

Abstract
AIMS
To examine possible effects of polymorphism in the SLCO1B1 gene, encoding the hepatic uptake transporter organic anion transporting polypeptide (OATP) 1B1, on the pharmacokinetics of rosiglitazone and pioglitazone in a prospective genotype panel study.
METHODS
Sixteen healthy volunteers with the homozygous SLCO1B1 c.521TT genotype (controls), 12 with the heterozygous c.521TC genotype and four with the homozygous c.521CC genotype ingested a single 4-mg dose of rosiglitazone and a single 15-mg dose of pioglitazone in a cross-over study with a wash-out period of at least 1 week.
RESULTS
SLCO1B1 polymorphism had no statistically significant effect on any of the pharmacokinetic variables of rosiglitazone, pioglitazone or their metabolites. The mean ± SD area under the plasma rosiglitazone concentration–time curve from time 0 to infinity (AUC0–∞) was 2024 ± 561 ng ml−1 h in the c.521TT subjects, 1763 ± 288 ng ml−1 h in the c.521TC subjects (geometric mean ratio c.521TC/c.521TT 0.89; 95% confidence interval 0.72, 1.11) and 1729 ± 346 ng ml−1 h in the c.521CC subjects (c.521CC/c.521TT 0.87; 0.63, 1.20). The AUC0–∞ of pioglitazone averaged 6244 ± 1909 ng ml−1 h in the c.521TT subjects, 5123 ± 1165 ng ml−1 h in the c.521TC subjects (c.521TC/c.521TT 0.83; 0.65, 1.06) and 4851 ± 1123 ng ml−1 h in the c.521CC subjects (c.521CC/c.521TT 0.79; 0.55, 1.14). There was a significant correlation between the AUC0–∞ of rosiglitazone and pioglitazone (r = 0.717, P < 0.001).
CONCLUSIONS
The SLCO1B1 c.521T→C SNP does not affect the pharmacokinetics of rosiglitazone or pioglitazone, indicating that OATP1B1 plays no significant role in the disposition of these drugs.
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
A common single nucleotide polymorphism (SNP) (c.521T→C) of the SLCO1B1 gene, encoding the hepatic uptake transporter organic anion transporting polypeptide (OATP) 1B1, has been associated with marked changes in the pharmacokinetics of the antidiabetic drug repaglinide.
Rosiglitazone and pioglitazone are competitive inhibitors of OATP1B1 and might thus be its substrates.
Gemfibrozil, an inhibitor of OATP1B1 in vitro, considerably increases the plasma concentrations of rosiglitazone and pioglitazone in vivo in humans.
WHAT THIS STUDY ADDS
The SLCO1B1 c.521T→C SNP was not associated with changes in rosiglitazone or pioglitazone pharmacokinetics in healthy volunteers.
OATP1B1 is thus unlikely to play an important role in the disposition of rosiglitazone or pioglitazone.
Keywords: OATP1B1, pharmacogenetics, pharmacokinetics, pioglitazone, rosiglitazone, SLCO1B1
Introduction
The thiazolidinediones rosiglitazone and pioglitazone (Figure 1) are peroxisome proliferator-activated receptor-γ agonists, with insulin-sensitizing properties, used in the treatment of Type 2 diabetes [1]. The oral bioavailability of rosiglitazone is nearly 100% and that of pioglitazone >80% [2, 3]. Both drugs are extensively metabolized in the liver. Rosiglitazone is mainly biotransformed by N-demethylation and pyridine ring hydroxylation and pioglitazone by hydroxylation and oxidation [2, 4]. In vitro studies suggest that these reactions are catalysed mainly by CYP2C8, with minor contributions from CYP2C9 for rosiglitazone and CYP3A4 for pioglitazone [5, 6]. All circulating metabolites of rosiglitazone are less potent than the parent drug and are not thought to have substantial effects on blood glucose concentrations [7], whereas the main metabolites of pioglitazone (M3 and M4) are pharmacologically active, and their plasma concentrations are equal to or greater than those of the parent pioglitazone [4, 8]. The elimination half-life of rosiglitazone is about 3–6 h and that of pioglitazone is about 4–9 h [2, 3, 7, 8].
Figure 1.
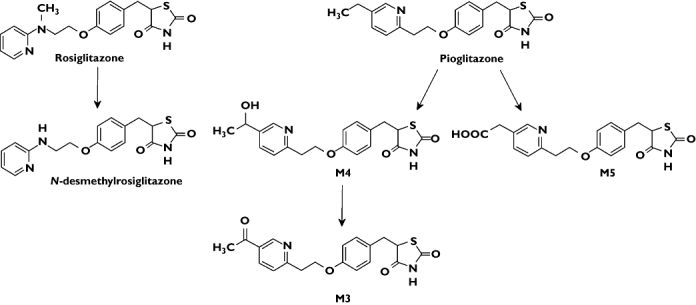
Chemical structures of rosiglitazone, its N-desmethyl metabolite and pioglitazone and its M3, M4 and M5 metabolites [3, 4]
SLCO1B1 encodes the organic anion transporting polypeptide 1B1 (OATP1B1) transporter, which is present at the basolateral membrane of hepatocytes and mediates uptake of its substrates from sinusoidal blood [9]. Its substrates include endogenous compounds, such as bilirubin and bile acids, as well as various drugs, such as statins [10–12]. A common single nucleotide polymorphism (SNP) in SLCO1B1, c.521T→C (p.Val174Ala), has been associated with reduced activity of OATP1B1 in vitro[13, 14]. Studies in humans have revealed that the pharmacokinetics of, for example, the antidiabetic repaglinide, as well as fexofenadine, simvastatin acid and pravastatin, have been significantly associated with SLCO1B1 polymorphism [15–21]. In particular, the AUC of these compounds has been markedly higher in subjects with the c.521CC genotype than in those with the c.521TT genotype [15–18]. In Whites, the c.521T→C SNP exists in four major haplotypes, differentiated by the g-11187G→A, g-10499A→C and c.388A→G SNPs: *16 (g-11187G/g-10499C/c.388G/c.521C), *17 (AAGC), *5 (GAAC) and *15 (GAGC) [22].
Rosiglitazone and pioglitazone are potent competitive inhibitors of OATP1B1 in vitro and could thus be its substrates [23]. Moreover, in an in silico pharmacophore modelling study, rosiglitazone and pioglitazone have been identified as possible substrates of OATP1B1 [24]. In vivo in humans, gemfibrozil, an inhibitor of CYP2C8 and OATP1B1, has considerably increased the plasma concentrations of rosiglitazone and pioglitazone [25, 26]. Although this evidence suggests that rosiglitazone and pioglitazone could be substrates of OATP1B1, it is not known whether SLCO1B1 genotype affects the pharmacokinetics of rosiglitazone or pioglitazone. The aim of this study was to investigate the effects of SLCO1B1 polymorphism on the pharmacokinetics of rosiglitazone and pioglitazone in a prospective genotype panel study. Because rosiglitazone and pioglitazone are metabolized via CYP2C8 and CYP2C9, the study was controlled for CYP2C8*3, CYP2C9*2 and CYP2C9*3, the major functionally significant alleles of these CYP enzymes [27].
Methods
Subjects
A total of 32 healthy White volunteers (19 men and 13 women) participated in this study after giving written informed consent. The participants were recruited from a pool of subjects genotyped for SLCO1B1, CYP2C8 and CYP2C9 SNPs [22]. All genotyping was performed by TaqMan allelic discrimination with an Applied Biosystems 7300 Real-Time Polymerase Chain Reaction System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions with a reaction volume of 10 µl. Genotyping for SLCO1B1 SNPs was carried out as described previously [22]. Genotyping for the CYP2C9*2 (c.430C→T) and CYP2C9*3 (c.1075A→C) alleles was performed using TaqMan® Pre-Developed Assay Reagents for Allelic Discrimination (Applied Biosystems). Genotyping for the CYP2C8*3 allele (c.416G→A, c.1196A→G) was carried out using Custom TaqMan® SNP genotyping assays (Applied Biosystems). CYP2C8 genotyping was validated against a previously described method [28]. Only noncarriers of the CYP2C8*3, CYP2C9*2 and CYP2C9*3 alleles were recruited. The participants were selected on the basis of the SLCO1B1 c.521T→C SNP as well as the g-11187G→A, g-10499A→C and c.388A→G SNPs and were allocated to one of three groups according to the genotype. Haplotypes were assigned as described previously [22]. The control group comprised 16 participants (five women, 11 men) with the homozygous reference genotype at each position (c.521TT group). Their mean ± SD age was 23 ± 2 years, height 177 ± 9 cm and weight 73 ± 10 kg. The second group included 12 participants (six women, six men) heterozygous for the c.521T→C SNP (c.521TC group). Their mean ± SD age was 22 ± 2 years, height 175 ± 11 cm and weight 68 ± 11 kg. Four c.521TC participants had the *15 haplotype (three *1A/*15, one *1B/*15 diplotype), four had the *16 haplotype (one *1A/*16, three *1B/*16) and four had the *17 haplotype (two *1A/*17, two *1B/*17). The third group consisted of four participants (one woman, three men) with the homozygous c.521CC genotype (c.521CC group). Their mean ± SD age was 23 ± 3 years, height 179 ± 9 cm and weight 78 ± 14 kg. The SLCO1B1 diplotypes of the participants with the c.521CC genotype were *5/*15, *15/*16, *16/*16 and *16/*17. The subjects were ascertained to be healthy by medical history, physical examination and routine laboratory tests before enrolment. None of the subjects was a tobacco smoker or taking any continuous medication, including oral contraceptives. Use of other drugs was prohibited for 1 week, use of grapefruit products for 3 days and use of alcohol for 1 day before the day of administration of rosiglitazone or pioglitazone.
Study design
The study protocol was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District and the National Agency for Medicines (Helsinki, Finland). Following an overnight fast, the subjects ingested a single 4-mg dose of rosiglitazone (one Avandia 4-mg tablet; GlaxoSmithKline, Brentford, UK) and, after a wash-out period of at least 1 week, a single 15-mg dose of pioglitazone (one Actos 15-mg tablet; Takeda, London, UK). The study drugs were administered with 150 ml water at 08.00 h. Subjects received a standardized warm meal 4 h after the administration of study medication and a standardized snack after 7 and 10 h. Timed blood samples (5–10 ml each) were drawn from a cannulated forearm vein into tubes containing ethylenediamine tetraaceticacid before administration of rosiglitazone or pioglitazone and at 15, 30, 60, 90 min and at 2, 3, 4, 5, 7, 9 and 12 h, after which the subjects were discharged from the clinical research unit. In addition, blood samples were drawn by venepuncture 24 and 48 h after rosiglitazone, and 24, 48 and 72 h after pioglitazone administration. Plasma was stored at −70°C until analysis.
Determination of drug concentrations
Plasma rosiglitazone and N-desmethylrosiglitazone were measured by use of an API 2000 Q TRAP liquid chromatography-tandem mass spectrometry system (Sciex Division of MDS Inc., Toronto, Ontario, Canada). The reversed-phase chromatographic separation was achieved on a XBridge C18 column (internal diameter 100 × 2.1 mm and particle size 3.5 µm) (Waters Corp., Milford, MA, USA) using a mobile phase consisting of 10 mmol l−1 ammonium acetate (pH 9.5, adjusted with 25% ammonium hydroxide solution) and acetonitrile at a ratio of 70 : 30 (v/v). A 10-µl aliquot was injected and the mobile phase was delivered at a flow rate of 200 µl min−1, yielding a total chromatographic run time of 7 min. Pioglitazone served as an internal standard for both analytes. The mass spectrometer was operated in positive TurboIonSpray® mode, and the samples were analysed via selected reaction monitoring by use of the transition of the [M+H]+ precursor ion to product ionfor each analyte and internal standard. The selected ion transitions monitored were as follows: m/z 358 to m/z 135 for rosiglitazone, m/z 344 to m/z 121 for N-desmethylrosiglitazone, and m/z 357 to m/z 134 for pioglitazone. The limit of quantification of plasma rosiglitazone was 0.25 ng ml−1 and the day-to-day coefficient of variation (CV) was 6.3% at 0.3 ng ml−1, 5.1% at 3.0 ng ml−1, 5.6% at 30 ng ml−1 and 3.9% at 300 ng ml−1 (n = 6). The plasma concentrations of pioglitazone and its metabolites were determined as described previously [29]. The limit of quantification of plasma pioglitazone and its M3 metabolite was 0.3 ng ml−1 and that of M4 was 1 ng ml−1. The day-to-day CV was 4.3% at 20 ng ml−1, 4.5% at 200 ng ml−1 and 4.9% at 2000 ng ml−1 of pioglitazone, 9.5% at 10 ng ml−1, 8.8% at 100 ng ml−1 and 5.5% at 1000 ng ml−1 of M3 and 14.1% at 10 ng ml−1 and 5.7% at 100 ng ml−1 of M4 (n = 16). Because authentic reference compounds were not available, the concentrations of N-desmethylrosiglitazone and the M5 metabolite of pioglitazone are given in arbitrary units relative to the ratio of the peak area of the metabolite to that of the internal standard. The detector response for these metabolites was confirmed to be linear over the relevant concentration range by means of sample dilution (1–125-fold dilutions, r > 0.995), and a signal to noise ratio (S/N) of 10 : 1 was used as the determination limit.
Pharmacokinetic analysis
The pharmacokinetics of rosiglitazone and pioglitazone were characterized by the peak concentration in plasma (Cmax), time to Cmax (tmax), elimination half-life (t1/2) and area under the plasma concentration–time curve (AUC) from 0 to 48 (rosiglitazone) or 72 h (pioglitazone) and AUC from time 0 to infinity (AUC0–∞). The terminal log-linear part of each concentration–time curve was identified visually, and the elimination rate constant (ke) was determined from natural log-transformed data with linear regression analysis. The t1/2 was calculated by the equation t1/2 = ln2/ke. AUC was calculated by a combination of the linear (for increasing concentrations) and log-linear (for decreasing concentrations) trapezoidal rules, with extrapolation to infinity, when appropriate, by division of the last measured concentration by ke. Weight-adjusted AUC0–∞ was calculated as follows: AUC0–∞ · weight (kg)/70 kg. The apparent formation rate constant (kf) of N-desmethylrosiglitazone and the metabolites of pioglitazone was determined by the method of residuals from the ascending part of the metabolite concentration–time curve.
Statistical analysis
Results are expressed as mean ± SD in the text and tables. Statistical comparisons of the pharmacokinetic variables of rosiglitazone and pioglitazone and their metabolites between subjects with the SLCO1B1 c.521TT, c.521TC and c.521CC genotypes were performed using analysis of variance (anova) and post hoc testing with the Tukey test (equal variances) or the Games–Howell test (unequal variances). Equality of group variances was tested with the Levene statistics and compatibility of the residuals with normal distribution with the Shapiro–Wilk test. AUC and Cmax data were logarithmically transformed before analysis. The 95% confidence intervals (CI) were calculated on the geometric mean ratio of the Cmax and AUC values for the c.521TC/c.521TT and c.521CC/c.521TT pairs, and on the mean difference of the t1/2 and kf values. The tmax data were analysed by the Kruskal–Wallis test. The number of subjects in each genotype group was estimated to be sufficient to detect a 50% greater mean AUC0–∞ of rosiglitazone and pioglitazone in subjects with the c.521TC genotype than in those with the c.521TT genotype, as well as a 100% greater mean AUC0–∞ of rosiglitazone and pioglitazone in subjects with the c.521CC genotype than in those with the c.521TT genotype, with a power of at least 80% (α level of 0.05). All data were analysed with the statistical program SPSS 11.0 for Windows (SPSS Inc., Chicago, IL, USA). Differences were considered statistically significant at P < 0.05.
Results
The AUC0–∞ and Cmax of rosiglitazone varied 2.4-fold (range 1293–3076 ng ml−1 h) and 2.6-fold (199–526 ng ml−1 h) between individual subjects. However, SLCO1B1 genotype had no significant effect on any of the pharmacokinetic variables of rosiglitazone or those of its N-desmethyl metabolite (Figure 2, Table 1). The mean ± SD area under the plasma rosiglitazone concentration–time curve from time 0 to infinity (AUC0–∞) was 2024 ± 561 ng ml−1 h in the c.521TT subjects, 1763 ± 288 ng ml−1 h in the c.521TC subjects (geometric mean ratio c.521TC/c.521TT 0.89; 95% CI 0.72, 1.11) and 1729 ± 346 ng ml−1 h in the c.521CC subjects (c.521CC/c.521TT 0.87; 0.63, 1.20). Adjusting the AUC0–∞ for weight gave similar results (Table 1).
Figure 2.
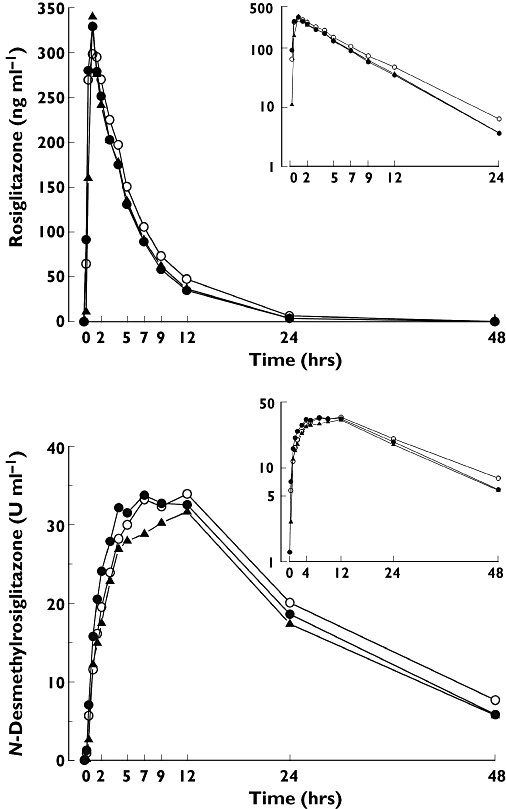
Mean plasma concentrations of rosiglitazone and N-desmethylrosiglitazone in 32 healthy Whites after a single 4-mg oral dose of rosiglitazone in relation to the SLCO1B1 c.521T→C single nucleotide polymorphism. For clarity, only mean values are presented. Insets depict the same data on a semilogarithmic scale. ○, Subjects with the SLCO1B1 c.521TT genotype (n = 16); •, subjects with the c.521TC genotype (n = 12); ▴, subjects with the c.521CC genotype (n = 4)
Table 1.
Pharmacokinetic variables of a single 4-mg oral dose of rosiglitazone and N-desmethylrosiglitazone in relation to the SLCO1B1 521T→C (Val174Ala) single nucleotide polymorphism
| c.521TT (n = 16) | c.521TC (n = 12) | Mean ratio/difference c.521TC vs. c.521TT* (95% CI) | c.521CC (n = 4) | Mean ratio/difference c.521CC vs. c.521TT (95% CI) | |
|---|---|---|---|---|---|
| Rosiglitazone | |||||
| Cmax (ng ml−1) | 361 ± 91 | 353 ± 48 | 1.00 (0.83, 1.21) | 341 ± 99 | 0.94 (0.49, 1.80) |
| AUC0−48 (ng ml−1 h) | 2022 ± 559 | 1763 ± 288 | 0.89 (0.72, 1.11) | 1729 ± 346 | 0.87 (0.63, 1.20) |
| AUC0–∞ (ng ml−1 h) | 2024 ± 561 | 1763 ± 288 | 0.89 (0.72, 1.11) | 1729 ± 346 | 0.87 (0.63, 1.20) |
| Weight-adjusted AUC0–∞ (ng ml−1 h) | 2067 ± 539 | 1693 ± 243 | 0.84 (0.69, 1.01) | 1877 ± 230 | 0.93 (0.71, 1.22) |
| tmax (h) | 1.0 (0.5–2.0) | 1.0 (0.5–1.0) | 1.0 (1.0–1.5) | ||
| t1/2 (h) | 4.1 ± 0.8 | 3.7 ± 0.5 | −0.3 (−1.0, 0.3) | 3.6 ± 0.1 | −0.4 (−1.3, 0.5) |
| N-desmethylrosiglitazone | |||||
| Cmax (U ml−1) | 35.6 ± 4.6 | 35.3 ± 4.7 | 0.99 (0.87, 1.13) | 31.8 ± 5.0 | 0.89 (0.74, 1.10) |
| AUC0−48 (U ml−1 h) | 950 ± 176 | 906 ± 130 | 0.96 (0.82, 1.13) | 836 ± 136 | 0.89 (0.70, 1.13) |
| AUC0–∞ (U ml−1 h) | 1147 ± 263 | 1032 ± 182 | 0.91 (0.75, 1.11) | 958 ± 160 | 0.85 (0.64, 1.13) |
| Weight-adjusted AUC0–∞ (U ml−1 h) | 1166 ± 216 | 994 ± 168 | 0.85 (0.73, 1.00) | 1046 ± 135 | 0.90 (0.72, 1.14) |
| kf | 0.32 ± 0.10 | 0.41 ± 0.21 | 0.09 (−0.09, 0.26) | 0.25 ± 0.03 | −0.08 (−0.15, −0.00) |
| tmax (h) | 8.0 (5.0–12.0) | 8.0 (4.0–12.0) | 12.0 (9.0–12.0) | ||
| t1/2 (h) | 16.9 ± 3.4 | 14.4 ± 2.4 | −2.5 (−5.3, 0.2) | 14.6 ± 1.5 | −2.3 (−6.3, 1.7) |
Data are given as mean ± SD, tmax data as median (range). CI, Confidence interval; Cmax, peak plasma concentration; AUC0−48, area under the plasma concentration–time curve from 0 to 48 h; AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; tmax, time to Cmax; t1/2, elimination half-life; kf, apparent formation rate constant.
These data are geometric mean ratio (95% CI) for the Cmax and AUC values and mean difference (95% CI) for the t1/2 and kf values.
The AUC0–∞ and Cmax of pioglitazone varied 3.9-fold (range 2672–10 504 ng ml−1 h) and 3.6-fold (248–894 ng ml−1 h) between individual subjects. SLCO1B1 polymorphism had no statistically significant effect on any of the pharmacokinetic variables of pioglitazone or its M3, M4 and M5 metabolites (Figure 3, Table 2). The mean ± SD AUC0–∞ of pioglitazone was 6244 ± 1909 ng ml−1 h in the c.521TT subjects, 5123 ± 1165 ng ml−1 h in the c.521TC subjects (c.521TC/c.521TT 0.83; 0.65, 1.06) and 4851 ± 1123 ng ml−1 h in the c.521CC subjects (c.521CC/c.521TT 0.79; 0.55, 1.14). Adjusting the AUC0–∞ for weight gave similar results (Table 2).
Figure 3.
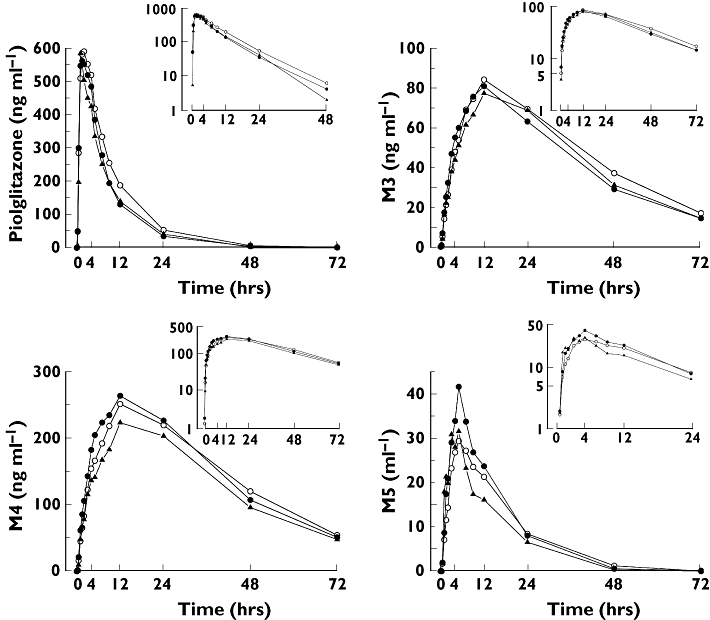
Mean plasma concentrations of pioglitazone and its M3, M4 and M5 metabolites in 32 healthy Whites after a single 15-mg oral dose of pioglitazone in relation to the SLCO1B1 c.521T→C single nucleotide polymorphism. For clarity, only mean values are presented. Insets depict the same data on a semilogarithmic scale. ○, Subjects with the SLCO1B1 c.521TT genotype (n = 16); •, subjects with the c.521TC genotype (n = 12); ▴, subjects with the c.521CC genotype (n = 4)
Table 2.
Pharmacokinetic variables of a single 15-mg oral dose of pioglitazone and its metabolites in relation to the SLCO1B1 521T→C (Val174Ala) single nucleotide poymorphism
| c.521TT (n = 16) | c.521TC (n = 12) | Mean ratio/difference c.521TC vs. c.521TT* (95% CI) | c.521CC (n = 4) | Mean ratio/difference c.521CC vs. c.521TT* (95% CI) | |
|---|---|---|---|---|---|
| Pioglitazone | |||||
| Cmax (ng ml−1) | 617 ± 129 | 610 ± 205 | 0.95 (0.73, 1.25) | 590 ± 165 | 0.95 (0.64, 1.42) |
| AUC0−72 (ng ml−1 h) | 6228 ± 1892 | 5117 ± 1165 | 0.83 (0.65, 1.06) | 4850 ± 1121 | 0.80 (0.55, 1.14) |
| AUC0–∞ (ng ml−1 h) | 6244 ± 1909 | 5123 ± 1165 | 0.83 (0.65, 1.06) | 4851 ± 1123 | 0.79 (0.55, 1.14) |
| Weight-adjusted AUC0–∞ (ng ml−1 h) | 6422 ± 2050 | 4922 ± 1062 | 0.78 (0.61, 1.00) | 5384 ± 1469 | 0.85 (0.59, 1.22) |
| tmax (h) | 1.8 (1.0–4.0) | 2.0 (1.0–4.0) | 1.3 (1.0–1.5) | ||
| t1/2 (h) | 7.9 ± 2.5 | 7.4 ± 1.5 | −0.5 (−2.4, 1.5) | 6.2 ± 0.5 | −1.7 (−4.5, 1.1) |
| M3 | |||||
| Cmax (ng ml−1) | 86 ± 31 | 84 ± 29 | 0.97 (0.73, 1.29) | 78 ± 16 | 0.93 (0.62, 1.41) |
| AUC0−72 (ng ml−1 h) | 3456 ± 1233 | 3104 ± 792 | 0.92 (0.70, 1.21) | 3210 ± 708 | 0.96 (0.64, 1.43) |
| AUC0–∞ (ng ml−1 h) | 4059 ± 1531 | 3594 ± 871 | 0.92 (0.68, 1.23) | 3649 ± 739 | 0.94 (0.61, 1.45) |
| Weight-adjusted AUC0–∞ (ng ml−1 h) | 4106 ± 1242 | 3457 ± 861 | 0.86 (0.66, 1.12) | 4017 ± 921 | 1.00 (0.68, 1.48) |
| kf | 0.21 ± 0.06 | 0.25 ± 0.11 | 0.04 (−0.04, 0.12) | 0.20 ± 0.06 | −0.02 (−0.13, 0.10) |
| tmax (h) | 12.0 (7.0–24.0) | 12.0 (4.0–24.0) | 12.0 (12.0–24.0) | ||
| t1/2 (h) | 23.2 ± 4.7 | 22.8 ± 4.2 | −0.4 (−4.5, 3.6) | 21.5 ± 1.9 | −1.7 (−7.7, 4.1) |
| M4 | |||||
| Cmax (ng ml−1) | 257 ± 39 | 266 ± 75 | 1.01 (0.83, 1.23) | 223 ± 38 | 0.87 (0.65, 1.16) |
| AUC0−72 (ng ml−1 h) | 10949 ± 1902 | 11061 ± 2745 | 1.00 (0.82, 1.21) | 9577 ± 2192 | 0.87 (0.65, 1.16) |
| AUC0–∞ (ng ml−1 h) | 12890 ± 2433 | 12802 ± 3252 | 0.98 (0.79, 1.21) | 11202 ± 3001 | 0.86 (0.63, 1.78) |
| Weight-adjusted AUC0–∞ (ng ml−1 h) | 13227 ± 2216 | 12307 ± 3096 | 0.92 (0.74, 1.14) | 12451 ± 3807 | 0.92 (0.67, 1.26) |
| kf | 0.18 ± 0.06 | 0.21 ± 0.08 | 0.03 (−0.03, 0.09) | 0.18 ± 0.02 | 0.01 (−0.08, 0.09) |
| tmax (h) | 12.0 (9.0–24.0) | 12.0 (5.0–24.0) | 12.0 (12.0–12.0) | ||
| t1/2 (h) | 24.0 ± 4.0 | 22.6 ± 4.4 | −1.4 (−5.3, 2.5) | 22.7 ± 3.4 | −1.2 (−6.9, 4.5) |
| M5 | |||||
| Cmax (U ml−1) | 32 ± 12 | 42 ± 28 | 1.22 (0.80, 1.85) | 34 ± 15 | 1.05 (0.56, 1.94) |
| AUC0−72 (U ml−1 h) | 568 ± 206 | 623 ± 326 | 1.06 (0.71, 1.58) | 476 ± 186 | 0.85 (0.47, 1.52) |
| AUC0–∞ (U ml−1 h) | 571 ± 208 | 629 ± 325 | 1.06 (0.71, 1.59) | 494 ± 210 | 0.86 (0.49, 1.56) |
| Weight-adjusted AUC0–∞ (ng ml−1 h) | 585 ± 211 | 584 ± 222 | 1.00 (0.69, 1.43) | 535 ± 212 | 0.92 (0.54, 1.57) |
| kf | 0.42 ± 0.13 | 0.46 ± 0.12 | 0.04 (−0.08, 0.15) | 0.72 ± 0.40 | 0.30 (−0.53, 1.13) |
| tmax (h) | 5.0 (3.0–12.0) | 5.0 (4.0–7.0) | 3.0 (1.5–5.0) | ||
| t1/2 (h) | 9.1 ± 1.7 | 8.5 ± 2.1 | −0.6 (−2.5, 1.4) | 8.6 ± 3.1 | −0.5 (−3.3, 2.3) |
Data are given as mean ± SD, tmax data as median (range). CI, Confidence interval; Cmax, peak plasma concentration; AUC0−72, area under the plasma concentration–time curve from 0 to 72 h; AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; tmax, time to Cmax; t1/2, elimination half-life; kf, apparent formation rate constant.
These data are geometric mean ratio (95% CI) for the Cmax and AUC values and mean difference (95% CI) for the t1/2 and kf values.
There was a significant linear relationship between the AUC0–∞ values of rosiglitazone and pioglitazone (r = 0.717, P < 0.001) (Figure 4).
Figure 4.
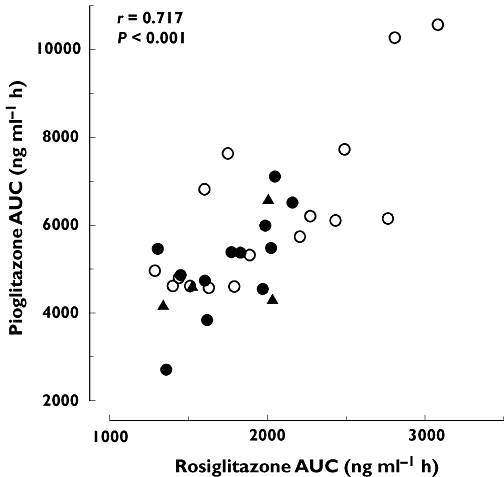
Relationship between the AUC0–∞ of rosiglitazone and pioglitazone. ○, Subjects with the SLCO1B1 c.521TT genotype (n = 16); •, subjects with the c.521TC genotype (n = 12); ▴, subjects with the c.521CC genotype (n = 4)
Discussion
In the present study, in which the pharmacokinetics of both rosiglitazone and pioglitazone were consistent with those observed earlier in young adult subjects [25, 26, 29], SLCO1B1 polymorphism had no significant effect on the pharmacokinetic variables of usual therapeutic doses of rosiglitazone (4 mg) or pioglitazone (15 mg); the Cmax, AUC and t1/2 of rosiglitazone, pioglitazone and their metabolites were similar in subjects with the c.521TT, c.521TC and c.521CC genotypes. Furthermore, adjusting the AUC0–∞ for weight did not change the results. These data strongly suggest that OATP1B1 does not have any clinical significance in the disposition of rosiglitazone and pioglitazone in vivo in humans.
Previous studies have suggested that rosiglitazone and pioglitazone might be substrates of OATP1B1 [23–26]. However, the lack of effect of SLCO1B1 polymorphism on their pharmacokinetics suggests that this may not be the case in vivo, or that the transport of these two drugs into the hepatocyte via OATP1B1 may not be a rate-limiting step in their pharmacokinetics. Other transporters (e.g. OATP1B3) might primarily mediate the hepatic uptake of rosiglitazone and pioglitazone, or the effect of SLCO1B1 c.521T→C polymorphism might be substrate specific, as has been seen, for example, with the CYP2C8*3 allele [28, 30]. Furthermore, rosiglitazone and pioglitazone may be sufficiently permeable to enter the hepatocytes efficiently without OATP1B1. Overall, the passive permeability of a lipid membrane is mainly determined by molecular size, lipophilicity and charge of the drug molecule [31]. The calculated octanol/water partition coefficients at pH 7.0 (logD7.0) of rosiglitazone and pioglitazone are 1.73 and 2.21, respectively (values obtained from the SciFinder Database, American Chemical Society), indicating that they are moderately lipophilic. However, this lipophilicity is unlikely to alone explain the lack of effect of SLCO1B1 polymorphism on their pharmacokinetics, as the AUC of simvastatin acid with a similar logD7.0 value (1.88) has been more than threefold larger in subjects with the SLCO1B1 c.521CC genotype than in those with the c.521TT genotype [17]. One possible explanation for the present findings is that the metabolism, rather than hepatic uptake, may be the rate-determining step in the clearance of the glitazones. Any alteration in hepatic uptake would therefore have minimal impact on the pharmacokinetics of rosiglitazone and pioglitazone.
The oral bioavailability of rosiglitazone is nearly 100% and that of pioglitazone >80%, indicating that they both have low hepatic extraction ratios [2, 3]. According to pharmacokinetic theory, decrease in the hepatic uptake of a low extraction drug should prolong its elimination t1/2[31], which is clearly not the case for rosiglitazone and pioglitazone (Figures 2 and 3). In our previous studies on repaglinide, simvastatin acid and pravastatin, subjects with the SLCO1B1 c.521CC genotype have had significantly higher Cmax and AUC0–∞ values than those with the c.521TT genotype, but the elimination t1/2 has remained unchanged [16–18], which might be explained by intermediate to high extraction ratios of these compounds and corresponding decreases in the clearance and volume of distribution due to the SLCO1B1 polymorphism.
Ingestion of gemfibrozil (600 mg twice daily for 3 days) has increased the AUC of rosiglitazone and pioglitazone by 2.3- and 3.2-fold, respectively [25, 26]. Gemfibrozil and its glucuronide metabolite inhibit both CYP2C8 and OATP1B1 in vitro[32]. However, as the pharmacokinetics of rosiglitazone and pioglitazone were not affected by SLCO1B1 polymorphism in the present study, it is likely that the interactions of gemfibrozil with rosiglitazone and pioglitazone are primarily due to inhibition of CYP2C8. Of note is that gemfibrozil has increased the AUC of repaglinide more than eightfold [33], and that the pharmacokinetics of repaglinide (unlike those of rosiglitazone and pioglitazone) are affected by polymorphisms in both SLCO1B1 and CYP2C8[16, 28].
Interestingly, the AUC0–∞ of rosiglitazone correlated well with that of pioglitazone (Figure 4), indicating that similar characteristics determine interindividual variability in the pharmacokinetics of these two drugs. This is in accordance with CYP2C8 being the major enzyme involved in the metabolism of both rosiglitazone and pioglitazone, although in vitro studies have suggested that CYP2C9 may contribute to the metabolism of rosiglitazone and CYP3A4 to that of pioglitazone [5, 6]. Itraconazole, an inhibitor of CYP3A4, did not affect the plasma concentrations of pioglitazone in vivo[26].
Although rosiglitazone and pioglitazone are usually well tolerated, they may induce adverse reactions such as fluid retention and pulmonary oedema [34–36]. These rare adverse reactions appear to be concentration dependent [7, 8]. Moreover, the blood glucose-lowering effect of thiazolidinediones varies considerably between individual subjects [37]. As the effects of rosiglitazone and pioglitazone are usually seen only after 1–2 months' treatment, it would be useful if their response could be predicted before initiation of treatment. However, because SLCO1B1 polymorphism had no effect on the pharmacokinetics of rosiglitazone and pioglitazone, it is unlikely that SLCO1B1 polymorphism would affect their efficacy or risk of adverse reactions.
In conclusion, genetic polymorphism in SLCO1B1, encoding the hepatic uptake transporter OATP1B1, had no significant effect on the pharmacokinetics of rosiglitazone, pioglitazone or their metabolites in healthy volunteers. It is reasonable to assume that SLCO1B1 polymorphism has no clinically relevant effect on the efficacy or risk of adverse reactions of these antidiabetic drugs.
Acknowledgments
We thank Mrs Kerttu Mårtensson, Mrs Eija Mäkinen-Pulli, Mrs Lisbet Partanen and Mr Jouko Laitila for skilful technical assistance. This study was supported by grants from the Helsinki University Central Hospital Research Fund (Helsinki, Finland) and the Sigrid Juselius Foundation (Helsinki).
References
- 1.Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–18. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 2.Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M, Cowley H. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–80. [PubMed] [Google Scholar]
- 3.Hanefeld M. Pharmacokinetics and clinical efficacy of pioglitazone. Int J Clin Pract Suppl. 2001;121:19–25. [PubMed] [Google Scholar]
- 4.Eckland DA, Danhof M. Clinical pharmacokinetics of pioglitazone. Exp Clin Endocrinol Diabetes. 2000;108(Suppl. 2):234–42. [Google Scholar]
- 5.Baldwin SJ, Clarke SE, Chenery RJ. Characterization of the cytochrome P450 enzymes involved in the in vitro metabolism of rosiglitazone. Br J Clin Pharmacol. 1999;48:424–32. doi: 10.1046/j.1365-2125.1999.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaakkola T, Laitila J, Neuvonen PJ, Backman JT. Pioglitazone is metabolised by CYP2C8 and CYP3A4 in vitro: potential for interactions with CYP2C8 inhibitors. Basic Clin Pharmacol Toxicol. 2006;99:44–51. doi: 10.1111/j.1742-7843.2006.pto_437.x. [DOI] [PubMed] [Google Scholar]
- 7. [2 February 2007]. Avandia Prescribing Information. Available at http://www.avandia.com.
- 8. [2 February 2007]. Actos Prescribing Information. Available at http://www.actos.com.
- 9.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–65. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 10.Nakai D, Nakagomi R, Furuta Y, Tokui T, Abe T, Ikeda T, Nishimura K. Human liver-specific organic anion transporter, LST-1, mediates uptake of pravastatin by human hepatocytes. J Pharmacol Exp Ther. 2001;297:861–7. [PubMed] [Google Scholar]
- 11.Simonson SG, Raza A, Martin PD, Mitchell PD, Jarcho JA, Brown CD, Windass AS, Schneck DW. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004;76:167–77. doi: 10.1016/j.clpt.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004;311:139–46. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- 13.Tirona RG, Leake BF, Merino G, Kim RB. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J Biol Chem. 2001;276:35669–75. doi: 10.1074/jbc.M103792200. [DOI] [PubMed] [Google Scholar]
- 14.Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005;15:513–22. doi: 10.1097/01.fpc.0000170913.73780.5f. [DOI] [PubMed] [Google Scholar]
- 15.Niemi M, Kivistö KT, Hofmann U, Schwab M, Eichelbaum M, Fromm MF. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1) Br J Clin Pharmacol. 2005;59:602–4. doi: 10.1111/j.1365-2125.2005.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, Eichelbaum M, Kivistö KT, Neuvonen PJ. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther. 2005;77:468–78. doi: 10.1016/j.clpt.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 17.Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16:873–9. doi: 10.1097/01.fpc.0000230416.82349.90. [DOI] [PubMed] [Google Scholar]
- 18.Niemi M, Pasanen MK, Neuvonen PJ. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin Pharmacol Ther. 2006;80:356–66. doi: 10.1016/j.clpt.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Nishizato Y, Ieiri I, Suzuki H, Kimura M, Kawabata K, Hirota T, Takane H, Irie S, Kusuhara H, Urasaki Y, Urae A, Higuchi S, Otsubo K, Sugiyama Y. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokinetics. Clin Pharmacol Ther. 2003;73:554–65. doi: 10.1016/S0009-9236(03)00060-2. [DOI] [PubMed] [Google Scholar]
- 20.Mwinyi J, Johne A, Bauer S, Roots I, Gerloff T. Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin Pharmacol Ther. 2004;75:415–21. doi: 10.1016/j.clpt.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, Backman JT, Kerb R, Schwab M, Neuvonen PJ, Eichelbaum M, Kivistö KT. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1) Pharmacogenetics. 2004;14:429–40. doi: 10.1097/01.fpc.0000114750.08559.32. [DOI] [PubMed] [Google Scholar]
- 22.Pasanen MK, Backman JT, Neuvonen PJ, Niemi M. Frequencies of single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide 1B1 SLCO1B1 gene in a Finnish population. Eur J Clin Pharmacol. 2006;62:409–15. doi: 10.1007/s00228-006-0123-1. [DOI] [PubMed] [Google Scholar]
- 23.Nozawa T, Sugiura S, Nakajima M, Goto A, Yokoi T, Nezu J, Tsuji A, Tamai I. Involvement of organic anion transporting polypeptides in the transport of troglitazone sulfate: implications for understanding troglitazone hepatotoxicity. Drug Metab Dispos. 2004;32:291–4. doi: 10.1124/dmd.32.3.291. [DOI] [PubMed] [Google Scholar]
- 24.Chang C, Pang KS, Swaan PW, Ekins S. Comparative pharmacophore modeling of organic anion transporting polypeptides: a meta-analysis of rat Oatp1a1 and human OATP1B1. J Pharmacol Exp Ther. 2005;314:533–41. doi: 10.1124/jpet.104.082370. [DOI] [PubMed] [Google Scholar]
- 25.Niemi M, Backman JT, Granfors M, Laitila J, Neuvonen M, Neuvonen PJ. Gemfibrozil considerably increases the plasma concentrations of rosiglitazone. Diabetologia. 2003;46:1319–23. doi: 10.1007/s00125-003-1181-x. [DOI] [PubMed] [Google Scholar]
- 26.Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther. 2005;77:404–14. doi: 10.1016/j.clpt.2004.12.266. [DOI] [PubMed] [Google Scholar]
- 27.Kirchheiner J, Roots I, Goldammer M, Rosenkranz B, Brockmöller J. Effect of genetic polymorphisms in cytochrome p450 (CYP) 2C9 and CYP2C8 on the pharmacokinetics of oral antidiabetic drugs: clinical relevance. Clin Pharmacokinet. 2005;44:1209–25. doi: 10.2165/00003088-200544120-00002. [DOI] [PubMed] [Google Scholar]
- 28.Niemi M, Leathart JB, Neuvonen M, Backman JT, Daly AK, Neuvonen PJ. Polymorphism in CYP2C8 is associated with reduced plasma concentrations of repaglinide. Clin Pharmacol Ther. 2003;74:380–7. doi: 10.1016/S0009-9236(03)00228-5. [DOI] [PubMed] [Google Scholar]
- 29.Jaakkola T, Backman JT, Neuvonen M, Laitila J, Neuvonen PJ. Effect of rifampicin on the pharmacokinetics of pioglitazone. Br J Clin Pharmacol. 2006;61:70–8. doi: 10.1111/j.1365-2125.2005.02515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez C, Garcia-Martin E, Blanco G, Gamito FJ, Ladero JM, Agundez JA. The effect of the cytochrome P450 CYP2C8 polymorphism on the disposition of (R)-ibuprofen enantiomer in healthy subjects. Br J Clin Pharmacol. 2005;59:62–9. doi: 10.1111/j.1365-2125.2004.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowland M, Tozer TN. Clinical Pharmacokinetics: Concepts and Applications. 3. Baltimore, MD: Williams & Wilkins; 1995. [Google Scholar]
- 32.Shitara Y, Hirano M, Sato H, Sugiyama Y. Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug–drug interaction between cerivastatin and gemfibrozil. J Pharmacol Exp Ther. 2004;311:228–36. doi: 10.1124/jpet.104.068536. [DOI] [PubMed] [Google Scholar]
- 33.Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46:347–51. doi: 10.1007/s00125-003-1034-7. [DOI] [PubMed] [Google Scholar]
- 34.Niemeyer NV, Janney LM. Thiazolidinedione-induced edema. Pharmacotherapy. 2002;22:924–9. doi: 10.1592/phco.22.11.924.33626. [DOI] [PubMed] [Google Scholar]
- 35.Cheng AY, Fantus IG. Thiazolidinedione-induced congestive heart failure. Ann Pharmacother. 2004;38:817–20. doi: 10.1345/aph.1D400. [DOI] [PubMed] [Google Scholar]
- 36.Cekmen N, Cesur M, Cetinbas R, Bedel P, Erdemli O. Acute pulmonary edema due to rosiglitazone use in a patient with diabetes mellitus. J Intensive Care Med. 2006;21:47–50. doi: 10.1177/0885066605283385. [DOI] [PubMed] [Google Scholar]
- 37.Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L, Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–89. doi: 10.1016/S0140-6736(05)67528-9. PROactive investigators. [DOI] [PubMed] [Google Scholar]