

Abstract
Aims
To determine the steady-state pharmacokinetics of perhexiline (PHX) enantiomers over one interdosing interval in CYP2D6 extensive and poor metabolizer (EM and PM, respectively) patients administered rac-PHX. To elucidate the processes responsible for enantioselectivity, particularly in PM patients.
Methods
Blood samples were taken over one interdosing interval from six EM and two PM patients at steady-state with respect to rac-PHX metabolism. Complete urine collections were taken from five EM patients. PHX concentrations in plasma and urine were determined with enantioselective high-performance liquid chromatography methods.
Results
EM patients had 16- and 10-fold greater median apparent oral clearances of (+)- and (−)-PHX, respectively, than PM patients (P < 0.05 for both) and required significantly larger doses of rac-PHX (69 vs. 4.2 µg kg−1 h−1, P < 0.05) to maintain therapeutic concentrations in plasma. Patient phenotypes were consistent with CYP2D6 genotypes. Both groups displayed enantioselective pharmacokinetics, with higher apparent oral clearances for (−)-PHX compared with (+)-PHX, although PM patients exhibited significantly greater enantioselectivity (P < 0.05). The renal clearance of PHX enantiomers was not enantioselective and accounted for <1% of the median apparent oral clearance of each enantiomer in EM patients. Assuming the same renal clearances for PM patients accounts for approximately 9 and 4% of their median apparent oral clearances of (+)- and (−)-PHX, respectively.
Conclusions
The enantioselective pharmacokinetics of PHX are primarily due to metabolism by CYP2D6 in EM patients. The mechanism responsible for the enantioselective pharmacokinetics of PHX in PM patients is unknown, but may be due to enantioselective biliary or intestinal excretion.
What is already known about this subject
Perhexiline (PHX) is administered as a racemic mixture and exhibits enantioselective pharmacokinetics in both poor and extensive metabolizers of CYP2D6 (PM and EM, respectively).
Extensive metabolism by CYP2D6 is primarily responsible for the observed enantioselectivity in EM, but the process responsible in PM is unknown.
Analysis of the steady-state plasma concentration–time profiles of the enantiomers of PHX in PM and EM was undertaken in order to elucidate the observed enantioselectivity, particularly with respect to PM.
What this study adds
This is the first study to examine the steady-state plasma concentration–time profiles of the enantiomers of PHX in EM and PM over the course of an interdosing interval.
The apparent oral clearance of each enantiomer was calculated from their respective AUC rather than from trough concentrations and was enantioselective in both phenotypes, with higher apparent oral clearances of (−)-than (+)-PHX.
Renal clearance, calculated for EM and subsequently assumed for PM, constitutes a greater proportion of the total apparent oral clearance of each enantiomer in PM than EM, but was not enantioselective and thus unable to explain the enantioselectivity observed in PM.
Keywords: CYP2D6, enantioselective, perhexiline, pharmacokinetics
Introduction
Perhexiline (PHX) was introduced to clinical use over 30 years ago as an efficacious monotherapy for the treatment of angina [1]. Its mode of action was originally considered to be mediated by calcium channel antagonism producing coronary artery vasodilation [2]. A metabolic rather than haemodynamic action is now favoured [3] as an explanation for its efficacy in patients receiving maximal conventional antianginal therapy [4, 5] or in patients suffering either ischaemic or non-ischaemic chronic heart failure [6]. The metabolic action of PHX was postulated to be through the inhibition of mitochondrial carnitine palmitoyltransferase-1 (CPT-1) [7] altering the substrate preference for myocardial energy metabolism from fatty acids to carbohydrates to beneficial effect during ischaemia [8]; new evidence suggests that PHX may increase myocardial efficiency by a mechanism largely independent of its effects on CPT-1 [9].
The use of PHX is limited by its potential to cause peripheral neuropathy and severe hepatotoxicity associated with elevated concentrations of PHX in plasma [10, 11]. Therapeutic drug monitoring is necessary in order to maintain a concentration range in plasma of 0.15–0.60 mg l−1[4, 5]. The dosage required to maintain this range in a group of 23 patients was reported to vary 50-fold and be the result of polymorphic metabolism by the cytochrome P450 2D6 (CYP2D6) [12]. CYP2D6 phenotype is assigned from the metabolic ratio of the concentration in plasma of the CYP2D6-dependent metabolite cis-OH-PHX to PHX. The frequency distribution of the metabolic ratio is bimodal, with a reported antimode of 0.3 [12] or 0.4 [13] segregating approximately 10% of the total population as poor metabolizers of CYP2D6 (PM). Skewing of the distribution of metabolic ratios within the population of extensive metabolizers of CYP2D6 (EM) suggests the presence of a subpopulation of ultrarapid metabolizers of CYP2D6 (UM) [12, 13]. We have previously reported that the metabolic ratio, as a measure of the relative CYP2D6 metabolic capacity of a patient, may be used as an estimate of apparent oral clearance for individualizing long-term maintenance regimens [12, 14, 15]. EM patients require 50–500 mg of PHX maleate administered daily to maintain therapeutic concentrations in plasma, whereas PM patients are typically maintained with one dose of 50–150 mg administered once weekly [12].
Individuals with two null CYP2D6 alleles have no detectable CYP2D6 activity. The most frequent null allele in Whites is CYP2D6*4 and homozygous individuals account for 70–90% of PM. CYP2D6*9, *10 and *41 are associated with decreased activity, but are not as frequent as CYP2D6*1 and *2, associated with normal activity. Individuals with alleles containing duplicated active genes account for approximately 20% of UM [16]. Several recent studies have demonstrated a direct relationship between CYP2D6 genotype and the clinical pharmacokinetics of PHX [13–15, 17].
PHX is a chiral molecule due to asymmetry of the second carbon of the piperidine ring (Figure 1) and is formulated for therapeutic use with a racemic mixture of the (+) and (−) enantiomers (Pexsig®; Sigma Pharmaceuticals, Clayton, Victoria, Australia). Both EM and PM exhibit distinct enantioselectivity with respect to rac-PHX metabolism, with a median ratio of (−)- to (+)-PHX apparent oral clearance of 1.5 and 2.3, respectively, calculated from the trough concentrations of each enantiomer in plasma [17]. For EM, in whom CYP2D6-mediated hepatic metabolism contributes approximately 90% to the total apparent oral clearance of each enantiomer of PHX [17], enantioselective apparent oral clearance is attributed to this enzyme, although no similar CYP-mediated intrinsic enantioselectivity has been described for PM [18]. The current study investigates the pharmacokinetics of the enantiomers of PHX over the course of one interdosing interval in six EM and two PM patients at steady-state with respect to rac-PHX metabolism. The results of the current study and published data are discussed in an attempt to elucidate the processes responsible for the observed enantioselectivity, particularly with respect to PM patients.
Figure 1.
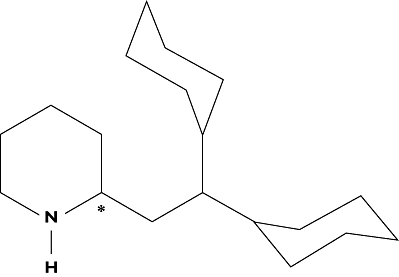
Chemical structure of perhexiline. The chiral carbon is indicated with an asterisk
Methods
Patients
Approval was obtained from the Ethics of Human Research Committee of The Queen Elizabeth Hospital. From patients admitted to the Cardiology Unit of The Queen Elizabeth Hospital, six EM and two PM were identified from their ratio of the concentration in plasma of cis-OH-(+/–)-PHX to (+/–)-PHX [12] whose dose of rac-PHX had not changed for at least 1 month; all gave written informed consent prior to participation. Six of the patients were male and two female. Body weights ranged from 45.9 to 121.0 kg (median 62.4 kg) and age ranged from 77 to 88 years (median 80 years). For the EM patients, the dose ranged from 50 mg of rac-PHX maleate once per day to 100 mg twice per day. It was necessary to split the dosing regimen of the first PM patient (PM1) over 2 weeks to achieve concentrations within the therapeutic range; 50 mg was administered as a single dose in the first week and 100 mg as a single dose in the alternate week. PM2 took 100 mg once each week. The dosage regimens and concomitant medications are presented in Table 1. Body weight is shown in Table 2. The length of continuous treatment with rac-PHX at the time of the study ranged from 1 month to 4 years and 11 months (median 1 year 10 months); the length of treatment with the current dose ranged from 33 to 589 days (median 195 days). EM1–5 and PM1 were also subjects for a previous study describing the determination of the 4-monohydroxy metabolites of PHX in human plasma, urine and liver microsomes by liquid chromatography [19].
Table 1.
Perhexiline and other drug therapy in six CYP2D6 EM and two CYP2D6 PM patients
| Patient | Rac-perhexiline maleate | Other drugs |
|---|---|---|
| EM1 | 100 mg a.m., 100 mg p.m. | Aspirin, clopidogrel, perindopril, metoprolol, simvastatin, frusemide, metformin, glicazide, isosorbide dinitrate |
| EM2 | 150 mg a.m. | Aspirin, insulin (s.c.), morphine (s.c.), verapamil, pantoprazole, prednisolone, spironolactone, venlafaxine, paracetamol, frusemide, isosorbide mononitrate, ipratropium (inhalation), salbutamol (inhalation) |
| EM3 | 100 mg a.m. | Frusemide, paroxetine, pantoprazole, theophylline, aspirin, spironolactone, salmeterol (inhalation), fluticasone (inhalation), prednisolone |
| EM4 | 50 mg a.m. | Diltiazem, amitriptyline, prednisolone, glimepiride, frusemide, tiotropium (inhalation), salmeterol (inhalation), fluticasone (inhalation), ramipril, aspirin, isosorbide mononitrate, theophylline |
| EM5 | 100 mg a.m., 100 mg p.m. | Clopidogrel, ramipril, pravastatin, isosorbide mononitrate, metoclopramide (i.v.) |
| EM6 | 100 mg a.m., 100 mg p.m. | Glyceryl trinitrate, aspirin, irbesartan, pantoprazole, clopidogrel, isosorbide dinitrate, pravastatin |
| PM1 | 50 and 100 mg, alternate weeks | Omeprazole, isosorbide mononitrate, trandolapril, clopidogrel, risedronate, allopurinol, frusemide |
| PM2 | 100 mg weekly | Aspirin, diltiazem, atorvastatin, gliclazide, thyroxine, indapamide, irbesartan, amitriptyline, isosorbide dinitrate, clopidogrel |
Administration was oral unless otherwise indicated. EM, extensive metabolizer; PM, poor metabolizer; s.c., subcutaneous; i.v., intravenous.
Table 2.
CYP2D6 genotype, metabolic ratio and the disposition of (+)- and (−)-perhexiline (PHX) at steady-state in six EM and two PM patients
| CL/F (ml h−1 kg−1) | |||||||
|---|---|---|---|---|---|---|---|
| Patient | CYP2D6 genotype | cis-OH-(+/–)-PHX:(+/–)-PHX | Body weight (kg) | Dose rate (µg kg−1 h−1)rac-PHX | (+)-PHX | (−)-PHX | CL/F (−)-PHX: CL/F (+)-PHX |
| EM1 | *2/*4 | 11.7 | 59.1 | 99 | 318 | 416 | 1.31 |
| EM2 | *1/*4 | 3.6 | 112.3 | 39 | 69 | 88 | 1.28 |
| EM3 | *2/*6 | 0.8 | 94.6 | 31 | 34 | 45 | 1.32 |
| EM4 | *4/*10 | 10.3 | 51.7 | 28 | 145 | 163 | 1.13 |
| EM5 | *1/*1 | 7.9 | 46.2 | 127 | 211 | 274 | 1.30 |
| EM6 | *2/*2 | 4.5 | 45.9 | 128 | 376 | 534 | 1.42 |
| Median | 69* | 178* | 219* | 1.30* | |||
| PM1 | *4/*4 | 0.0 | 65.6 | 4.8 | 10.7 | 21.9 | 2.05 |
| PM2 | *4/*4 | 0.0 | 121.0 | 3.5 | 11.2 | 23.9 | 2.15 |
| Median | 4.2* | 11.0* | 23.0* | 2.10* | |||
Indicates a statistically significant difference (P < 0.05) between the EM and PM patient groups. EM, extensive metabolizer; PM, poor metabolizer.
Blood samples were collected over the duration of one interdosing interval. For the EM patients, samples were drawn from an indwelling venous catheter at times 0 (predose), 0.5, 1, 2, 4, 8 and 12 h. Three of the patients were taking rac-PHX maleate only once daily and thus had a subsequent sample drawn at 24 h by venepuncture. Patency of the catheter was maintained by instilling 1.5 ml of heparinized saline (15 U heparin) after each sample was drawn. The first 1.5 ml of blood drawn was discarded to prevent dilution by the heparinized saline. For the PM patients, blood samples were drawn by venepuncture at times 0 (predose), 8, 24, 72, 120 and 168 h. Because PM1 took 50 and 100 mg of rac-PHX maleate on alternating weeks, the 168-h sample was drawn before the 100-mg dose was given and further samples were drawn at times 176, 192, 240, 288 and 336 h. Blood was collected in ethylenediamine tetraaceticacid tubes, centrifuged immediately and the plasma collected. All of the urine of EM1–5 was collected over their respective dosing intervals. The urine collection for EM6 was incomplete. Urine was not collected from the PM patients because the collection time was prohibitively long. Plasma and urine samples were stored at −20°C pending assay for the enantiomers of PHX.
Genomic DNA was isolated from whole blood using a QIAamp® DNA mini kit, according to the manufacturer's protocol (Qiagen Pty Ltd, Clifton Hill, Australia). CYP2D6 genotyping was performed as previously reported [15, 20]. The genotypes of the patients are presented in Table 2.
Determination of the enantiomers of PHX in plasma and urine
Concentrations of each enantiomer of PHX in plasma were determined by a previously published method [21]. Concentrations in urine were determined by substituting urine for plasma after validation according to the protocol described by Shah et al.[22]. The intra-assay (n = 6) bias and coefficient of variation (CV) were <10% for the top calibrator (2.0 mg l−1) and <15% for the bottom calibrator (0.010 mg l−1, lowest limit of quantification) for each PHX enantiomer. The interassay (n = 7) bias and CV were <15% for the low-concentration quality control (QC) samples (0.030 mg l−1) and <10% for the medium- and high-concentration QC samples (0.40 and 0.75 mg l−1, respectively). The relative extraction efficiencies from urine for both enantiomers and the internal standard were >90%. Drugs coadministered to patients receiving treatment with rac-PHX maleate were screened for possible chromatographic interference by analysis of urine from patients admitted to the Cardiology Unit of The Queen Elizabeth Hospital who were not taking rac-PHX maleate.
Steady-state pharmacokinetic and statistical analysis
The areas under the plasma (+)- and (−)-PHX concentration–time curves during the interdosing interval at steady-state (AUCτss) were calculated by the linear trapezoidal method. The predose steady-state concentration in plasma (Cinitialss) and the final steady-state concentration in plasma (Cfinalss) were determined by direct measurement. Apparent oral clearance at steady-state (CL/F) was calculated as the dose of enantiomer (as free base) divided by the corresponding AUCτss and the renal clearance (CLR) as the amount recovered in urine (as free base) over the interdosing interval divided by the corresponding AUCτss. Creatinine clearance reported previously [19] was used as an estimate of glomerular filtration rate (GFR). Renal clearance of (+)- or (−)-PHX by glomerular filtration (CLGF) was estimated as the product of GFR and the average unbound fraction of (+)- or (−)-PHX in plasma, determined previously by equilibrium dialysis of spiked pooled plasma samples [17]. Dose rate, CL/F, CLR and CLGF were corrected for body weight. Cinitialss and Cfinalss for all patients were compared with a two-tailed paired t-test. Comparisons between the EM and PM patient groups were by one-tailed Mann–Whitney tests. P-values <0.05 were considered statistically significant.
Results
The median (range) trough concentration in plasma of rac-PHX for all the patients was 0.29 mg l−1 (0.18–0.76). Only EM3 was maintained outside the therapeutic range. The plasma (+)- and (−)-PHX concentration–time profiles for the EM and PM patients are presented in Figures 2 and 3, respectively. Cinitialss and Cfinalss were not significantly different [P-value; mean difference; 95% confidence interval (CI) of difference] for (+)- (0.111; −0.009 mg l−1; −0.020, 0.003) or (−)-PHX (0.095; −0.006 mg l−1; −0.014, 0.001).
Figure 2.
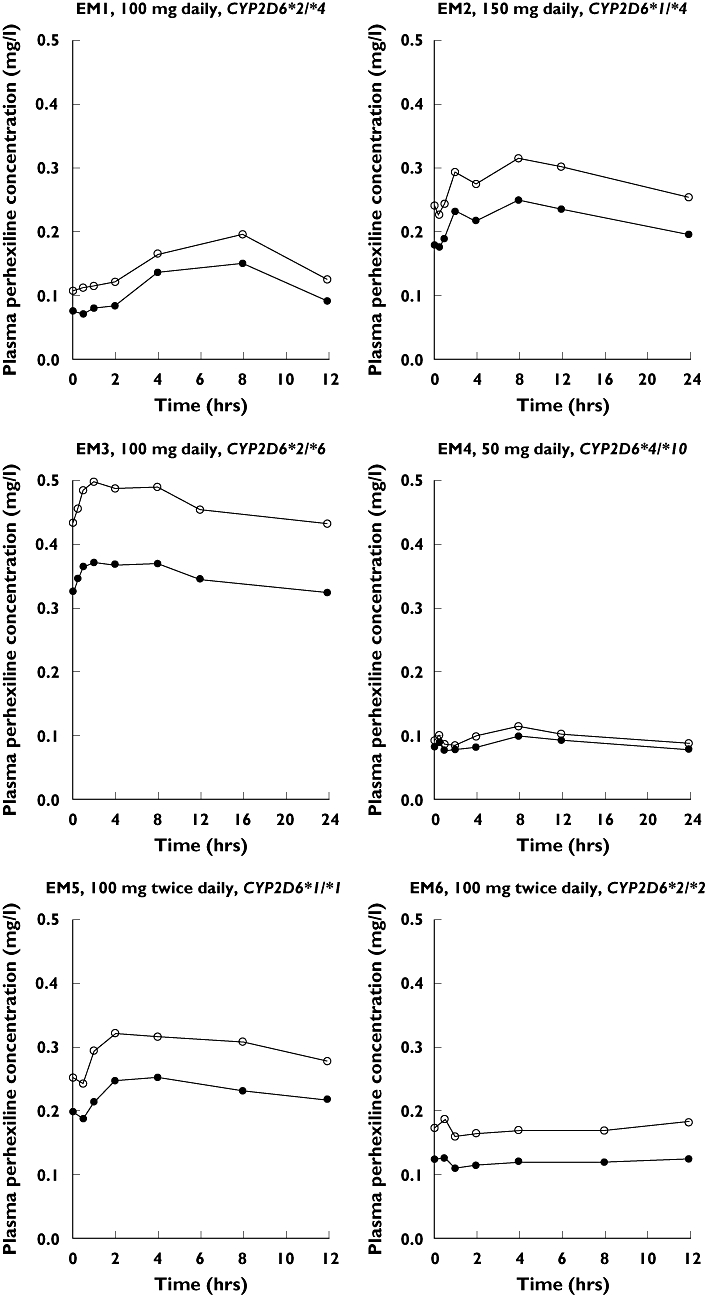
Rac-perhexiline (PHX) maleate maintenance dosage regimen, CYP2D6 genotype and plasma (+)- (○) and (−)-PHX ( ) concentration–time profiles for the six extensive metabolizer (EM) patients. Note that the time axis is dependent on the dosage regimen
) concentration–time profiles for the six extensive metabolizer (EM) patients. Note that the time axis is dependent on the dosage regimen
Figure 3.
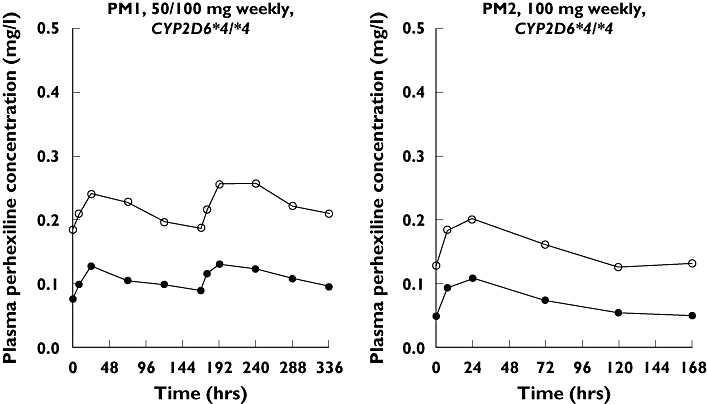
Rac-perhexiline (PHX) maleate maintenance dosage regimen, CYP2D6 genotype and plasma (+)- (○) and (−)-PHX ( ) concentration–time profiles for the two poor metabolizer (PM) patients. Note that the time axis is dependent on the dosage regimen
) concentration–time profiles for the two poor metabolizer (PM) patients. Note that the time axis is dependent on the dosage regimen
Patient phenotypes were consistent with CYP2D6 genotypes (Table 2). Both PM patients had no functional CYP2D6 alleles. EM1–4 each had one functional CYP2D6 allele and EM5 and 6 each had two functional CYP2D6 alleles. The median dose rate of rac-PHX and the CL/F of (+)- and (−)-PHX was significantly higher (P < 0.05 for all) for the EM patient group than the PM patient group. In both patient groups the CL/F of (−)-PHX was greater than that of (+)-PHX, although the median ratio of the CL/F of (−)-PHX to the CL/F of (+)-PHX was significantly greater (P < 0.05) for the PM patient group than for the EM patient group (Table 2).
The mean (95% CI) recovery of the dose of rac-PHX (as free base) in urine of the EM patients studied over the dosing interval was 0.74% (−0.17, 1.66). The CLR of (+)- and (−)-PHX exhibited no enantioselectivity. The estimated CLGF of (+)- or (−)-PHX was lower than the corresponding CLR for EM1, 2 and 4 and higher for EM3 and 5 (Table 3).
Table 3.
Renal excretion of (+)- and (−)-PHX at steady-state in five EM patients
| Patient | Dose recovered (%) (+)-PHX | (−)-PHX: | CLGF (ml h−1 kg−1) (+)-PHX | (+)-PHX | CLR (ml h−1 kg−1) | (−)-PHX: | CLR (−)-PHX CLR (+)-PHX |
|---|---|---|---|---|---|---|---|
| EM1 | 0.75 | 0.55 | 0.22 | 0.16 | 2.40 | 2.30 | 0.96 |
| EM2 | 2.28 | 1.73 | 0.36 | 0.26 | 1.58 | 1.53 | 0.97 |
| EM3 | 0.48 | 0.37 | 0.74 | 0.54 | 0.16 | 0.16 | 1.00 |
| EM4 | 0.57 | 0.51 | 0.44 | 0.33 | 0.83 | 0.83 | 1.00 |
| EM5 | 0.09 | 0.08 | 0.51 | 0.38 | 0.20 | 0.21 | 1.05 |
| Mean | 0.84 | 0.65 | 0.45 | 0.33 | 1.03 | 1.01 | 1.00 |
| (95% CI) | (−0.21, 1.88) | (−0.14, 1.43) | (0.21, 0.69) | (0.16, 0.51) | (−0.15, 2.22) | (−0.13, 2.14) | (0.95, 1.04) |
PHX, Perhexiline. EM, extensive metabolizer.
Discussion
The results of the current study confirm the importance of CYP2D6 in the in vivo metabolism of both (+)- and (−)-PHX. EM patients, with at least one functional CYP2D6 allele, had significantly greater apparent oral clearances of both PHX enantiomers than PM patients, with no functional CYP2D6 alleles, and accordingly required significantly larger doses of rac-PHX in order to maintain similar therapeutic rac-PHX concentrations in plasma.
The current study also confirms that significant enantioselectivity in the pharmacokinetics of PHX occurs in both EM and PM patients. In EM patients, the greater CL/F of (−)- than (+)-PHX is consistent with the recently reported enantioselective in vitro metabolism by EM human liver microsomes, catalysed almost entirely by CYP2D6 [18]. In contrast, no enantioselectivity was observed in the in vitro metabolism by PM human liver microsomes. Thus, the significant in vivo enantioselectivity in the pharmacokinetics of rac-PHX in PM patients is unlikely to be due to enantioselective CYP-mediated hepatic metabolism. From the difference in median CL/F between EM and PM patient groups in the current study and that of Inglis et al.[17], it is clear that CYP2D6-catalysed metabolism accounts for approximately 90–95% of the total apparent oral clearance of each enantiomer of PHX in EM patients. There must be another process, unmasked by the absence of CYP2D6 in PM patients, which is more enantioselective. Obviously, this pathway could contribute only a small fraction of total apparent oral clearance in EM patients, yet represent a much greater proportion in PM patients.
Similar to an observation reported for rac-PHX [15], apparent oral clearance of both PHX enantiomers was greater in patients possessing at least one CYP2D6*2 allele than in patients possessing any other combination of functional CYP2D6 alleles, with the exception of EM3. In this patient the apparent oral clearance of both PHX enantiomers by CYP2D6 was probably inhibited by the coadministration of paroxetine. The potential for a clinically significant pharmacokinetic interaction between rac-PHX and the selective serotonin reuptake inhibitors paroxetine, fluoxetine [23] and citalopram [24] has been documented previously. Prior to commencing daily treatment with 20 mg of paroxetine, EM3 was stably maintained within the therapeutic range of rac-PHX concentrations in plasma (data not shown). The subsequent interaction produced an increase in steady-state trough rac-PHX concentration of 38%, 27% above the therapeutic range, and a similar decrease in metabolic ratio, although the observed inhibition of CYP2D6 was not sufficient to produce phenocopying from EM to PM. At therapeutic doses, rac-PHX [14], paroxetine and fluoxetine [25] have been shown to produce phenocopying of EM to PM using the dextromethorphan to dextrorphan urinary metabolic ratio in approximately 40–80% of EM. It is unlikely that other drugs taken by the patients would be sufficiently potent in vivo inhibitors of CYP2D6 to impact rac-PHX pharmacokinetics significantly, although the reverse is possible.
In the five EM patients for whom a complete urine collection was possible, the mean proportion of a dose of rac-PHX recovered unchanged was <1%, consistent with the recoveries reported by Singlas et al.[26] for EM patients at steady state. Recovery of unchanged rac-PHX from the urine of EM patients following a single dose is an order of magnitude lower [27] because hepatic metabolism by CYP2D6 is less likely to be saturated than at the therapeutic concentrations attained in plasma following repeated dosing [12, 14, 17].
The CLR of (+)- and (−)-PHX varied more than 10-fold among the five EM patients, accounted for ≤2% of the apparent oral clearance of each enantiomer and exhibited no stereoselectivity. Renal clearance of both enantiomers of PHX may have been subject to active tubular secretion and passive tubular reabsorption due to the differences between CLR and CLGF, although this may have been due in part to the use of an average unbound fraction of (+)- or (−)-PHX, rather than determining it for individual patients. Although not determined, an assumed mean renal clearance of 1 ml h−1 kg−1 for each enantiomer in PM1 and 2 would equate to approximately 9 and 4% of the total apparent oral clearances of (+)- and (−)-PHX, respectively. However, this is not a major contributor to total apparent oral clearance and thus is unable to explain the observed enantioselective pharmacokinetics in PM patients.
PHX may be subject to enantioselective biliary or intestinal excretion, although there are few data to support these hypotheses. Cooper et al.[28] have reported that in two patients with biliary T-tubes, the biliary excretion rate of unchanged rac-PHX was two to three orders of magnitude greater than the excretion rate of unchanged rac-PHX in urine, despite the fact that the T-tubes collected only a proportion of the bile draining via the common bile duct. If the renal clearance of unchanged (+)- and (−)-PHX is <10% of their respective total apparent oral clearances in PM patients, it follows that biliary excretion of rac-PHX may be greater than this and a major contributor to total apparent oral clearance in PM patients. The absolute clearance by this route would be the net difference between canalicular secretion into bile and subsequent reabsorption and efflux from and into the gastrointestinal tract, and could be highly enantioselective. This could account for the significant enantioselectivity in the pharmacokinetics of (+)- and (−)-PHX in PM patients, yet still be an order of magnitude smaller than the less enantioselective apparent oral clearance accounted for by CYP2D6 in EM patients. There was no evidence of secondary peaks in the (+)- and (−)-PHX plasma concentration–time profiles of the two PM patients to support the notion of enterohepatic recirculation, although this may have been an artefact of the relatively large intervals in the sampling protocol for the PM patients. Given the advanced age, cardiovascular disease and frequent comorbidities of most patients receiving treatment with rac-PHX maleate, animal experiments and in vitro models are necessary to test these hypotheses.
In conclusion, although both EM and PM patient groups display enantioselective pharmacokinetics, the ratio of the apparent oral clearance of (−)- to (+)-PHX is significantly greater in PM. Because therapeutic drug monitoring targets a racemic concentration range in plasma, PM patients are effectively exposed to greater concentrations of (+)-PHX and lower concentrations of (−)-PHX than EM patients. To date, the pharmacodynamic activities and adverse effects of the individual enantiomers of PHX have not been determined. Once this has been established, therapeutic drug monitoring may be improved by developing specific enantiomer target concentration ranges in plasma for racemic preparations of PHX.
Acknowledgments
This work was funded by a project grant from the National Heart Foundation of Australia. B.J.D. is the recipient of the MF and MH Joyner Scholarship in Medicine and the Freemasons Medical Research Scholarship of the Faculty of Health Sciences, University of Adelaide.
References
- 1.Armstrong ML. A comparative study of perhexiline, beta-adrenergic blocking agents and placebos in the management of angina pectoris. Postgrad Med J. 1973;49(Suppl. 3):108–11. [PubMed] [Google Scholar]
- 2.Opie LH. Drugs and the heart. III. Calcium antagonists. Lancet. 1980;1:806–10. doi: 10.1016/s0140-6736(80)91303-3. [DOI] [PubMed] [Google Scholar]
- 3.Lee L, Horowitz J, Frenneaux M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur Heart J. 2004;25:634–41. doi: 10.1016/j.ehj.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Horowitz JD, Sia STB, Macdonald PS, Goble AJ, Louis WJ. Perhexiline maleate for severe angina pectoris—correlations with pharmacokinetics. Int J Cardiol. 1986;13:219–29. doi: 10.1016/0167-5273(86)90146-4. [DOI] [PubMed] [Google Scholar]
- 5.Cole PL, Beamer AD, McGowan N, Cantillon CO, Benfell K, Kelly RA, Hartley LH, Smith TW, Antman EM. Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation. 1990;81:1260–70. doi: 10.1161/01.cir.81.4.1260. [DOI] [PubMed] [Google Scholar]
- 6.Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, Ashrafian H, Horowitz J, Fraser AG, Clarke K, Frenneaux M. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112:3280–8. doi: 10.1161/CIRCULATIONAHA.105.551457. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol. 1996;52:273–80. doi: 10.1016/0006-2952(96)00204-3. [DOI] [PubMed] [Google Scholar]
- 8.Jeffrey FM, Alvarez L, Diczku V, Sherry AD, Malloy CR. Direct evidence that perhexiline modifies myocardial substrate utilisation from fatty acids to lactate. J Cardiovasc Pharmacol. 1995;25:469–72. doi: 10.1097/00005344-199503000-00018. [DOI] [PubMed] [Google Scholar]
- 9.Unger SA, Kennedy JA, McFadden-Lewis K, Minerds K, Murphy GA, Horowitz JD. Dissociation between metabolic and efficiency effects of perhexiline in normoxic rat myocardium. J Cardiovasc Pharmacol. 2005;46:849–55. doi: 10.1097/01.fjc.0000190488.77434.f1. [DOI] [PubMed] [Google Scholar]
- 10.Morgan MY, Reshef R, Shah RR, Oates NS, Smith RL, Sherlock S. Impaired oxidation of debrisoquine in patients with perhexiline liver injury. Gut. 1984;25:1057–64. doi: 10.1136/gut.25.10.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah RR, Oates NS, Idle JR, Smith RL, Lockhart JD. Impaired oxidation of debrisoquine in patients with perhexiline neuropathy. BMJ. 1982;284:295–9. doi: 10.1136/bmj.284.6312.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sallustio BC, Westley IC, Morris RG. Pharmacokinetics of the antianginal agent perhexiline: relationship between metabolic ratio and steady state dose. Br J Clin Pharmacol. 2002;54:107–14. doi: 10.1046/j.1365-2125.2002.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barclay ML, Sawyers SM, Begg EJ, Zhang M, Roberts RL, Kennedy MA, Elliott JM. Correlation of CYP2D6 genotype with perhexiline phenotypic metabolizer status. Pharmacogenetics. 2003;13:627–32. doi: 10.1097/00008571-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Davies BJ, Coller JK, James HM, Gillis D, Somogyi AA, Horowitz JD, Morris RG, Sallustio BC. Clinical inhibition of CYP2D6-catalysed metabolism by the antianginal agent perhexiline. Br J Clin Pharmacol. 2004;57:456–63. doi: 10.1046/j.1365-2125.2003.02033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies BJ, Coller JK, James HM, Somogyi AA, Horowitz JD, Sallustio BC. The influence of CYP2D6 genotype on trough plasma perhexiline and cis-OH-perhexiline concentrations following a standard loading regimen in patients with myocardial ischaemia. Br J Clin Pharmacol. 2006;61:321–5. doi: 10.1111/j.1365-2125.2005.02570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:23–37. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 17.Inglis SC, Herbert MK, Davies BJ, Coller JK, James HM, Horowitz JD, Morris RG, Milne RW, Somogyi AA, Sallustio BC. Effect of CYP2D6 metabolizer status on the disposition of the (+) and (−) enantiomers of perhexiline in patients with myocardial ischaemia. Pharmacogenet Genomics. 2007;17:305–12. doi: 10.1097/FPC.0b013e32800ffba0. [DOI] [PubMed] [Google Scholar]
- 18.Davies BJ, Coller JK, Somogyi AA, Milne RW, Sallustio BC. CYP2B6, CYP2D6, and CYP3A4 catalyze the primary oxidative metabolism of perhexiline enantiomers by human liver microsomes. Drug Metab Dispos. 2007;35:128–38. doi: 10.1124/dmd.106.012252. [DOI] [PubMed] [Google Scholar]
- 19.Davies BJ, Herbert MK, Coller JK, Somogyi AA, Milne RW, Sallustio BC. Determination of the 4-monohydroxy metabolites of perhexiline in human plasma, urine and liver microsomes by liquid chromatography. J Chromatogr B. 2006;843:302–9. doi: 10.1016/j.jchromb.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 20.James HM, Coller JK, Gillis D, Bahnisch J, Sallustio BC, Somogyi AA. A new simple diagnostic assay for the identification of the major CYP2D6 genotypes by DNA sequencing analysis. Int J Clin Pharmacol Ther. 2004;42:719–23. doi: 10.5414/cpp42719. [DOI] [PubMed] [Google Scholar]
- 21.Davies BJ, Herbert MK, Culbert JA, Pyke SM, Coller JK, Somogyi AA, Milne RW, Sallustio BC. Enantioselective assay for the determination of perhexiline enantiomers in human plasma by liquid chromatography. J Chromatogr B. 2006;832:114–20. doi: 10.1016/j.jchromb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 22.Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, Yacobi A, Layloff T, Viswanathan CT, Cook CE, McDowall RD, Pittman KA, Spector S. Analytical methods validation: bioavailability, bioequivalence, and pharmacokinetic studies. J Pharm Sci. 1992;81:309–12. doi: 10.1007/BF03189968. [DOI] [PubMed] [Google Scholar]
- 23.Alderman CP, Hundertmark JD, Soetratma TW. Interaction of serotonin re-uptake inhibitors with perhexiline. Aust NZ J Psychiatry. 1997;31:601–3. doi: 10.3109/00048679709065084. [DOI] [PubMed] [Google Scholar]
- 24.Nyfort-Hansen K. Perhexiline toxicity related to citalopram use. Med J Aust. 2002;176:560–1. doi: 10.5694/j.1326-5377.2002.tb04558.x. [DOI] [PubMed] [Google Scholar]
- 25.Alfaro CL, Lam YW, Simpson J, Ereshefsky L. CYP2D6 inhibition by fluoxetine, paroxetine, sertraline, and venlafaxine in a crossover study: intraindividual variability and plasma concentration correlations. J Clin Pharmacol. 2000;40:58–66. doi: 10.1177/00912700022008702. [DOI] [PubMed] [Google Scholar]
- 26.Singlas E, Goujet MA, Simon P. Pharmacokinetics of perhexiline maleate in anginal patients with and without peripheral neuropathy. Eur J Clin Pharmacol. 1978;14:195–201. doi: 10.1007/BF02089960. [DOI] [PubMed] [Google Scholar]
- 27.Amoah AG, Gould BJ, Parke DV, Lockhart JD. Further studies on the pharmacokinetics of perhexiline maleate in humans. Xenobiotica. 1986;16:63–8. doi: 10.3109/00498258609043506. [DOI] [PubMed] [Google Scholar]
- 28.Cooper RG, Evans DA, Price AH. Studies on the metabolism of perhexiline in man. Eur J Clin Pharmacol. 1987;32:569–76. doi: 10.1007/BF02455990. [DOI] [PubMed] [Google Scholar]