

Abstract
Menin is a tumor suppressor required to prevent multiple endocrine neoplasia in humans. Mammalian menin protein is associated with chromatin modifying complexes and has been shown to bind a number of nuclear proteins, including the transcription factor JunD. Menin shows bidirectional effects acting positively on c-Jun and negatively on JunD. We have produced protein null alleles of Drosophila menin (mnn1) and have over expressed the Mnn1 protein. Flies homozygous for protein-null mnn1 alleles are viable and fertile. Localized over-expression of Mnn1 causes defects in thoracic closure, a phenotype that sometimes results from insufficient Jun activity. We observed complex genetic interactions between mnn1 and jun in different developmental settings. Our data support the idea that one function of menin is to modulate Jun activity in a manner dependent on the cellular context.
Keywords: AP1, Oxidative stress, Paraquat, Life span, Cleft thorax, Tumor suppressor, Multiple endocrine neoplasia type I
Introduction
Human multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant cancer syndrome characterized by tumors occurring prevalently in endocrine tissues. Common features of most MEN1 tumors are low proliferation rates, well-differentiated morphology and excessive hormone secretion. Hereditary tumors arise in individuals heterozygous for a loss-of-function MEN1 allele followed by somatic loss of wild type alleles. Sporadic tumors also show bi-allelic loss of MEN1 (Agarwal et al., 2004). The MEN1 locus encodes menin, a nuclear protein with two nuclear-localization sites at the C-terminal quarter of the protein, but no other overt sequence motifs (Chandrasekharappa et al., 1997; Guru et al., 1998). Menin is ubiquitously expressed, but only shows a loss of heterozygosity phenotype in a highly restricted set of cells (Scacheri et al., 2004). This context dependency suggests that regulated co-factors or modifiers act in conjunction with menin for cell-type specific function. Menin has also been found in a SET1-like histone methylation complex (Hughes et al., 2004; Karnik et al., 2005; Milne et al., 2005; Yokoyama et al., 2004, 2005). The mouse menin gene is required for embryonic viability and, like in humans, inactivation of both alleles results in endocrine tumors (Crabtree et al., 2001, 2003). Therefore, menin is a classic tumor suppressor in the endocrine system. Interestingly, there is also recent evidence that menin is an oncogenic co-factor in Mixed Lineage Leukemia (Yokoyama et al., 2005). The nature of this dual growth suppressing and enhancing role in the regulation of proper cell number and differentiation has not been clarified.
Multiple potential transcription factor partners for mammalian menin protein have been identified (Agarwal et al., 2004) including JunD, which has been shown to interact directly with menin (Agarwal et al., 1999). It is unclear how these protein–protein interactions relate to menin in the SET-1 like histone methylation complex, although it is possible that menin association with many different nuclear proteins helps target the complex to appropriate regions of chromatin. Experiments performed in immortalized mouse embryo fibroblasts have shown that menin binding to JunD is necessary for JunD to act as a growth suppressor (Agarwal et al., 2003). Menin functions to reduce JunD activity (Agarwal et al., 1999; Gobl et al., 1999; Kim et al., 2005; Naito et al., 2005) and has been shown to inhibit the accumulation of active phosphorylated JunD or c-Jun (Gallo et al., 2002). Even though menin does not directly bind c-Jun, it augments the transcriptional activity of this transcriptional factor (Knapp et al., 2000). Thus, menin is strongly implicated in regulating Jun function. Interestingly, according to the potential roles of menin to promote or suppress tumorigenesis, menin can act in turn negatively on JunD or positively on c-Jun function.
Jun and Fos heterodimers are well-known regulators of tumorigenesis, differentiation, apoptosis, immune and stress responses in both vertebrates and Drosophila (Kockel et al., 2001; Mechta-Grigoriou et al., 2001). There are a number of mammalian homodimers and heterodimers consisting in c-Jun, JunB or JunD and c-Fos, FosB, Fra1 or Fra2 combinations (Mechta-Grigoriou et al., 2001). Unlike mammals, Drosophila has a single Jun and a single Fos (Kockel et al., 2001). Drosophila Jun has features of both JunD and c-Jun. This makes Drosophila a good reductionist model for learning more about Jun/menin interactions. While experiments to see if Drosophila menin binds Jun have been negative (Guru et al., 2001), genetic interactions have not been explored. In this study, we have specifically investigated the functional connection between Drosophila menin and Jun.
The Drosophila melanogaster menin protein (Mnn1) is 47% identical to the human protein, including 69% of the amino acid residues that are required for tumor suppression in human endocrine tissues (Guru et al., 2001; Maruyama et al., 2000). The ongoing sequencing of multiple species in Drosophila reveals that menin is highly conserved among them (Fig. 1). Despite this high degree of conservation, menin is not required for viability in D. melanogaster. Flies lacking mnn1 expression are viable and fertile (Busygina et al., 2004; Papaconstantinou et al., 2005). One report suggests that mnn1 is required for a wild type life span and some aspect of either chromosome stability or DNA repair (Busygina et al., 2004), while another report suggests that mnn1 is required for a robust response to various types of stress (Papaconstantinou et al., 2005).
Fig. 1.

Alignment of predicted menin polypeptides in Drosophila species. Multiple alignment of predicted menin protein sequences from seven species of Drosophila compared to human performed by ClustalW. Conserved residues (shaded) and Drosophila melnaogaster splice junctions (brackets). Human (gi|18860839, NP_000235.2) and Drosophila melanogaster (gi:28574051, NP_523498.2) sequences are from Genbank (Benson et al., 2005). Drosophila pseudoobscura sequence is from FlyBase (FlyBase, 2003). Remaining sequences are from draft annotations (V. N. Iyer, D. A. Pollard and M. B. Eisen, personal communication) of assemblies generated by Agencourt (D. Smith, personal communication) and Washington University (R. Wilson, personal communication) genome sequencing centers.
We have isolated two protein-null mnn1 alleles and have generated transgenic flies for the controlled over-expression of Drosophila Mnn1 protein. As previously reported, mnn1− Drosophila is viable and fertile. It has been reported that uniform over-expression of mnn1 has no effect on development or viability in flies (Papaconstantinou et al., 2005). We find that over-expression results in pharate-adult phenotype, proboscis ablation and a cleft thorax. These over-expression phenotypes are modified by both gain-of-function and loss-of-function alleles of jun. Dominant-negative alleles of fos are enhanced by loss-of-function alleles of mnn1. The finding that both Drosophila and mammalian menin (Agarwal et al., 1999, 2003) are capable of interacting with Jun suggests that an evolutionarily conserved menin function in normal development and disease is linked to the Jun/Fos family of transcriptional regulators. Interestingly, as in mammals, Drosophila menin shows bidirectional modulation of Jun function.
Materials and methods
Flies
A P-element insertion at the mnn1 locus, P{wHy}30G01 (Huet et al., 2002), is located in the 5′ untranslated region (at + 601 bp from transcription start, 503 bp upstream of the start codon) of the gene models based on two studies directed at mnn1 characterization (Guru et al., 2001; Maruyama et al., 2000). We have not found evidence to support the slightly different gene models annotated at FlyBase (FlyBase, 2003). Imprecise excision alleles were generated by crossing P{wHy}30G01 to Δ2–3 flies. We screened 14,984 chromosomes identifying y− w+ lines (159) and w− y− lines (141). Excision rearrangements were detected by Southern blots, diagnostic PCR reactions and DNA sequencing. We obtained 14 lines with rearrangements in mnn1 DNA sequences and 12 precise-excision alleles. Two of the precise-excision lines were saved and used as isogenic controls. The mnn1Δ46 allele has a deletion of 573 bp of the mnn1 gene and the entire P{wHy}30G01 insertion. This allele is missing 70 bp downstream of the first ATG of the mnn1 ORF. The mnn1Δ79 allele has a more extensive deletion of 186 bp downstream of the start codon of mnn1, but retains part of the 5′ sequence of P{wHy}30G01 (and is w+). The final new aberration, Df(2L)mnn1, is a large deficiency removing several vital genes proximal to mnn1 (Fig. 2).
Fig. 2.

Map of mnn1 locus and description of mnn1 alleles. (A) Molecular map of the mnn1 genomic region showing the flanking gene milton (milt) and the CG31907 gene nested in mnn1. Big arrows indicate the 5′ to 3′ orientation of the genes. Genomic maps of mnn1 mutants are reported in scale below. The black filled triangle localizes the P{wHy}30G01 insertion. Gaps in lines indicate the deleted genomic sequences described for each mnn1 mutant allele. (B) Molecular map of the mnn1 transcripts. Filled boxes show exons, thin lines show introns, black filling indicates the open reading frame, gray filling indicates untranslated regions and alternative poly-A. Mnn1-RA and mnn1-RB indicate the long and the short transcript annotated in FlyBase. Y sign indicates the antigenic sequence used to generate anti-Mnn1. Interrupted lines show genomic map of mnn1Δ46 and mnn1Δ79 alleles in scale with the mnn1 transcripts. Small arrows localize the sites and directions of RT-PCR primers. (C) Amino acid sequence of N-terminus of Mnn1 protein. Stars indicate alternative translational start sites. Filled circles indicate amino acid residues mutated in MEN1 patients. Sequences in gray show the amino acid residues missing in mnn1Δ46 and mnn1Δ79 due to the absence of the ATG (D) RT-PCR analysis of RNA extracted from mnn1+84 precise-excision and mnn1Δ46 and mnn1Δ79 mutant strains, using a primer overlapping the ATG sequence (upper panel) or primers spanning intron 4 sequence (middle panel). Control for genomic DNA contamination is shown (lower panel). (E) Western blot anti-Mnn1 on bacterial expressed pure Mnn1 and protein extracts from wild type (mnn1+113 and mnn1+84) and mnn1 mutant (mnn1Δ46 and mnn1Δ79) adult heads. All lanes are from a single blot. Expected mobility of Mnn1 and an anonymous (Anon) cross-reacting material is shown.
UAS-mnn1 transgenes were generated by subcloning the full-length Drosophila mnn1 cDNA (Guru et al., 2001) into the pUAST vector (Brand and Perrimon, 1993). Flies were transformed using standard techniques (Rubin and Spradling, 1982). Induction of mnn1 with assorted drivers was verified by Western blotting or immunostaining with anti-Mnn1. All six UAS-mnn1 transgenic lines showed similar results. In some cases (as noted in the text), crosses showing a strong phenotype were retested at 18°, 25° and 29°C to allow us to look for effects of different levels of Gal4 activity.
Flies were grown on GIF medium, under uncrowded conditions, at 25°C (KD Medical, Columbia MD). Extensive descriptions of mutant and GAL4 lines can be found at FlyBase (FlyBase, 2003).
RT-PCR
RT-PCR reactions were performed on flies homozygous for mnn1 alleles. Total RNAwas isolated from 50 homozygous female flies, aged 3–5 days and fed overnight on yeast paste. Females were chosen to increase the probability of detecting both zygotic and maternally loaded mnn1 transcripts. Total RNA was isolated with Trizol reagent (Invitrogen, Carlsbad CA). Approximately 250 μg of total RNA was treated with 10U RQ1 DNAse (Promega, Madison WI) at 37°C, for 15 min followed by two phenol extractions and ethanol precipitation. Total RNA (25 μg) was used for first strand synthesis using 2 μg dT16–18 and three different concentrations of random hexamers that ranged to 100-fold dilutions. RNA/primers were heated 70°C for 3 min and DNA synthesis was done with Superscript according to manufacturer protocols (Invitrogen, Carlsbad CA). Thirty cycle PCRs were done using a PTC-225 gradient thermocycler (Bio-Rad, Hercules CA).
Forward primer 5′TGTCCACGATTACCAGAAGCG overlapping the ATG sequence was designed in pair with a reverse primer 5′AGCGAGTTCCAGATCACATCCG designed in a retained sequence of exon 3 of mnn1; forward primer 5′TACGACATTAGGTCCCAGGTGG and reverse primer 5′TTGCTTGTGGTTGTTGCGTTAG were designed to amplify sequence spanning intron 4.
Additional primer sequences used for transcript analysis are available upon request.
Immunoblotting and microscopy
Rabbit polyclonal anti-Mnn1 was produced using a 23 a.a. peptide, LPEDLEAEQAKAELARAEQEAKE, corresponding to a.a. 464–487 as an antigen (BioCon, Bangalore India). Antiserum was affinity purified on the peptide as described (Goldsmith et al., 1987). Whole flies or tissues were directly homogenized in Laemmli buffer and separated by 8% SDS-PAGE. Blots were developed with Enhanced Chemiluminescence (Amersham, Piscataway NJ). For cell staining, tissues were dissected in PBS buffer, fixed in 2% paraformaldehyde in PBS, permeabilized in PBS with 0.1% Triton X-100, blocked in PBS with 0.1% Triton X-100 and 0.5% BSA for 2 h and incubated in rabbit anti-Mnn1 or anti-Actin (Sigma-Aldrich, St. Louis MO) overnight. After rinsing in TBS, tissues were incubated with a secondary antibody conjugated to fluorescein or rhodamine (Jackson ImmunoResearch Laboratory, West Grove PA), rinsed in TBS, counterstained with DAPI (Invitrogen, Carlsbad CA), mounted in 70% glycerol containing 2.5% DABCO (Sigma-Aldrich, St. Louis MO) and observed on a Zeiss confocal microscope.
For scanning electron microscopy, flies were fixed in 4% glutaraldehyde in PBS for 2 h at room temperature, then washed in PBS and dehydrated in a graded series of acetone-PBS (50, 70 and 100%) at room temperature. Afterwards, the specimens were critical point dried, mounted in stubs head up, sputter coated, scanned at 200, 1000 and 2500 × and photographed in a Zeiss scanning electron microscope.
Life span and paraquat resistance determinations
For life span and paraquat experiments, 0–2 day post-eclosion progeny were collected and were allowed to mate freely for 3 days. Sexes were then separated and used in the two assays. For paraquat treatments, 10 mM paraquat (Sigma-Aldrich, St. Louis MO) solution was freshly prepared in 1% sucrose. 400 μL was used to saturate two filter paper disks (Whatman, Florham Park NJ) in an otherwise empty vial. 100 flies/genotype were starved for 2 h and then transferred in groups of 20 to the disk-containing vials. Flies immediately started feeding on the sucrose-paraquat solution and their viability was scored at 5 h intervals. The experiment was performed on both sexes and repeated three times giving similar results each time. Life span determination was examined using 50 males/genotype. Flies were separated in two groups of 25 flies and placed on fresh vials of standard food every second day (starting at day 5). Replicate life span determinations were performed. Data are not pooled in the results shown. Mnn1 mutants always showed reduced viability vs. controls. Statistical analysis was performed in BioConductor (Gentleman et al., 2004).
Results and discussion
Generation of mnn1 mutants
The mnn1 locus is tightly flanked upstream by the milton gene (milt) and the CG31907 gene is nested in a mnn1 intron (Fig. 2A). Previously identified deletion alleles of mnn1 (Busygina et al., 2004; Papaconstantinou et al., 2005) are likely to disrupt the function of flanking genes in addition to mnn1 (Fig. 2A). The mnn1e200 allele is also mutant for milt (Busygina et al., 2004). The mnn1e173 allele potentially disrupts milt. Both mnn1e173 and mnn1e30 delete sequences that approach the 3′ end of CG31907 (Papaconstantinou et al., 2005). We have mobilized a P-element, P{wHy}30G01, inserted in the 5′UTR of the mnn1 locus (Fig. 2A), and screened for small deletions in order to identify new alleles of mnn1 that would not affect other genes.
FlyBase (2003) annotates two mnn1 transcripts, but neither of these transcripts have been isolated in previous molecular studies of the mnn1 locus (Guru et al., 2001; Maruyama et al., 2000). Maruyama and Guru, independently, describe mnn1 transcripts that differ from the two FlyBase annotations in the UTRs and in the terminal coding exon (Fig. 2B). A developmental profile of mnn1 expression has revealed two mnn1 transcripts (Guru et al., 2001) that are due to alternative poly-A sites (Fig. 2B). The shorter transcript annotated in FlyBase (mnn1-RB) may have been primed from an A-rich sequence in the intron of an unprocessed message rather than from the poly-A tail. Additionally, none of the 18 amino acids specific to Mnn1-PB are present in each of mnn1 genes of the Drosophila species that we have reported in Fig. 1. Thus, the Muruyama and Guru gene models are our reference throughout this manuscript.
We generated two deletion alleles, mnn1Δ46 and mnn1Δ79. RT-PCR and sequence analysis indicate that the mnn1Δ46 allele has a deletion of 573 bp of the mnn1 locus missing 70 bp downstream of the first ATG of the mnn1 ORF (Agarwal et al., 2004), while the mnn1Δ79 allele has a more extensive deletion of 186 bp downstream of the start codon (Fig. 2B). However, all distal genes appeared to be intact, including CG31907 which is nested in mnn1 intron 4. A third new allele, Df(2L)mnn1Δ65 deletes at least 14 kb proximal to the 3′ end of the Muruyama and Guru mnn1 gene model (Fig. 2B). This deletion, along with Df(2L)JH, removes several additional complementation groups required for viability (Fig. 2A).
Flies homozygous for either mnn1Δ46 or mnn1Δ79 are viable and fertile and can be readily maintained as homozygous stocks. Hemizygous mnn1Δ46 or mnn1Δ79 flies are also viable and fertile over Df(2L)mnn1 or Df(2L)JH. In addition to the mutant alleles, we selected two precise excision lines, mnn1+84 and mnn1+113, as wild type isogenic controls for further experiments. The mnn1Δ46 and mnn1Δ79 chromosomes were extensively backcrossed to control y− w− flies to remove any undetected mutations associated with transposon mobilization.
Mnn1 mRNA isoforms are expressed in wild type early embryos and in adult females. The longer isoform is detected throughout development (Guru et al., 2001). To determine if the deletion alleles express mnn1 mRNA, as might be expected given the presence of residual promoter region and upstream sequences, we performed RT-PCR reactions on total RNA extracts from mnn1Δ46 to mnn1Δ79 homozygous adult females and on perfect excision lines using multiple primer pairs also overlapping the ATG sequence and spanning mnn1 intronic sequences (Fig. 2B, additional data not shown). The use of intron-spanning primers in the absence of reverse transcriptase allowed us to distinguish between transcripts and any contaminating genomic DNA. RT-PCR results obtained on wild type flies supported the first exon structure in the Muruyama and Guru gene model (Fig. 2D, additional data not shown). While no transcripts were detected with primers directed against deleted sequence in homozygous mnn1Δ46 and mnn1Δ79, transcripts were detected using primers downstream from those deletions (Fig. 2D). These results indicate that mnn1 mRNAs are produced from the mutant alleles.
Both mnn1Δ46 and mnn1Δ79 alleles delete mnn1 sequence coding for residues homologous to those known to be required for menin function in humans (Agarwal et al., 2004). In both mnn1Δ46 or mnn1Δ79, the first two in-frame ATGs of mnn1 are deleted, such that homologs of at least five amino acids required to prevent disease in humans are deleted due to a downstream translational start site utilization (Fig. 2C). To determine if mnn1Δ46 and mnn1Δ79 alleles encode defective menin proteins initiated from downstream AUGs in the mutant mRNAs (Fig. 2C), we performed immunoblots (Fig. 2E) and cell staining experiments (Figs. 3A–D). While these putative mutant polypeptides would be missing critical Mnn1 residues, they might retain some function (wild type or even dominant negative). Mnn1 proteins initiated by alternative downstream AUGs present in mnn1 mutant mRNAs should migrate faster on SDS-PAGE. Immunoblot analysis performed with an antibody produced against an epitope mapping in exon 4 (Fig. 2B) showed a species migrating at ~ 95 kDa in extracts from wild type flies and extracts from bacteria expressing Drosophila Mnn1, but not from homozygous mnn1Δ46 or mnn1Δ79 flies (Fig. 2E). These results indicate that the antibody recognizes wild type Mnn1. We have analyzed protein extracts of whole adult females or males, third instar larvae and Central Nervous System (CNS) from third instar larvae and in no case did we detect a shorter isoform. Thus, in mnn1Δ46 or mnn1Δ79 larvae or adults, there is no evidence of N-terminally truncated Mnn1 proteins. Western blot results therefore simultaneously confirm that the bands in the wild type lanes correspond to endogenous Mnn1, not a cross-reacting species of similar mobility, and that the deletion alleles encode undetectable levels of N-terminally deleted Mnn1 protein. We did observe an anonymous slower migrating band in some of the Western blots (Fig. 2E); however, it is difficult to envision how a protein with this mobility could be encoded by mnn1. This band is almost certainly due to a cross-reacting species. We conclude that the mnn1Δ46 and mnn1Δ79 alleles are protein nulls. Previously reported mnn1 alleles are also likely to be protein nulls (Busygina etal.,2004; Papaconstantinouetal., 2005). In no case has mnn1 been shown to be required for viability or fertility. All these data strongly suggest that mnn1 is not an essential gene in Drosophila.
Fig. 3.

Anti-Mnn1 tissue staining in wild type, over-expressing and Mnn1 null mutants. (A–D) Anti-Mnn1 immunofluorescence (green) of third instar larval brain, in wild type (B, D) and protein null (A, C). Immunofluorescent (A, B) and differential contrast channels (C, D) of the same preparation are shown. Staining is observed in the brain hemispheres (arrow) and ring gland (arrow head). (E–H) Multiple channel view of magnified ring gland polyploid tissue. Mnn1 is stained in green (E), actin in red (F) and DNA in blue (G). Merge of the three channels (H) reveals weak but consistent nuclear staining of Mnn1. Nuclear staining appears non-uniform. (I–P) Mnn1 staining (green) in third instar larvae following UAS-mnn1 induction by AB1-GAL4 driver in salivary gland (I), insulin dilp2-GAL4 driver in brain lobe (K), How24-GAL4 driver in CNS (M), motor neuron OK6-GAL4 driver in CNS (O). (J, L, N, P) Magnification of groups of cells showing intense Mnn1 staining (empty boxes in upper panel). Double staining for DNA (blue) clearly shows Mnn1 in the nucleus of polytene salivary cells (J) and nuclei of groups of neurons (L, N, P). Weaker Mnn1 staining appears punctate (arrowheads in panel L) and may represent endogenous Mnn1 in surrounding cells.
Human menin is a nuclear protein (Guru et al., 1998). The Drosophila Mnn1 protein has a potential nuclear localization signal (KRTRR) in the region corresponding to the NLS-2 of human menin (Guru et al., 2001). The mnn1 gene is broadly expressed throughout development (Guru et al., 2001). As expected, we detected Mnn1 immunoreactivity in the nuclei of the central nervous system (Fig. 3B), and many other larval tissues of wild type flies. Such staining was absent in flies homozygous for mnn1Δ46 or mnn1Δ79 (Fig. 3A). The weak anti-Mnn1 staining of endogenous protein was enriched in the nucleus and showed sub nuclear localization (Figs. 3E–H). To further evaluate the cellular localization of Drosophila Mnn1, we detected over-expressed Mnn1 following induction of UAS-mnn1 with any number of Gal4 drivers (e.g. AB1, 69B, How24, dilp2 and OK6). In all cases, over-expressed Mnn1 is nuclear (Figs. 3I–P, additional data not shown). This staining is robust, again suggesting that endogenous Mnn1 is not abundant. Over-expressed Mnn1 from Drosophila extracts also co-migrates with bacterially expressed Mnn1 at ~ 95 kDa (not shown). These data suggest that, like mammalian menin, Drosophila menin is nuclear.
mnn1 loss-of-function phenotype
Flies lacking mnn1 are viable and fertile as also shown by others (Busygina et al., 2004; Papaconstantinou et al., 2005). Our mutants show no overt and consistent phenotype as homozygote, trans-heterozygote or in trans to Df(2L)mnn1Δ65 or Df(2L)J–H. As expected for a protein null allele, they behave as genetic amorphs, with the amorphic condition being viable, fertile, with no visible phenotype. Mnn1e200 flies were stated to have a reduced life span (Busygina et al., 2004). The mnn1e200 allele is deleted for both mnn1 and milt (Fig. 2A), and the milt locus is required for viability. Thus, incomplete rescue with a milt+ transgene could cause a reduction in life span (Busygina et al., 2004). However, our observations support the idea that mnn1 is required for a wild type life span (Fig. 4A). A slight, but highly significant reduction in viability was observed both in homozygous mnn1− flies and in the trans-allelic mnn1− flies (D = 0.35, P < 3 × 10−4 by two-sample Kolmogorov–Smirnov test). There were no significant differences between different genotypes of mnn1− (P > 0.99). Interestingly, the reduction in viability was due to a constant rate of early mortality of mnn1− males in days 1–30 (D = 0.65, P < 2 × 10−7 by two-sample Kolmogorov–Smirnov test).
Fig. 4.

Loss-of-function mnn1 phenotypes. Analysis of mnn1− mutant (red lines) and mnn1+ control (black lines) for survival on standard media (A) and on 10 mM paraquat in sucrose (B). Genotypes are indicated. (A) Red vs. black lines show a significant survival difference (P < 3 × 10−4 K–S test) more evident in the first 30 days (P < 2 × 10−7 K–S test) while red vs. red lines are almost identical (P > 0.99 K–S test). (B) Red vs. black lines show a slight difference (P < 3 × 10−4 K–S test) in oxidative stress response.
It has also been reported that mnn1e173 and mnn1e30 mutants are sensitive to a range of stressors (Papaconstantinou et al., 2005), but again the results obtained might have been confounded by the more extensive deletions (Fig. 2A). We tested our mnn1 mutant alleles for oxidative stress sensitivity using the herbicide paraquat, a powerful generator of reactive oxygen species. We find that flies either homozygous for the mnn1− alleles or trans-heterozygous for those alleles appear to be slightly more resistant to paraquat than mnn1+ or mnn1+/mnn1− flies (D = 0.35, P < 3 × 10−4 by two-sample Kolmogorov–Smirnov test) (Fig. 4B). This observation is more striking given the increased mortality of young mnn1− flies. Our results appear to be in contrast to what was reported previously by Papaconstantinou indicating that mnn1e178 or mnn1e30 flies are more sensitive to paraquat, not resistant. Determining if this inconsistency is due to the different nature of the generated mutants will require further investigation. The salient agreement among all the mnn1 functional studies is that mnn1 is a non-essential gene in Drosophila. The lack of a developmental defect in the more streamlined Drosophila genome is surprising as mice homozygous for menin null alleles die as embryos (Crabtree et al., 2001). There are no obvious additional mnn1-like genes in Drosophila suggesting that the absence of a developmental defect is not due to the function of a second mnn1 gene. Perhaps menin has acquired non-conditional function only in the vertebrate lineage.
mnn1 over-expression phenotypes
We explored the consequences of excess mnn1 expression by generating transgenic lines bearing the full-length mnn1+ cDNA under the control of the yeast Gal4 inducible UAS promoter and a wide range of Gal4 drivers. It has been reported that uniform over-expression of Mnn1 does not alter development or viability (Papaconstantinou et al., 2005). We see distinct and dramatic effects of Mnn1 over-expression in a subset of tissues.
To begin systematically exploring the effect of Mnn1 over-expression on Drosophila development, we drove mnn1 expression with Gal4 in a series of distinct spatiotemporal patterns (Table 1). Because the distribution of endogenous Mnn1 is quite broad, this is likely to increase the levels of Mnn1 in cells (cf. Figs. 3I–P), rather than altering the spatial distribution of Mnn1. The over-expression of Mnn1 protein (as determined by cell staining and/or immunoblotting) with any of five different Gal4 drivers (Table 1) resulted in a adult-pharate lethal phenotype (Figs. 5B–D). Interestingly, we found that all of these drivers are expressed in subsets of neurons in addition to the reported expression patterns. Development was arrested during late pupal morphogenesis at stage P14. Dissection of dead pupae shows deletion of distal elements of the proboscis and a melanotic mass at that location. The melanotic mass is evident prior to lethality (stage P7) as shown in Fig. 5C. Exceptional flies that escape adult-pharate lethality when UAS-mnn1 is driven by How24-GAL4 (~ 5%) or 69B-GAL4 (~ 25%) show a melanotic mass at the anterior proboscis following eclosion. In the rare eclosing flies, the presence of a proboscis defect is not compatible with adult life, flies die 2–3 days later probably because of hindered intake of food and water. Wing inflation also failed in these escaping flies. Experiments performed at a lower temperature (22°) show an increased percentage of escaped flies and a reduced severity in the proboscis and wing defects. Gal4 is known to be less active at lower temperatures (Duffy, 2002), suggesting that the level of Mnn1 induction correlates with the intensity of the phenotype observed.
Table 1.
UAS-mnn1 phenotypes induced by Gal4 drivers
| Gal4 driver | Reported expression pattern a | Phenotype with UAS-mnn1 |
|---|---|---|
| How24 | Mesoderm b | Pharate lethal, cleft thorax |
| elavC155 | CNS | Pharate lethal |
| daG32UH1 | Ubiquitous | Pharate lethal |
| 69B | Ectoderm b | Pharate lethal, cleft thorax |
| OK6 | Motor neurons | Pharate lethal |
| twiG108.4 | Mesoderm | wt |
| ninaE.GMR12 | Morphogenic eye | wt |
| ey.H3–8 | Eye primordia | wt |
| C1003 | Ectoderm, CNS | wt |
| AB1 | Salivary gland | wt |
| crc929 | Neuroendocrine | wt |
| dilp2 | Insulin secreting neurons | wt |
| 48Y | Endoderm | wt |
| dpp.blk140C6 | dpp pattern | wt |
| pnrMD237 | pnr pattern | Cleft thorax |
See Flybase (2003) for references.
Also expressed in a subset of neurons.
Fig. 5.
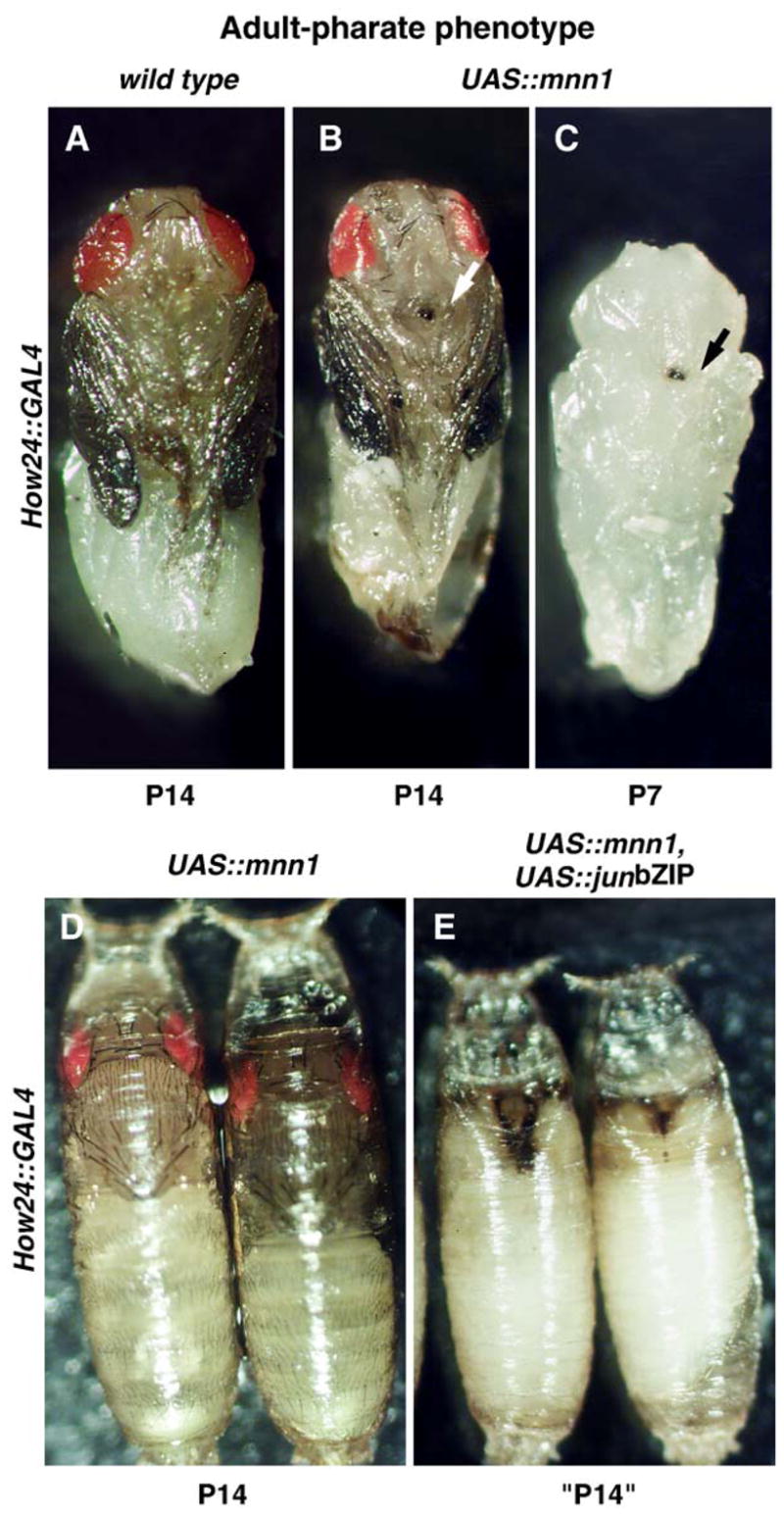
Over-expression of mnn1 results in adult-pharate phenotype enhanced by jun dominant negative. (A) Wild type pupae dissected at developmental stage P14 corresponding to the time of death caused by mnn1 induction. (B) Adult-pharate phenotype observed when UAS-mnn1 is driven by How24-GAL4. Note the dark spot at the end of the proboscis (arrow). (C) The dark spot is detectable as early as pupal stage P7 (arrow), same genotype as panel B. (D) Pupae over-expressing mnn1 die later than (E) pupae over-expressing mnn1 and dominant negative jun contextually (UAS-mnn1 and UAS-junbZIP driven by How24-GAL4).
We occasionally observed a cleft thorax phenotype in flies when UAS-mnn1 is driven with 69B-GAL4. To further investigate the role of mnn1 in the developing Drosophila thorax, we expressed UAS-mnn1 using the pnr-GAL4 driver, which is expressed specifically in the leading edge cells of the wing disc and the medial region of the thorax in adults (Calleja et al., 1996). These cells participate in thorax closure during metamorphosis. Mnn1 protein is expressed throughout the wing disc in wild type flies and is clearly over-expressed in the leading edge cells in pnr > mnn1 (not shown). The thorax of adult flies carrying one copy each of both pnr-GAL4 and the UAS-mnn1 transgene always showed a dorsal cleft along the entire thorax with disrupted chaetae orientation (Figs. 6B, D) whereas the thorax of flies carrying either one copy of pnr-GAL4 or one copy of UAS-mnn1 alone was wild type (Figs. 6A, C). This thoracic defect was 100% penetrant. Furthermore, the severity of the phenotype was modulated by the number of copies of UAS-mnn1 expressed in the thorax (more copies result in a more extreme phenotype) and by the growth temperature, indicating that the phenotype is proportional to the degree of Mnn1 over expression (data not shown).
Fig. 6.

Thoracic over-expression of mnn1 and interactions between Mnn1 and Jun/Fos in the thorax. (A) Dorsal view of thorax in wild type. (B) Dorsal view of thorax when UAS-mnn1 is driven by pnr-GAL4. The thorax is reduced in the anterior–posterior dimension, the scutum is slightly enlarged, the scutellum is shortened, intrascutal and scutoscutellar sutures are less evident, scutellar bristles are curved and shorter. (D) SEM view of the thoracic dorsal midline showing an enlarged space between the dorsocentral bristles. (C) Wild type bristle distribution. (E) Absence of thoracic abnormalities in Jun defective flies (jra1/+). (F) Thoracic defect observed when UAS-mnn1 is driven by pnr-GAL4 in the thorax. (G) Enhanced thoracic cleft in flies with reduced dose of jun and over-expressing mnn1. (H) Mild defect of thoracic closure produced by over-expression of a jun dominant-negative when UAS-junbZIP is driven by pnr-GAL4. (I) Enhanced thoracic cleft when UAS-mnn1 and UAS-junbZIP are driven simultaneously by pnr-GAL4. (J) Absence of thoracic defect in flies over-expressing a dominant negative fos (UAS-fosbZIP) with the How24 driver. (K) Moderate thoracic defect detected in eclosed flies expressing fos dominant negative with reduced dose of mnn1. Absence of mnn1 in How24 > fosbZIP flies results in pupae lethality (Table 2).
The cleft thorax phenotype raises the possibility that Drosophila menin can act in the jun/fos pathway. Thorax formation occurs by fusion of hemithoraces during pupal development as the result of spreading and fusion of two lateral groups of cells in the midline. This event requires the coordinated action of the Jun/Fos signaling pathway. Too little or too much Jun/Fos activity results in failure to properly suture imaginal discs during metamorphosis (Agnes et al., 1999; Martin-Blanco et al., 2000). Jun/Fos activity is also required for embryonic dorsal closure, but we never observed an overt dorsal closure phenotype associated with loss-of-function or over expression of mnn1. The latter may be due in part to the abundant maternally deposited mnn1 transcript in embryos (Guru et al., 2001).
jun/fos interactions with mnn1 in the thorax
The cleft thorax phenotype is consistent with the idea that Mnn1 interacts with Jun/Fos, although this does not imply that the only function of mnn1 is in the jun/fos pathway. We tested the idea of Mnn1 interacting with Jun/Fos by using an extensive set of crosses designed to explore genetic interactions between mnn1 over-expressed with pnr-GAL4 and jun/fos alleles (Figs. 6E–I). We over expressed Mnn1 in a heterozygous jun background (jra1/+) and observed a more severe thoracic cleft phenotype. Similarly, the contextual over-expression of a dominant-negative jun (UAS-junbZIP) and mnn1 enhanced the thorax defect. We also tested for an effect of the induction of UAS-junbZIP on the adult-pharate lethal phenotype observed in How24 > mnn1 flies. While flies over-expressing UAS-junbZIP were wild type (Table 2), the flies expressing both mnn1 and junbZIP showed a much earlier arrest of the pupal development (Fig. 5E), rather than the adult-pharate phenotype seen when only mnn1 was over-expressed (Fig. 5D), again indicating that mnn1 over-expression is exacerbated by dominant negative jun activity.
Table 2.
Over-expression of mnn1 in jun or fos backgrounds
| mnn1 genotype | jun genotype | fos genotype | pnr-GAL4 phenotype a | ey-GAL4 phenotype | How24-GAL4 phenotype |
|---|---|---|---|---|---|
| UAS-jun | wt | wt | wt | ||
| UAS-fos | Weak cleft thorax (class I/II) | wt | wt | ||
| UAS-mnn1 | Moderate cleft thorax (class II) | wt | Pharate lethal (P14) | ||
| UAS-mnn1 | UAS-fos | Strong cleft thorax (class III/IV) | wt | Pharate lethal (P14) | |
| UAS-mnn1 | UAS-jun | Moderate cleft thorax (class II) | wt | Pharate lethal (P14) | |
| jra1/+ | wt | wt | wt | ||
| UAS-mnn1 | jra1/+ | Strong cleft thorax (class III) | n.d. | n.d. | |
| UAS-junbZIP | Weak cleft thorax (class I) | wt | wt | ||
| UAS-mnn1 | UAS-junbZIP | Cleft thorax (strong) | wt | Pupal lethal (P7) | |
| UAS-junasp | Lethal b | Lethal b | Lethal b | ||
| UAS-mnn1 | UAS-junasp | Semi-lethal c | Semi-lethal d | Lethal | |
| UAS-fosbZIP | Strong cleft thorax (class III) | Small rough eye | wt | ||
| UAS-mnn1 | UAS-fosbZIP | Strong cleft thorax (class III) | wt | Lethal | |
| mnn1Δ46/+ | UAS-fosbZIP | n.d. | n.d. | Moderate cleft thorax (class II) | |
| mnn1Δ46/mnn1Δ79 | UAS-fosbZIP | n.d. | n.d. | Pre-pupal lethal |
Description and classifications (Tateno et al., 2000) of thorax phenotypes are given.
Fully penetrant lethality. Thorax and eye phenotypes unscorable.
5% eclosion. Viable flies have a severe cleft thorax (class IV).
< 5% eclosion. Viable flies have small rough eyes.
The synergistic effect of mnn1 transgene and dominant negative alleles of jun along with the effect of altered jun dose suggests that menin acts to antagonize Jun protein function. If this is the case, then menin might suppress the effect of excess Jun activity. Expression of constitutively active, phosphomimetic jun (UAS-junasp) using pnr-GAL4 results in fully penetrant embryonic lethality. In contrast, the simultaneous expression of UAS-junasp and UAS-mnn1 driven from pnr-GAL4 results in 1–5% of flies escaping to eclosion (Table 2). Thus, expression of Mnn1 suppresses the lethality associated with constitutive expression of active Jun. These data are consistent with mnn1 acting as a negative regulator of jun function.
As mnn1 shows a genetic interaction with jun, we also tested for interaction with fos (kay), the other component of the heterodimer. Heterozygosity for kay1 had no effect on the mnn1 over-expression phenotype. However, expression of UAS-fos from pnr-GAL4 results in a weak cleft thorax phenotype and this phenotype is greatly enhanced by simultaneous expression from UAS-mnn1 (Table 2). Thus, excessive Fos is deleterious to Jun/Fos function, perhaps by affecting the homodimer/heterodimer ratio. These data suggest that menin and Fos have a negative synergistic effect on Jun/Fos function. However, genetic interactions between loss-of-function mnn1 alleles and dominant-negative alleles of fos suggest that the interaction is complex. In mnn1−/mnn1+ flies, over-expression of fosbZIP with the How24 driver has a more dramatic effect on thorax closure (Figs. 6J, K), and in the complete absence of mnn1, How24-GAL4 induction of fosbZIP results in pupal lethality (Table 2). Thus, in some experiments, Mnn1 is behaving as a positive regulator of Jun/Fos and in other experiments Mnn1 is acting as a negative regulator of Jun/Fos.
We also tested for dominant interactions between mnn1 over-expression and loss-of-function alleles of other members of the Jun kinase (JNK) cascade, hemipterous (hep) or basket (bsk), that lead to activated Jun and the negative regulator puckered (puc), but saw no interaction. We also examined the phenotypic effects of simultaneous over-expression of mnn1 with hep, bsk or puc and observed no interaction. Thus, there is no evidence that the interaction between mnn1 and jun/fos is mediated by these components of the JNK signaling pathway. However, this does not imply that there is direct contact between Mnn1 and Jun/Fos proteins, and indeed, direct testing for physical interaction between Mnn1 and Drosophila Jun or Fos has revealed no interaction (Guru et al., 2001; and data not shown).
jun/fos interactions with mnn1 in eye development
Jun/Fos is also required for Drosophila eye morphogenesis (Kockel et al., 2001). Analysis of interactions between Mnn1 and Fos in eye development also suggests that Mnn1 can negatively or positively modulate Jun/Fos. While we observed no effect of mnn1 over-expression in otherwise wild type eyes (Table 1), we did observe an interaction with Fos. Over-expression of a fos dominant negative, UAS-fosbZIP, in the eye using ey-GAL4 results in a small rough eye phenotype (Fig. 7B). The simultaneous induction of mnn1 expression dramatically suppresses this severe small eye phenotype (Fig. 7C). Thus, even though loss-of-function and gain-of function of mnn1 are not overtly deleterious to eye development, interaction with dominant negative fos reveals a genetic interaction of mnn1 with Jun/Fos in the eye. Thus, wild type Mnn1 appears to augment Jun/Fos activity in the eye, or to negatively regulate the dominant negative activity of FosbZIP.
Fig. 7.
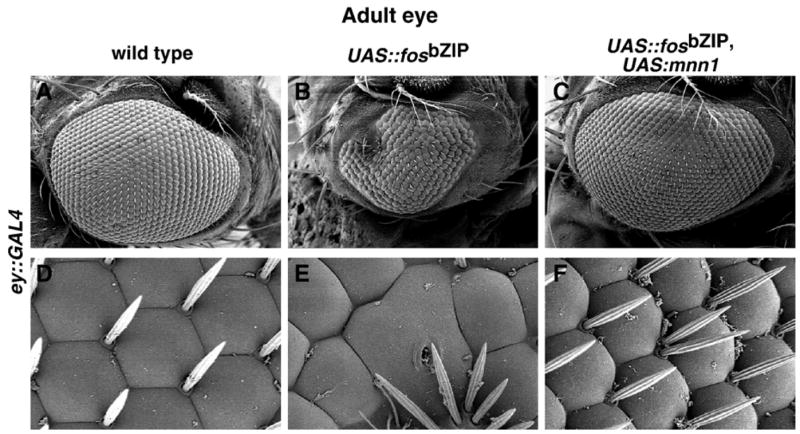
Interactions between Mnn1 and Fos in the eye. (A, D) Wild type eye showing the organization of normal ommatidia. (B, E) Small-rough eye phenotype resulting from the expression of fos dominant negative (UAS-fosbZIP driven by the ey-GAL4 driver). The number and hexagonal shape of ommatidia, as well as the distribution of the mechanical bristles are altered. (C, F) Mnn1 over-expression reverts the phenotype when UAS-mnn1 and UAS-fosbZIP are driven by ey-GAL4. (A–C) × 125; (D–F) × 1000.
If Mnn1 is a positive regulator of Jun/Fos in the eye, then it might also enhance the effect of constitutive active jun in that tissue. We found the opposite. Induction of UAS-junasp by ey-GAL4 resulted in pre-pupae lethality, but this was partially rescued by contextual over-expression of Mnn1. Flies expressing both UAS-junasp and UAS-mnn1 driven by ey-GAL4 eclose (1%), suggesting that Mnn1 can inhibit active Jun/Fos.
Conclusions
Mnn1 is highly conserved in Drosophila. Our experiments show that over-expressed Mnn1 can functionally interact with either wild type or over-expressed Jun/Fos. Furthermore, the absence of Mnn1 also modulates the activity of over-expressed Fos. Interestingly, these interactions result in defects consistent with both positive and negative influences of Mnn1 on Jun/Fos. While these results are unsatisfying for placing Mnn1 squarely in a particular and invariant position in the Jun/Fos pathway, it is clear that Jun/Fos is differently regulated in the eye and thorax of Drosophila (Kockel et al., 2001). It is also clear that mammalian Menin can function as either a positive or negative regulator of Jun family members (Agarwal et al., 2003). We suggest that there are contextual influences that allow Mnn1 to be both an activator or suppressor of Jun/Fos in Drosophila. This context-dependent effect might also underlie the opposing tumor suppressing (Chandrasekharappa et al., 1997; Crabtree et al., 2001) and tumor promoting (Yokoyama et al., 2005) effects of menin in mammals. Finally, while we have seen strong interactions between mnn1 and jun/fos, this does not rule out a role for mnn1 in other nuclear events. Indeed, mammalian menin is in a complex which is involved in modifying the histones at a large number of genes (Hughes et al., 2004;Yokoyama et al., 2004) and a large number of transcription factors have been reported to physically contact menin (Agarwal et al., 2004). Why such a broad biochemical activity is associated with such modest phenotypic effects is not well understood in mammals or in Drosophila. Perhaps Mnn1 has a more subtle role in fine tuning gene expression.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH (NIDDK, NHGRI, NICHD and OD). We acknowledge our colleagues Virginia Boulais, Diego Caro, Sandra Farkas and Jamileh Jemison for technical assistance, and members of the MEN1 consortium at the NIH. We are also grateful to the Drosophila community and the Bloomington Drosophila Stock Center, for generously and expeditiously sharing strains. Finally, we thank the Drosophila sequencing projects for making data public prior to formal publication.
References
- Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- Agarwal SK, Novotny EA, Crabtree JS, Weitzman JB, Yaniv M, Burns AL, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc Natl Acad Sci U S A. 2003;100:10770–10775. doi: 10.1073/pnas.1834524100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK, Lee Burns A, Sukhodolets KE, Kennedy PA, Obungu VH, Hickman AB, Mullendore ME, Whitten I, Skarulis MC, Simonds WF, Mateo C, Crabtree JS, Scacheri PC, Ji Y, Novotny EA, Garrett-Beal L, Ward JM, Libutti SK, Richard Alexander H, Cerrato A, Parisi MJ, Santa Anna AS, Oliver B, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. Molecular pathology of the MEN1 gene. Ann N Y Acad Sci. 2004;1014:189–198. doi: 10.1196/annals.1294.020. [DOI] [PubMed] [Google Scholar]
- Agnes F, Suzanne M, Noselli S. The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development. 1999;126:5453–5462. doi: 10.1242/dev.126.23.5453. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2005;33:D34–D38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Busygina V, Suphapeetiporn K, Marek LR, Stowers RS, Xu T, Bale AE. Hypermutability in a Drosophila model for multiple endocrine neoplasia type 1. Hum Mol Genet. 2004;13:2399–2408. doi: 10.1093/hmg/ddh271. [DOI] [PubMed] [Google Scholar]
- Calleja M, Moreno E, Pelaz S, Morata G. Visualization of gene expression in living adult Drosophila. Science. 1996;274:252–255. doi: 10.1126/science.274.5285.252. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98:1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JS, Scacheri PC, Ward JM, McNally SR, Swain GP, Montagna C, Hager JH, Hanahan D, Edlund H, Magnuson MA, Garrett-Beal L, Burns AL, Ried T, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS. Of mice and MEN1: insulinomas in a conditional mouse knockout. Mol Cell Biol. 2003;23:6075–6085. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy JB. GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis. 2002;34:1–15. doi: 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- FlyBase The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2003;31:172–175. doi: 10.1093/nar/gkg094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo A, Cuozzo C, Esposito I, Maggiolini M, Bonofiglio D, Vivacqua A, Garramone M, Weiss C, Bohmann D, Musti AM. Menin uncouples Elk-1, JunD and c-Jun phosphorylation from MAP kinase activation. Oncogene. 2002;21:6434–6445. doi: 10.1038/sj.onc.1205822. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobl AE, Berg M, Lopez-Egido JR, Oberg K, Skogseid B, Westin G. Menin represses JunD-activated transcription by a histone deacety-lase-dependent mechanism. Biochim Biophys Acta. 1999;1447:51–56. doi: 10.1016/s0167-4781(99)00132-3. [DOI] [PubMed] [Google Scholar]
- Goldsmith P, Gierschik P, Milligan G, Unson CG, Vinitsky R, Malech HL, Spiegel AM. Antibodies directed against synthetic peptides distinguish between GTP-binding proteins in neutrophil and brain. J Biol Chem. 1987;262:14683–14688. [PubMed] [Google Scholar]
- Guru SC, Goldsmith PK, Burns AL, Marx SJ, Spiegel AM, Collins FS, Chandrasekharappa SC. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci U S A. 1998;95:1630–1634. doi: 10.1073/pnas.95.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guru SC, Prasad NB, Shin EJ, Hemavathy K, Lu J, Ip YT, Agarwal SK, Marx SJ, Spiegel AM, Collins FS, Oliver B, Chandrasekharappa SC. Characterization of a MEN1 ortholog from Drosophila melanogaster. Gene. 2001;263:31–38. doi: 10.1016/s0378-1119(00)00562-x. [DOI] [PubMed] [Google Scholar]
- Huet F, Lu JT, Myrick KV, Baugh LR, Crosby MA, Gelbart WM. A deletion-generator compound element allows deletion saturation analysis for genomewide phenotypic annotation. Proc Natl Acad Sci U S A. 2002;99:9948–9953. doi: 10.1073/pnas.142310099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee JE, Kim BY, Cho EJ, Kim ST, Youn HD. Menin represses JunD transcriptional activity in protein kinase Ctheta-mediated Nur77 expression. Exp Mol Med. 2005;37:466–475. doi: 10.1038/emm.2005.57. [DOI] [PubMed] [Google Scholar]
- Knapp JI, Heppner C, Hickman AB, Burns AL, Chandrasekharappa SC, Collins FS, Marx SJ, Spiegel AM, Agarwal SK. Identification and characterization of JunD missense mutants that lack menin binding. Oncogene. 2000;19:4706–4712. doi: 10.1038/sj.onc.1203832. [DOI] [PubMed] [Google Scholar]
- Kockel L, Homsy JG, Bohmann D. Drosophila AP-1: lessons from an invertebrate. Oncogene. 2001;20:2347–2364. doi: 10.1038/sj.onc.1204300. [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Pastor-Pareja JC, Garcia-Bellido A. JNK and decapentaplegic signaling control adhesiveness and cytoskeleton dynamics during thorax closure in Drosophila. Proc Natl Acad Sci U S A. 2000;97:7888–7893. doi: 10.1073/pnas.97.14.7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K, Tsukada T, Honda M, Nara-Ashizawa N, Noguchi K, Cheng J, Ohkura N, Sasaki K, Yamaguchi K. Complementary DNA structure and genomic organization of Drosophila menin. Mol Cell Endocrinol. 2000;168:135–140. doi: 10.1016/s0303-7207(00)00307-5. [DOI] [PubMed] [Google Scholar]
- Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian Jun proteins: redundancy and specificity. Oncogene. 2001;20:2378–2389. doi: 10.1038/sj.onc.1204381. [DOI] [PubMed] [Google Scholar]
- Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, Hua X, Roeder RG, Meyerson M, Hess JL. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito J, Kaji H, Sowa H, Hendy GN, Sugimoto T, Chihara K. Menin suppresses osteoblast differentiation by antagonizing the AP-1 factor, JunD. J Biol Chem. 2005;280:4785–4791. doi: 10.1074/jbc.M408143200. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou M, Wu Y, Pretorius HN, Singh N, Gianfelice G, Tanguay RM, Campos AR, Bedard PA. Menin is a regulator of the stress response in Drosophila melanogaster. Mol Cell Biol. 2005;25:9960–9972. doi: 10.1128/MCB.25.22.9960-9972.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Crabtree JS, Kennedy AL, Swain GP, Ward JM, Marx SJ, Spiegel AM, Collins FS. Homozygous loss of menin is well tolerated in liver, a tissue not affected in MEN1. Mamm Genome. 2004;15:872–877. doi: 10.1007/s00335-004-2395-z. [DOI] [PubMed] [Google Scholar]
- Tateno M, Nishida Y, Adachi-Yamada T. Regulation of JNK by Src during Drosophila development. Science. 2000;287:324–327. doi: 10.1126/science.287.5451.324. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]