006/viro.1997.9020. [DOI] [PubMed] [Google Scholar]