

Abstract
Aims
The drug-metabolizing capacity of transplanted liver highly influences drug efficacy or toxicity, particularly in the early postoperative period. The aim of our study was to predict therapeutic failures or severe adverse drug reactions by phenotyping for cytochrome P450 (P450) polymorphism resulting in reduced or no activity of the key drug-metabolizing enzymes.
Methods
A validated analytical system with metabolomic tools has been developed for estimation of the drug-metabolizing capacity of transplanted liver, which allows the prediction of potential poor metabolizer phenotypes of donors and facilitates improvement of the individual recipient therapy.
Results
Of the 109 liver donors in Hungary, the frequency of poor metabolizers was found to be 0.92%, 5.5% and 8.3% for CYP2C9, CYP2C19 and CYP2D6, respectively. In the present study, two liver grafts transplanted in paediatric recipients were reported to be poor metabolizer phenotypes. The liver grafts presented normal function in the early postoperative days; 2 weeks after transplantation, however, increasing liver enzymes were detected. Histological investigation of a liver biopsy suggested drug toxicity. The test of drug metabolizing status showed one of the liver grafts to be a CYP2C9 poor metabolizer, and the other was found to be a CYP2C19 poor metabolizer. Rationalization of the medication resulted in the recovery of both the grafts and the recipients within 1 week.
Conclusions
Prospective investigation of the P450 status may lead to the optimization of drug choice and/or dose for a more effective therapy, avoid serious adverse effects, and decrease medical costs. Phenotyping donor livers and tailored medication can contribute to the improvement of graft and recipient survival.
What is already known about this subject
The activity of drug-metabolizing enzymes, primarily cytochrome P450 enzymes, can determine a patient's response to a drug.
Therapeutic failure or drug toxicity in the postoperative period after liver transplantation is influenced by the drug metabolizing capacity of the graft.
Dose adjustment or selection of an alternative drug, which is not a substrate for the polymorphic enzyme may prevent the development of side-effects in recipients of poor metabolizer liver grafts.
What this study adds
A validated analytical system with metabolomic tools has been developed to estimate the drug-metabolizing capacity of transplanted liver, which allows the prediction of potential poor metabolizer phenotypes of donors and facilitates the improvement of individual recipient therapy.
In the test of drug-metabolizing status, one of the liver grafts was found to be a CYP2C9 poor metabolizer, while the other was a CYP2C19 poor metabolizer.
Rationalization of the medication resulted in the recovery of both the grafts and the recipients within 1 week.
Keywords: cytochrome P450, drug-induced toxicity, drug metabolizing capacity, liver-graft, poor-metabolizer phenotype
Introduction
Liver transplantation (LTx) is an effective treatment for patients with end stage liver disease. However, the widening gap between the waiting list and available cadaveric livers continues to affect adversely mortality rates of candidates on the waiting list. On the other hand, improvement in surgical techniques, anaesthesiology, intensive care and immunosuppression have led to increasing recipient and graft survival, and further efforts are made to minimize the risk of graft dysfunction after LTx. Early graft dysfunction is associated with the quality and type of liver graft, type of preservation fluid and length of cold and warm ischaemic time [1]. Late graft dysfunction is manifested by rejection, infection, or recurrence of liver disease [2]. One of the causes scarcely considered as a factor contributing to graft and patient survival is the drug-metabolizing capacity of the donor liver [3].
Therapeutic failure or drug toxicity in the postoperative period is influenced by the drug-metabolizing capacity of the graft. This capacity depends primarily on the levels and activities of the cytochrome P450 enzymes (P450). P450 enzymes play a central role in the biotransformation of various xenobiotics (drugs, pesticides, food additives, and chemical pollutants) to more polar compounds, which are readily excreted [4]. The metabolites of a drug can be inactive or less active than the parent compound, although some biotransformation products show enhanced pharmacological or toxicological activity. The activity of P450 enzymes can determine the recipient's response to a drug. Any change in the activity of P450 isoforms influences the rate of activation or inactivation of drugs. The metabolic conversion and excretion rate of drugs vary between individuals, from extremely slow to ultrafast. The most important reason for interindividual variation in drug metabolism is genetic polymorphism of P450 genes. Some P450 genes (CYP2C9, CYP2C19, CYP2D6, CYP3A5) are highly polymorphic, resulting in enzyme variants with reduced or even no activity [5]. The genetically determined variance in enzyme activity is further modulated by environmental factors (comedication, nutrition) resulting in different drug metabolism phenotypes [6]. For many drugs, three major phenotypes can be distinguished: poor metabolizers, intermediate metabolizers and extensive metabolizers [7]. As is well known, a significant proportion of adverse drug reactions as well as therapeutic failures is caused by genetically based differences in drug metabolism and elimination [8, 9]. Individuals with inactivating mutations and lack of functional activity in P450 enzymes need tailored drug treatment in order to avoid excessive drug concentration and toxic effects. Dose adjustment or selection of an alternative drug, which is not a substrate for the polymorphic enzyme, can prevent the development of side-effects in poor metabolizers [8].
Donor livers can be screened for drug-metabolizing capacity using phenotyping approaches. A validated analytical system with metabolomic tools has been established for estimation of the drug-metabolizing capacity of liver grafts. This system allows the prediction of donors with potential poor metabolizer phenotypes. Our knowledge of poor metabolizer status facilitates tailoring of the individual recipient therapy in the early postoperative period and also advances the rationalization of medication after liver transplantation. A well-designed therapeutic strategy adjusted to the drug-metabolizing capacity of the graft may also contribute to the reduction of hospitalization of the recipients. The main goal of this study was to determine the incidence of poor metabolizer phenotypes in liver donors in Hungary and to analyze the clinical impact of P450 polymorphism in the case of 16 donor-recipient pairs of reduced-size LTx.
Methods
Study design
Liver tissues of 109 donors retrieved during the period 1997–2006 by the liver team of Semmelweis University were used for phenotyping of the P450 status. Post-operative drug therapy for 16 patients who underwent split liver-graft transplantation was adjusted to the P450 phenotype of the donor liver in order to avoid drug-toxicity caused by poor metabolizing capacity.
Liver tissue donors
The drug-metabolizing capacity of livers (n = 93) not selected for transplantation or liver tissue remaining after reduced-size LTx (n = 16) was determined by P450 phenotyping approaches. Demographic data of all donors, subdivided as groups selected and not selected for transplantation are shown in Table 1. The livers were retrieved from haemodynamically stable brain dead donors with normal or near normal liver function. Organ procurement was performed according to well described techniques [10]. The livers were perfused and stored in HTK (Fresenius AG, Bad Homburg v.d.H., Germany). The use of human tissue for scientific research was approved by the Hungarian Regional Committee of Science and Research Ethics. All experimental activities were carried out under the regulation of Act CLIV of 1997 on Health and the decree 23/2002 of the Minister of Health of Hungary.
Table 1.
Demographic data of liver donors (n = 109)
| Non-transplanted (n = 93) | Transplanted (n = 16) | |
|---|---|---|
| Sex: | ||
| Male | 60.2% | 31.3% |
| Female | 39.8% | 68.7% |
| Age (years) | 43 (16–74) | 25 (13–63) |
| Cause of death | ||
| Intracranial bleeding | 68.7% | 43.7% |
| Cerebral contusion | 31.3% | 56.3% |
Human liver microsomes
Tissues were homogenized in 0.1 m Tris-HCl buffer (pH 7.4) containing 1 mm EDTA and 154 mm KCl. The hepatic microsomal fraction was prepared by differential centrifugation [11]. All procedures of preparation were performed at 0–4°C. The protein content of microsomes was determined by the method of Lowry et al.[12], with bovine serum albumin as the standard.
P450 enzyme assays
Published methods were followed to determine selective enzyme activities: phenacetin O-dealkylation for CYP1A2 [13], coumarin 7-hydroxylation for CYP2A6 [14], mephenytoin N-demethylation for CYP2B6 [15], tolbutamide 4-hydroxylation for CYP2C9 [16], mephenytoin 4′-hydroxylation for CYP2C19 [17], dextromethorphan O-demethylation for CYP2D6 [18], chlorzoxazone 6-hydroxylation for CYP2E1 [19], and nifedipine oxidation for CYP3A4/5 [20]. The incubation mixture contained a NADPH-generating system (1 mm NADPH, 10 mm glucose 6-phosphate, 5 mm MgCl2 and 2 units ml−1 glucose 6-phosphate dehydrogenase), human liver microsomes and various substrates selective for P450 forms (phenacetin, coumarin, mephenytoin, tolbutamide, dextromethorphan, chlorzoxazone, or nifedipine). The amount of microsomal protein used in enzymatic reactions was 0.8 mg ml−1 except for coumarin 7-hydroxylation (0.4 mg ml−1) and phenacetin O-dealkylation (1 mg ml−1). Microsomal P450 enzymes worked linearly in a 10–30 min incubation period. Enzyme reactions were terminated by the addition of ice-cold methanol. HPLC or fluorometric analyses were performed according to published methods [13–20]. Donor livers with P450 activity under the limit of quantification for the corresponding metabolite were considered to be of poor metabolizer phenotype. The lower limit of quantification was 1 pmol ml−1 for 7-hydroxy-coumarin (CYP2A6), 50 pmol ml−1 for acetaminophen (CYP1A2) and hydroxy-tolbutamide (CYP2C9), 100 pmol ml−1 for 4-hydroxy-mephenytoin (CYP2C19), 200 pmol ml−1 for nirvanol (CYP2B6), 6-hydroxy-chlorzoxazone (CYP2E1) and oxidized nifedipine (CYP3A4), and 500 pmol ml−1 for dextrorphan (CYP2D6). All measurements were performed in duplicate with <5% inter- and intraday precision.
Liver transplantation and recipient therapy
Sixteen patients who underwent partial LTx at the Transplantation and Surgical Clinic were enrolled in the study. The donor liver was split along the falciform ligament by standard liver resection techniques [21], separating the extended right lobe for transplantation and the left lateral segments for P450 phenotyping. Immunosuppressive therapy of the patients included cyclosporin A, mycophenolic acid and prednisone. In the case of acute rejection, the patient was treated with prednisone for 3–4 days. Trough concentrations of cyclosporin A were determined every day. Infection prophylaxis (co-amoxicillin and cyprofloxacin) continued for 48 h after transplantation. Routine treatments for ulcer prevention (omeprazole or pantoprazole), Pneumocystis jiroveci pneumonia prophylaxis (sulfamethoxazole-trimethoprim) and herpes prophylaxis (acyclovir) were administered. Fluconazole was used as an antifungal agent when necessary. Valgancyclovir was given for cytomegalovirus prophylaxis. Liver and kidney function were monitored routinely. Liver biopsy was performed, depending on the clinical picture, to assess the degree of rejection, drug toxicity, necrosis, cholestasis, steatosis or other pathological signs in the early and late postoperative period. Histological diagnosis was based on haematoxylin and eosin, PAS and sirius red staining.
Data analysis
P450 enzyme activities were expressed as mean ± SD values. Data analysis was performed using GraphPad InStat version 3.05 (GraphPad Software, San Diego, CA). Frequencies of poor metabolizers were calculated on the basis of phenotyping tests.
Results
Variations in P450 enzyme activities in Hungarian donors
Maximal catalytic activity assays of various P450 enzymes participating in drug metabolism were determined in hepatic microsomal fractions of 109 Hungarian (Caucasian) cadaveric donors. The choices of selective substrates to identify P450 activities were based on published reports [13–20]. The Vmax values represent the drug-metabolizing capacity of the liver grafts at the time of explantation. P450 activities towards selective marker substrates varied in the range of 10- or 100-fold (Table 2). In some cases, the activities ranged from nonquantifiable to a high value. Figure 1 shows the frequency distributions of CYP2C9, CYP2C19 and CYP2D6 activities. Liver donors with functional deficiency were considered to be poor metabolizers. After phenotyping the 109 liver donors, the frequency of poor metabolizers was found to be 0.92%, 5.5% and 8.3% for CYP2C9, CYP2C19 and CYP2D6, respectively. Ultrarapid metabolizers of the CYP2D6 substrate comprised 5.5% of the donors (Figure 1C). The drug-metabolizing capacity of the rest of the donors ranged between the extremes.
Table 2.
Characterization of human liver tissues (n = 109) for selective substrates of P450 enzymes. The values are expressed as pmol product mg−1 microsomal protein min−1
| P450 | Enzyme activity Mean ± SD | Minimum | Maximum |
|---|---|---|---|
| CYP1A2 | 267.9 ± 209.38 | blq | 1107.1 |
| CYP2A6 | 922.3 ± 735.23 | 34.6 | 3806.7 |
| CYP2B6 | 77.1 ± 96.37 | 9.74 | 538.3 |
| CYP2C9 | 245.3 ± 194.19 | blq | 1056.0 |
| CYP2C19 | 37.4 ± 33.79 | blq | 160.1 |
| CYP2D6 | 371.3 ± 282.45 | blq | 1124.1 |
| CYP2E1 | 1472.5 ± 887.58 | 328.6 | 4983.1 |
| CYP3A4/5 | 676.8 ± 617.54 | 3.79 | 2861.5 |
blq, below the limit of quantification.
Figure 1.
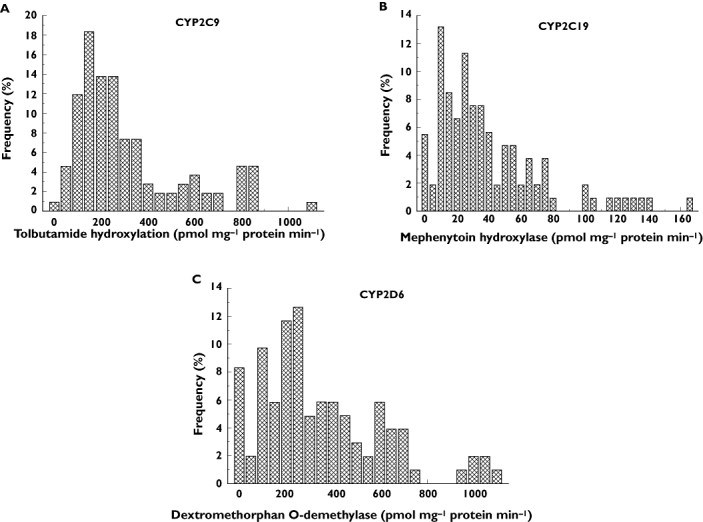
Frequency distribution of CYP2C9 (A), CYP2C19 (B), and CYP2D6 (C) activities in Hungarian liver donors (n = 109)
Transplantation of partial liver grafts
From January 1997 to July 2006, 16 (14 paediatric and two adult) patients underwent partial LTx at the Transplantation and Surgical Clinic. The primary liver diseases of recipients included congenital hepatic fibrosis (eight patients), fulminant liver failure (five patients), glycogenosis (one patient), primary sclerosing cholangitis (one patient) and primary biliary cirrhosis (one patient). The male : female ratio was 9 : 7. The mean Child-Pugh score was 9 (ranging between 5 and 15). The mean cold ischaemic time was 380 min (ranging between 340 and 615 min) and the mean warm ischaemic time was 58 min (ranging between 30 and 110 min).
All liver grafts were implanted uneventfully and presented normal liver function after 1 week. There were no vascular complications. One case with biliary tract complication was observed (6.2%). Re-operation for bleeding was performed in three cases (18.7%). One paediatric patient was succesfully retransplanted because of late biliary tract complications. Acute rejection was detected and treated successfully with steroid shot therapy (prednisone for 3–4 days) in seven cases (43.7%). Ten patients (62.5%) received fluconazole-based antifungal therapy for 7–18 days, depending on the clinical picture. The narrow therapeutic range of cyclosporin A and the high potential of metabolic drug interaction required daily determination of blood concentrations of cyclosporin A and careful dose adjustment. At present, 14 patients are alive with normal graft function. The mortality rate was 12.5% (two patients with fulminant liver failure). The 3-month and actual patient survival rate was 87.5%. In two paediatric patients (12.5%) transplanted with the HH-069 and HH-074 liver grafts, significant elevation of the liver enzymes was observed 2 weeks after LTx.
Clinical outcome for the recipients of grafts HH-069 and HH-074
Two paediatric recipients, a 12-year-old girl with congenital liver fibrosis and a 16-year-old girl with primary sclerosing cholangitis, were transplanted with donor livers HH-069 and HH-074, respectively. The donor HH-069 was an 18-year-old woman who died in a traffic accident, whereas HH-074 was a 25-year-old man who died from a cerebral haemorrhage. After transplantation of the partial liver grafts, the postoperative course in both cases was uneventful without surgical complications. Doppler ultrasound examination showed normal graft circulation and morphology. Two weeks after transplantation, a significant increase in concentrations of transaminases, alkaline phosphatase or bilirubin indicated liver function failure (Figure 2). A biopsy of the graft HH-069 showed liver necrosis, fatty degeneration, eosinophyl bodies and apoptosis probably due to drug toxicity, without any signs of rejection or infection (Figure 3).
Figure 2.
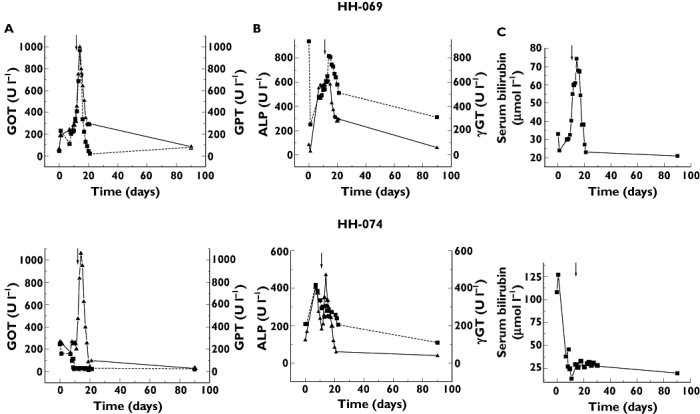
Liver transaminases (A) (GOT, (▪); GPT, (▴)), alkaline phosphatase (▪), γ-glutamyl transferase (▴) (B) and serum bilirubin (C) concentrations of recipients transplanted with grafts HH-069 and HH-074. Arrows show the modification in medication
Figure 3.
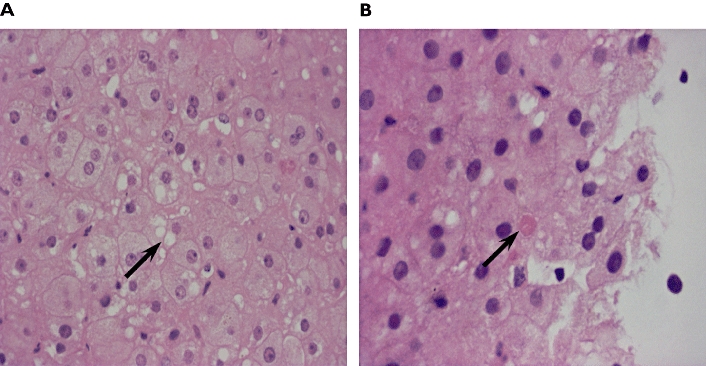
Histology (haematoxylin and eosin staining) of biopsy taken from HH-069 14 days after transplantation. Arrows point to a group of swollen ‘ground glass’ hepatocytes (A) and to an acidophilic (Councilman) body (B). (original magnification 400×)
In the test of drug-metabolizing status of the liver grafts at the time of explantation, the HH-069 liver showed strongly reduced activity of CYP2C9, and HH-074 was found to be a poor metabolizer of CYP2C19 substrates. Both liver grafts had normal CYP3A4 activity (Table 3). Withdrawal of omeprazole, sulfamethoxazole-trimethoprim and fluconazole was suggested because of the functional CYP2C9 deficiency of the graft HH-069. Rationalization of medication, withdrawal of fluconazole and replacement of pantoprazole with famotidine, was also proposed for the recipient of graft HH-074. As a consequence of these alterations in medication, both liver grafts recovered within 1 week (Figure 2) and have retained normal function for more than 1 year.
Table 3.
P450 phenotyping of liver grafts. Drug-metabolizing capacity of grafts showing normal function (n = 14) was compared with P450 activities of grafts HH-069 and HH-074
| Enzyme activity (pmol mg−1 min−1) | |||
|---|---|---|---|
| P450 | Liver grafts (n = 14) | HH-069 | HH-074 |
| CYP1A2 | 286.3 ± 373.66 | 421.0 | 1107.1 |
| CYP2A6 | 718.8 ± 532.48 | 716.2 | 1354.1 |
| CYP2B6 | 52.7 ± 43.00 | 538.3 | 215.1 |
| CYP2C9 | 271.2 ± 89.70 | blq | 572.5 |
| CYP2C19 | 29.5 ± 17.25 | 32.8 | blq |
| CYP2D6 | 442.2 ± 116.64 | 604.8 | 1069.7 |
| CYP2E1 | 1105.4 ± 335.03 | 905.2 | 2135.5 |
| CYP3A4/5 | 892.5 ± 645.96 | 1013.9 | 1197.8 |
blq, below the limit of quantification.
Discussion
Lifelong monitoring of liver graft function is essential and inevitable, since graft dysfunction may occur at any time after transplantation. Low synthetic functions of the graft and increasing or persistent elevations in liver transaminases, alkaline phosphatase, or bilirubin indicate hepatic allograft dysfunction. Since the pattern of hepatic enzyme elevations is frequently not helpful in discovering the causes of allograft dysfunction, histological examination of a liver biopsy may contribute to a distinct diagnosis (infection or rejection) [22]. Hepatic allograft dysfunction may also occur as a consequence of drug therapy [23]. Medications commonly used for the treatment of transplant patients, including azathioprine, sulfamethoxazole-trimethoprim and antifungal agents (amphotericin B, fluconazole) have been reported to cause hepatotoxicity [24–26]. Macrolide antibiotics, such as erythromycin, have been associated with cholestatic hepatitis [27]. Furthermore, lipid-lowering agents frequently used in liver recipients can be associated with chronic liver abnormalities [28]. The relationship between the onset of symptoms and commencement of drug administration may provide a clue to the presence of drug-related hepatotoxicity, although it is usually difficult or impossible to exclude unequivocally drug toxicity as the cause for liver graft dysfunction. Drug-related hepatic injury is manifested by hyperbilirubinaemia associated with elevations in transaminase and alkaline phosphatase concentrations. The fact that a liver biopsy demonstrates a rather nonspecific picture as a consequence of drug toxicity, may point to some limitations of histological evaluation [22]. Drug-induced hepatitis often reverses upon reduction or discontinuation of the offending medication. However, it should be noted that hepatic injury caused by drugs, e.g. amoxicillin, may occasionally result in fulminant hepatic failure and may lead to destruction of the liver graft in spite of withdrawal of the drug [29].
The effectiveness of the postoperative drug therapy is highly influenced by the drug-metabolizing capacity of the donor liver [3]. In humans, there is a large interindividual variability in drug metabolism, which results in different pharmacological effects and toxicity of drugs in the population. Variability is basically due to genetic polymorphisms of the drug-metabolizing enzymes. CYP3A enzymes, which account for almost 50% of total hepatic P450s, are the main catalysts of many drugs including immunosuppressive agents. Although no genetic polymorphism with clinical relevance has been described for the CYP3A4 gene, a greater than 30-fold interindividual variability was found in nifedipine oxidase activity measured in the 109 grafts in our study. CYP3A4 is abundantly expressed in liver and the main cause for interindividual variation is induction or inhibition due to concomitant drug therapies or environmental factors. In contrast, CYP3A5 expression is highly polymorphic. The frequency of defective CYP3A5 alleles resulting in the absence of functional CYP3A5 in the liver is 90–93% in Caucasians [30]. Since CYP3A4 and CYP3A5 have largely overlapping substrate specificities, patients carrying functional CYP3A5 alleles are expected to metabolize CYP3A substrates at higher rates and they require a higher daily dose. Owing to this and to the narrow therapeutic range, continuous monitoring of immunosuppressive drug concentrations is essential.
Much has been learned about the genetic polymorphism of CYP2C9, CYP2C19 and CYP2D6, which have a significant effect on the elimination of many drugs. Treatment of poor metabolizers with average doses of drugs leads to higher drug exposure, which may increase the risk of adverse drug reactions or drug toxicity. This means that we must take into consideration the effects of different drugs routinely used after transplantion. Screening the liver donors for poor metabolizer phenotypes may improve the prospects of survival. In our study, the results of 109 Hungarian (Caucasian) donors revealed considerable interindividual variations in the activities of the P450 enzymes which are in agreement with those previously reported [31–33]. P450 activities varied by one or even two orders of magnitude. In the case of CYP1A2, CYP2C9, CYP2C19 and CYP2D6, the activities ranged from undetectable to high Vmax values. These results can be partly explained by genetic polymorphism, mainly of CYP2C9, CYP2C19 and CYP2D6, which display different functional allele variants. Poor metabolizer phenotypes of CYP2C9, CYP2C19 and CYP2D6 are found in 1–2%, 2–6% and 5–10% of Europeans, respectively [8, 34]. In our study, liver donors with P450 activities under the limit of quantification were considered to be poor metabolizers. The rates of poor metabolizers for CYP2C9, CYP2C19 and CYP2D6 in 109 Hungarian donors (0.92%, 5.5% and 8.3%, respectively) were similar to rates in the Caucasian population. The frequency of ultrarapid metabolizers with extremely high microsomal dextromethorphan O-demethylation activity was 5.5% in Hungarian donors. This is in agreement with the variation reported by Lundqvist et al.[35] who showed that the extreme increase in CYP2D6 activity was a consequence of duplication or multiplication of the active CYP2D6 gene.
Sixteen patients transplanted with partial liver grafts were included in our clinical study. In the case of two paediatric patients transplanted with grafts HH-069 and HH-074, liver dysfunction was observed 11–14 days after transplantation. The rest of the patients presented almost normal liver function and no signs of drug toxicity in this period. On the basis of phenotyping analyses of donor livers for these 14 recipients, none of the donors displayed poor drug-metabolizing capacity. P450 phenotyping of the livers HH-069 and HH-074 revealed that the donors were poor metabolizers for CYP2C9 and CYP2C19 substrates, respectively. This was assumed to lead eventually to drug-induced liver toxicity. Withdrawal of the drugs involved in metabolic drug interactions via CYP2C9, namely sulfamethoxazole-trimethoprim, fluconazole and omeprazole, resulted in normalization of the HH-069 liver function within 1 week. CYP2C9 is the principal enzyme involved in drug interactions caused by sulfamethoxazole or fluconazole [36–38]. CYP2C9 is a minor enzyme in omeprazole metabolism [39]. All of these drugs have been reported to cause acute hepatitis, which resolves spontaneously on discontinuation of the drugs [26, 40, 41]. Rationalization of the drug therapy by withdrawing fluconazole and replacing pantoprazole with famitidine also reversed the dysfunction of the HH-074 liver graft carrying defective CYP2C19. CYP2C19 is the major enzyme in pantoprazole biotransformation [42], whereas no P450 is involved in famotidine metabolism and famotidine does not inhibit the activity of any P450 [43]. It should be mentioned that fluconazole has metabolic drug interaction potential with substrates selective for not only CYP2C, but also for CYP3A [37, 38]. The inhibition of CYP3A activity by fluconazole leads to elevated blood concentrations of the immunosuppressive drug, cyclosporin A. For the paediatric recipients transplanted with HH-069 and HH-074 liver grafts, the trough concentrations of cyclosporin A were doubled 2 days after administration of fluconazole. P450 inhibition occurs almost immediately at the beginning of treatment and lasts while fluconazole is present; careful tailoring (reduction) of the daily dose of cyclosporin A is essential during treatment. Steroid shot therapy for acute rejection also influences cyclosporin A concentrations. Since prednisone is a potent CYP3A inducer resulting in more rapid elimination of cyclosporin A [44], increase in the daily dose of the immunosuppressive drug was required during prednisone treatment and for some days after withdrawal of the steroid.
P450 phenotyping of liver grafts on the basis of P450 enzyme activities provides information on the current drug-metabolizing capacity. The measurement of P450 activities requires liver tissue (at least 0.5 g for eight P450 activities), which is easily obtained in the case of reduced-size LTx. For recipients who undergo full-size LTx, drug-metabolizing capacity can be predicted by P450 genotyping and phenotyping with transcriptomic tools. Screening drug metabolizing function of the liver grafts regarding poor metabolizer phenotypes can be an important factor in patient and graft survival. As also verified by these two cases, P450 phenotyping may contribute to the evaluation of clinical and histological data and to the determination of a proper diagnosis. Although the incidence of poor metabolizer liver donors is not frequent, about 1–10% in Caucasians for CYP2C9, CYP2C19 and CYP2D6 enzymes, reduced drug metabolizing capacity requires individual drug therapy.
Acknowledgments
This work was supported by János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00413/05).
References
- 1.Ploeg RJ, D'Alessandro AM, Hoffmann RM, Groshek M, Gange SJ, Knechtle SJ, Stegall MD, Eckhoff DE, Pirsch JD, Sollinger HW, Belzer FO. Impact of donor factors and preservation on function and survival after liver transplantation. Transplant Proc. 1993;25:3031–3. [PubMed] [Google Scholar]
- 2.Wiesner RH, Menon KV. Late hepatic allograft dysfunction. Liver Transpl. 2001;7:S60–73. doi: 10.1053/jlts.2001.29094. [DOI] [PubMed] [Google Scholar]
- 3.Liu S, Frye RF, Branch RA, Venkataramanan R, Fung JJ, Burckart GJ. Effect of age and postoperative time on cytochrome P450 enzyme activity following liver transplantation. J Clin Pharmacol. 2005;45:666–73. doi: 10.1177/0091270005276202. [DOI] [PubMed] [Google Scholar]
- 4.Lewis DFV. 57 varieties: the human cytochromes P450. Pharmacogenomics. 2004;5:305–18. doi: 10.1517/phgs.5.3.305.29827. [DOI] [PubMed] [Google Scholar]
- 5.Solus JF, Arietta BJ, Harris JR, Sexton DP, Steward JQ, McMunn C, Ihrie P, Mehall JM, Edwards EP. Genetic variation in eleven phase I drug metabolism genes in an ethnically diverse population. Pharmacogenomics. 2004;5:895–931. doi: 10.1517/14622416.5.7.895. [DOI] [PubMed] [Google Scholar]
- 6.Tamási V, Vereczkey L, Falus A, Monostory K. Some aspects of interindividual variations in the metabolism of xenobiotics. Inflamm Res. 2003;52:323–33. doi: 10.1007/s00011-003-1186-4. [DOI] [PubMed] [Google Scholar]
- 7.Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med. 2001;250:186–200. doi: 10.1046/j.1365-2796.2001.00879.x. [DOI] [PubMed] [Google Scholar]
- 8.Brockmöller J, Kirchheiner J, Meisel C, Roots I. Pharmacogenetic diagnostics of cytochrome P450 polymorphisms in clinical drug development and in drug treatment. Pharmacogenomics. 2000;1:125–51. doi: 10.1517/14622416.1.2.125. [DOI] [PubMed] [Google Scholar]
- 9.Wilke RA, Musana AK, Weber WW. Cytochrome P450 gene-based drug prescribing and factors impacting translation into routine clinical practice. Personalized Med. 2005;2:213–24. doi: 10.2217/17410541.2.3.213. [DOI] [PubMed] [Google Scholar]
- 10.Karam G, Compagnon P, Hourmant M, Despins P, Duveau D, Noury D, Boudjema K. A single solution for multiple organ procurement and preservation. Transpl Int. 2005;18:657–63. doi: 10.1111/j.1432-2277.2005.00083.x. [DOI] [PubMed] [Google Scholar]
- 11.van der Hoeven TA, Coon MJ. Preparation and properties of partially purified cytochrome P-450 and reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase from rabbit liver microsomes. J Biol Chem. 1974;249:6302–10. [PubMed] [Google Scholar]
- 12.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 13.Butler MA, Iwasaki M, Guengerich FP, Kadlubar FF. Human cytochrome P-450PA (P-450IA2), the phenacetin O-deethylase, is primarily responsible for the hepatic 3-demethylation of caffeine and N-oxidation of carcinogenic arylamines. Proc Natl Acad Sci USA. 1989;86:7696–700. doi: 10.1073/pnas.86.20.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raunio H, Syngelma T, Pasanen M, Juvonen R, Honkakoski P, Kairaluoma MA, Sotaniemi E, Lang MA, Pelkonen O. Immunochemical and catalytical studies on hepatic coumarin 7-hydroxylase in man, rat, and mouse. Biochem Pharmacol. 1988;37:3889–95. doi: 10.1016/0006-2952(88)90070-6. [DOI] [PubMed] [Google Scholar]
- 15.Heyn H, White RB, Stevens JC. Catalytic role of cytochrome P4502B6 in the N-demethylation of S-mephenytoin. Drug Metab Dispos. 1996;24:948–54. [PubMed] [Google Scholar]
- 16.Miners JO, Birkett DJ. Use of tolbutamide as a substrate probe for human hepatic cytochrome P450 2C9. Methods Enzymol. 1996;272:139–45. doi: 10.1016/s0076-6879(96)72017-7. [DOI] [PubMed] [Google Scholar]
- 17.Srivastava PK, Yun C-H, Beaune PH, Ged C, Guengerich FP. Separation of human liver microsomal tolbutamide hydroxylase and (S) -mephenytoin 4′-hydroxylase cytochrome P-450 enzymes. Mol Pharmacol. 1991;40:69–79. [PubMed] [Google Scholar]
- 18.Kronbach T, Mathys D, Gut J, Catin T, Meyer UA. High-performance liquid chromatographic assays for bufuralol 1′-hydroxylase, debrisoquine 4-hydroxylase, and dextromethorphan O-demethylase in microsomes and purified cytochrome P-450 isozymes of human liver. Anal Biochem. 1987;162:24–32. doi: 10.1016/0003-2697(87)90006-6. [DOI] [PubMed] [Google Scholar]
- 19.Peter R, Böcker R, Beaune PH, Iwasaki M, Guengerich FP, Yang CS. Hydroxylation of chlorzoxazone as a specific probe for human liver cytochrome P-450IIE1. Chem Res Toxicol. 1990;3:566–73. doi: 10.1021/tx00018a012. [DOI] [PubMed] [Google Scholar]
- 20.Guengerich FP, Martin MV, Beaune PH, Kremers P, Wolff T, Waxman DJ. Characterization of rat and human liver microsomal cytochrome P-450 forms involved in nifedipine oxidation, a prototype for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1986;261:5051–60. [PubMed] [Google Scholar]
- 21.Slooff MJ. Reduced size liver transplantation, split liver transplantation, and living related liver transplantation in relation to the donor organ shortage. Transpl Int. 1995;8:65–8. doi: 10.1007/BF00366715. [DOI] [PubMed] [Google Scholar]
- 22.Portmann B. Liver allograft pathology and biopsy interpretation. Verh Dtsch Ges Pathol. 2004;88:29–38. [PubMed] [Google Scholar]
- 23.Bissell DM, Gores GJ, Laskin DL, Hoofnagle JH. Drug-induced liver injury: mechanisms and test sytems. Hepatology. 2001;33:1009–13. doi: 10.1053/jhep.2001.23505. [DOI] [PubMed] [Google Scholar]
- 24.de Boer NKH, Mulder CJJ, van Bodegraven AA. Myelotoxicity and hepatotoxicity during azathioprine therapy. Neth J Med. 2005;63:444–6. [PubMed] [Google Scholar]
- 25.Galan MV, Potts JA, Silverman AL, Gordon SC. The burden of acute nonfulminant drug-induced hepatitis in a United States tertiary referral center. J Clin Gastroenterol. 2005;39:64–7. [PubMed] [Google Scholar]
- 26.Fischer MA, Winkelmayer WC, Rubin RH, Avorn J. The hepatotoxicity of antifungal medications in bone marrow transplant recipients. Clin Infect Dis. 2005;41:301–7. doi: 10.1086/431586. [DOI] [PubMed] [Google Scholar]
- 27.Kaplowitz N. Drug-induced liver injury. Clin Infect Dis. 2004;38:S44–8. doi: 10.1086/381446. [DOI] [PubMed] [Google Scholar]
- 28.Olyaei A, de Mattas AM, Bennett WM. Commonly used drugs and drug interactions. In: Norman DJ, Turka LA, editors. Primer on Transplantation. Mt. Laurel, NJ: American Society of Transplantation; 2001. pp. 99–113. [Google Scholar]
- 29.Gresser U. Amoxicillin-clavulanic acid therapy may be associated with severe side effects – review of the literature. Eur J Med Res. 2001;6:139–49. [PubMed] [Google Scholar]
- 30.Thervet E, Legendre C, Beaune P, Anglicheau D. Cytochrome P450 3A polymorphisms and immunosuppressive drugs. Pharmacogenomics. 2005;6:37–47. doi: 10.1517/14622416.6.1.37. [DOI] [PubMed] [Google Scholar]
- 31.Transon C, Lecoeur S, Leemann T, Beaunne P, Dayer P. Interindividual variability in catalytic activity and immunreactivity of three major human liver cytochrome P450 isozymes. Eur J Clin Pharmacol. 1996;51:79–85. doi: 10.1007/s002280050164. [DOI] [PubMed] [Google Scholar]
- 32.Blanco JG, Harrison PL, Evans WE, Relling MV. Human cytochrome P450 maximal activities in pediatric versus adult liver. Drug Metab Dispos. 2000;28:379–82. [PubMed] [Google Scholar]
- 33.Shu Y, Cheng Z-N, Liu Z-Q, Wang L-S, Zhu B, Huang S-L, Ou-Yang D-S, Zhou H-H. Interindividual variations in levels and activities of cytochrome P-450 in liver microsomes of Chinese subjects. Acta Pharmacol Sin. 2001;22:283–8. [PubMed] [Google Scholar]
- 34.Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci. 1999;20:342–9. doi: 10.1016/s0165-6147(99)01363-2. [DOI] [PubMed] [Google Scholar]
- 35.Lundqvist E, Johansson I, Ingelman-Sundberg M. Genetic mechanisms for duplication of the human CYP2D6 gene and methods for detection of duplicated CYP2D6 genes. Gene. 1999;226:327–38. doi: 10.1016/s0378-1119(98)00567-8. [DOI] [PubMed] [Google Scholar]
- 36.Wen X, Wang J-S, Backman JT, Laitila J, Neuvonen PJ. Trimethoprim and sulfamethoxazole are selective inhibitors of CYP2C8 and CYP2C9, respectively. Drug Metab Dispos. 2002;30:631–5. doi: 10.1124/dmd.30.6.631. [DOI] [PubMed] [Google Scholar]
- 37.Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokin. 2000;38:111–80. doi: 10.2165/00003088-200038020-00002. [DOI] [PubMed] [Google Scholar]
- 38.Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1813–6. doi: 10.1248/bpb.28.1805. [DOI] [PubMed] [Google Scholar]
- 39.Yamazaki H, Inoue K, Shaw PM, Checovich WJ, Guengerich FP, Shimada T. Different contributions of cytochrome P450 2C19 and 3A4 in the oxidation of omeprazole by human liver microsomes: effects of contents of these two forms in individual human samples. J Pharmacol Exp Therap. 1997;283:434–42. [PubMed] [Google Scholar]
- 40.Koury SI, Stone CK, La Charite DD. Omeprazole and the development of acute hepatitis. Eur J Emerg Med. 1998;5:467–9. [PubMed] [Google Scholar]
- 41.Kouklakis G, Mpoumponaris A, Zezos O, Moschos J, Koulaouzidis A, Nakos A, Pehlivanidis A, Iosiphidis M, Molyvas E, Nikolaidis N. Cholestatic hepatitis with severe systemic reactions induced by trimethoprim-sulfamethoxazole. Ann Hepatol. 2007;6:63–5. [PubMed] [Google Scholar]
- 42.Tanaka M, Ohkubo T, Otani K, Suzuki A, Kaneko S, Sugawara K, Ryokawa Y, Ishizaki T. Stereoselective pharmacokinetics of pantoprazole, a proton pump inhibitor, in extensive and poor metabolizers of S-mephenytoin. Clin Pharmacol Ther. 2001;69:108–13. doi: 10.1067/mcp.2001.113723. [DOI] [PubMed] [Google Scholar]
- 43.Satoh T, Munakata H, Fujita K, Itoh S, Kamataki T, Yoshizawa I. Studies on the interactions between drug and estrogen. II. On the inhibitory effect of 29 drugs reported to induce gynecomastia on the oxidation of estradiol at C-2 or C-17. Biol Pharm Bul. 2003;26:695–700. doi: 10.1248/bpb.26.695. [DOI] [PubMed] [Google Scholar]
- 44.Pichard L, Fabre I, Daujat M, Domergue J, Joyeux H, Maurel P. Effect of corticosteroids on the expression of cytochrome P450 and on cyclosporine A oxidase activity in primary cultures of human hepatocytes. Mol Pharmacol. 1992;41:1047–55. [PubMed] [Google Scholar]