

Abstract
The phenomenon of ischemic preconditioning has been recognized as one of the most potent mechanisms to protect against myocardial ischemic injury. In experimental animals and humans, a brief period of ischemia has been shown to protect the heart from more prolonged episodes of ischemia, reducing infarct size, attenuating the incidence, and severity of reperfusion-induced arrhythmias, and preventing endothelial cell dysfunction. Although the exact mechanism of ischemic preconditioning remains obscure, several reports indicate that this phenomenon may be a form of receptor-mediated cardiac protection and that the underlying intracellular signal transduction pathways involve activation of a number of protein kinases, including protein kinase C, and mitochondrial KATP channels. Apoptosis, a genetically programmed form of cell death, has been associated with cardiomyocyte cell loss in a variety of cardiac pathologies, including cardiac failure and those related to ischemia/reperfusion injury. While ischemic preconditioning significantly reduces DNA fragmentation and apoptotic myocyte death associated with ischemia-reperfusion, the potential mechanisms underlying this effect have not been fully clarified. A comprehensive understanding of these mechanisms and application to clinical scenarios will provide new directions in research and translate this information into new treatment approaches for reducing the extent of ischemia/reperfusion injury.
Keywords: preconditioning, ischemia, reperfusion, necrosis, apoptosis
Preconditioning in experimental studies
In 1986, a group of investigators studying the intracellular changes of glycolytic products at different time points of myocardial ischemia in a canine model, observed that four cycles of 5 minutes ischemia and reperfusion prior to a more sustained episode of 40 min ischemia reduced considerably myocardial infarction compared with the control (Murry et al 1986). This was the first description of an endogenous protective phenomenon called ischemic preconditioning. The same beneficial effect was since then confirmed in every species that was studied, independently of the presence of collaterals in the coronary circulation and independently of the size of the animal model (Schott et al 1990; Liu et al 1991; Liu and Downey 1992). Early studies mostly focused on the investigation of the natural history of preconditioning in terms of the number of brief ischemic episodes, the duration of each ischemic insult, the duration of the reperfusion interval that would separate the last cycle of preconditioning from the following sustained episode of ischemia and finally the duration of the prolonged ischemia in order the preconditioning to be effective (van Winkle et al 1991; Iliodromitis et al 1996). Species differences were observed regarding the number of brief episodes of ischemia: increased number of ischemic bursts was effective in protecting the dog heart (Li et al 1990) whereas protection was lost after a certain number of ischemic-reperfusion stimuli in the rabbit heart (Cohen et al 1994; Iliodromitis et al 1997).
The preconditioned state is very transient following a preconditioning protocol and lasts for only 1–2 h in anesthetized animals (van Winkle et al 1991; Jenkins et al 1995; Iliodromitis et al 1996). This constitutes the classic preconditioning or first window of myocardial protection. Of note, later experimental studies revealed that, while the provided protection is lost after this reperfusion interval and the heart remains unprotected for several hours, the protective effect re-appears after 24 hours and lasts up to 72 hours (Yellon and Baxter 1995; Yang et al 1996). This is the delayed phase of preconditioning or the second window of protection, which starts with no additional intervention, it is more prolonged although less robust.
The golden standard of the effectiveness of preconditioning is the reduction of the infarct size (Figure 1). However, there is a series of other benefits obtained from preconditioning and among these are the reduction of lethal arrhythmias and the better recovery of post-ischemic left ventricular function (Shiki and Hearse 1987; Przyklenk 2000; Yellon and Downey 2003).
Figure 1.
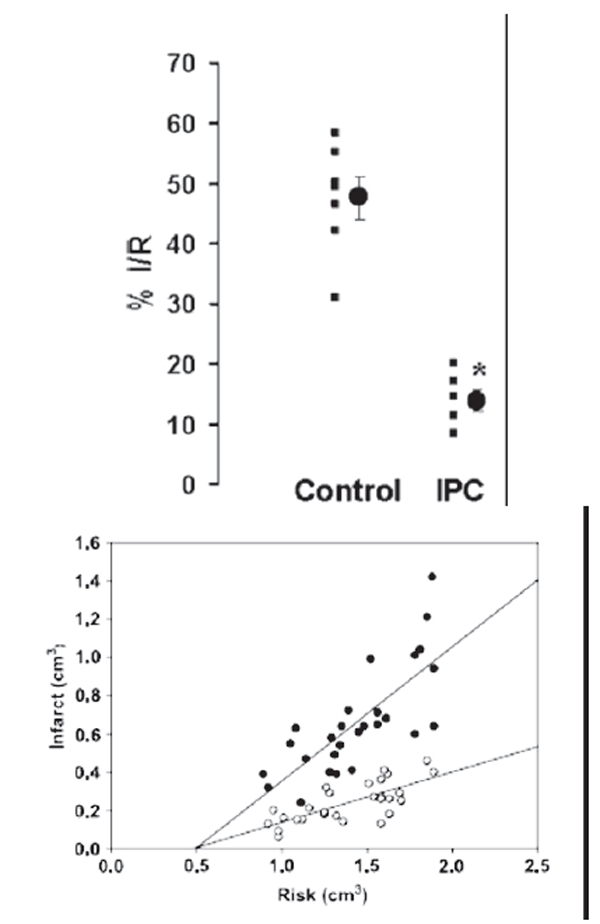
The effect of ischemic preconditioning on infarct size after ischemia/reperfusion. Upper panel:infarct size expressed as percent of risk zone size in control and preconditioned (IPC) rabbit hearts (from Iliodromitis et al 2006); lower panel: the absolute infarct volume plotted against absolute risk zone volume from control (closed symbols) and preconditioned (open symbols) hearts (from Iliodromitis et al 2004).
After the initial studies that elucidated the natural history of preconditioning in terms of the proper ischemic stimuli and time intervals, later experimental studies investigated the mechanisms that are involved in the protection mediated by preconditioning. The first breakthrough in this direction was the report by Downey and colleagues (Liu et al 1991) that the adenosine A1 receptor acts to trigger ischemic preconditioning’s protection in the rabbit heart, revealing that ischemic preconditioning is receptor mediated. It has now become established that the protection is receptor mediated, and a major objective in recent years has been the identification of the triggers, mediators and end effectors in the myocyte that are activated during ischemic preconditioning.
Activation of specific receptors on the cardiomyocyte cell membrane such as adenosine A1 and A3, bradykinin B2, α1-adrenergic, δ-opioid, and others, initiate a series of signal transduction cascades that carry the signal for protection and those presumably terminate on one or more end-effectors (Liu et al 1991; Cohen et al 2001; Schulz et al 2001). In recent years, much research has focused on the key role of the mitochondrial KATP channel as both a trigger and distal effector in preconditioning with equivocal results (Pain et al 2000; Fryer et al 2000; Sato et al 2000; Iliodromitis et al 2003; O’Rourke 2004). The signal transduction pathways downstream of the trigger are complex and a detailed description is beyond the scope of this review. However, it is clear that multiple protein kinases are involved, notably one or more isoforms of protein kinase C, at least two tyrosine kinases (eg, Src and PI3 kinase), as well as mitogen-activated protein kinases (Baines et al 1998; Iliodromitis et al 1998; Harada et al 2004; Armstrong 2004; Hausenloy et al 2005). Reperfusion injury salvage kinase (RISK) pathway has emerged as a concept and describes a group of survival protein kinases, which include Akt and Erk1/2 that confer powerful cardioprotection against lethal reperfusion injury when specifically activated at the time of reperfusion (Hausenloy and Yellon 2007). The cardioprotective effect of ischemic preconditioning is completely aborted by inhibiting RISK pathway (Hausenloy et al 2005).
More recently another endogenous mechanism of protection, called postconditioning, was described. In brief, a series of very short-lived episodes of ischemia and reperfusion applied at the onset of reperfusion after an index ischemic event reduce the infarct size. Postconditioning is more easily applicable and does not require fore-knowledge of a potentially lethal ischemic event. RISK pathway is also involved in the cardioprotective mechanism of postconditioning and the inhibition of PI3 kinase abrogates protection conferred by postconditioning (Tsang et al 2004). It has been suggested that the RISK pathway “unites” both ischemic preconditioning and postconditioning at the time of reperfusion (Hausenloy and Yellon 2007).
Preconditioning in humans
The acquired experience from the experimental studies was transferred to humans and the protective effect of preconditioning was examined using direct or indirect observations (Kloner et al 1995a, 1995b; Andreotti et al 1996; Kobayashi et al 1997; Karbanda et al 2001). It is difficult to transfer the strict laboratory conditions regarding the number and the duration of episodes of ischemia and reperfusion, into the clinical reality, so much for practical reasons as well as for ethical reasons. Thus, the final end points for the assessment of protection in humans are more or less indirect. Furthermore, the precise estimation of the infarct size and the area at risk is not very easy to be determined in humans and therefore the beneficial effect of preconditioning is difficult to be assessed. However, as in the experimental models, other surrogate end-points are used for the expression of the salutary effect of preconditioning and among these are the reduction of the episodes of angina after an initial episode, the reduction of ST deviation on the electrocardiogram, the reduction of lethal arrhythmias, a smaller peri-operative infarction, a better survival of the patients with pre-infarction angina and others (Tomai et al 1996, 1997; Ishihara et al 1997; Gheeraert et al 2001).
The preconditioning studies can be divided into observational, provocative, and pharmacological. In the first category belong those studies where the patients describe alleviation of their symptoms or there is an objective method which can confirm the benefit of preconditioning. In fact, many patients report an episode of angina early in the morning, called walk-through or warm up angina and they remain free of symptoms later and for the rest of the day (Przyklenk and Kloner 1999; Schwarz et al 1999). There are a large number of clinical studies which have shown that pre-infarction angina acts as preconditioning stimulus and the patients develop smaller CK-determined myocardial infarction in comparison to the patients without pre-infarction angina. In the objective studies, it is feasible to describe the effect of an intervention that may act as a preconditioning analogue. In this regard, less ST segment shift, less extent of angina and less lactate production at the second balloon inflation compared to the first are some surrogate end-points attributed to an effect called coronary angioplasty-related preconditioning (Tomai et al 1996, 1997). Furthermore, there is evidence that the second exercise treadmill test becomes better in comparison to the first and this may express an early or delayed preconditioning effect (Paraskevaidis et al 2005). Objective description of preconditioning has been also obtained from histological studies of tissue samples like atrial trabeculae or ventricular tissue taken at the time of operation or by the peripheral markers that have been used for the expression of preconditioning (Yellon et al 1993; Jenkins et al 1997).
Another interesting issue is the pharmacological stimulation of preconditioning and the expectation that several agents involved in the mechanism of protection could become safe and easily applicable preconditioning mimetics. Adenosine, acadesine, ACE inhibitors which prevent bradykinin degradation, nicorandil, and others have been used in clinical studies. In the AMISTAD study, adenosine has been used in parallel with thrombolysis in patients with evolving myocardial infarction and resulted in reduction of the infarct size in anterior myocardial infarction despite the fact that it was administered after the onset of chest pain (Mahaffey et al 1999). In the AMISTAD II study, the adenosine was given in anterior myocardial infarctions only at two different doses and again it was shown that at a dose of 70 μg/kg min for 3 hours and in parallel with thrombolysis, it reduced the infarct size especially if the thrombolysis started within the first 3 hours (Ross et al 2005). Acadesine, which is an agent that facilitates the effect of adenosine, was used in patients who underwent CABG. Patients treated with acadesine had fewer postoperative myocardial infarctions and better clinical outcome (Mangano et al 2006). Nicorandil, which opens the mitochondrial KATP channels, has been used in two multicenter clinical trials, CEASAR-2 and IONA, with very promising results (Patel et al 1999; the IONA study group 2002). These clinical results suggest that the cardioprotective effects of preconditioning, well described in animal studies, do appear to translate to clinical setting. However, direct translation of experimental observations to therapeutic intervention is hampered by factors encountered in clinical practice such as the necessity of proper time of intervention, the proper duration of the treatment and of course the appropriate dosage of drugs.
Apoptosis in the heart
While ischemic injury has long been considered to result in necrotic tissue damage, studies over the past 15 years have focused attention on apoptosis as a significant component of cell loss during reperfusion injury following myocardial infarction (Gottlieb et al 1994; Fliss and Gattinger 1996; Gottlieb and Engler 1999; Elsasser et al 2001). Myocardial apoptosis has also been documented in response to a variety of other cardiac stresses including pressure or volume overload, heart failure, diabetic cardiomyopathy, atherosclerosis and anti-cancer agents (Haunstetter and Izumo 1998; Feuerstein and Young 2000; Valen 2003; Singal et al 2000).
Cardiomyocytes undergoing necrosis and apoptosis show characteristic but morphologically and biologically distinct features. Necrosis, also called oncosis in the setting of ischemia/reperfusion, is an uncontrolled, irreversible process characterized by severe cell swelling, ATP depletion, denaturation and coagulation of cytoplasmic proteins, breakdown of cell organelles and disruption of cell membrane (Searle et al 1982; Majno and Joris 1995). It is a destructive process as release of cellular content into the surrounding environment provokes inflammation and can cause further damage or death to neighboring cells. In contrast, apoptosis is a genetically controlled, highly regulated process whereby the cell commits suicide without inducing inflammatory response. Apoptosis usually requires energy and it is characterized by cell shrinkage, chromatin condensation, DNA fragmentation, membrane blebbing and formation of apoptotic bodies (Majno and Joris 1995; Saraste and Pulkki 2000; Buja 2005). The hallmark of apoptosis in intact cells is endonucleolytic digestion of nuclear DNA into oligonucleosome-sized fragments (200 bp), in contrast to nonspecific degradation of DNA into pieces of random size after loss of membrane integrity.
The regulated disassembling of cellular contents, which is a feature of apoptosis, is largely conducted by a family of cysteine dependent aspartate-directed proteases, the caspases (Wang and Lenardo 2000). Fourteen members of this family have been identified in mammals. Caspases-2, -9, -8, and -10 are considered apoptosis initiators, and serve to cleave and activate apoptosis effectors (caspases-3, -6, and -7). All caspases are synthesized as zymogens that contain a prodomain, a large subunit and a small subunit, and activation of these enzymes is by proteolytic cleavage. Once activated, initiator caspases cleave and activate effector caspases and a proteolytic cascade is initiated.
There are two principal pathways that lead to caspase activation: the extrinsic and intrinsic cell death pathways (Danial and Korshmeyer 2004). The extrinsic pathway requires the activation of cell surface death receptors (DRs) by specific ligands, and allows the cell to respond directly to the immediate environment (Figure 2). The vast majority of death receptors belong to the tumor necrosis factor receptor (TNFR) superfamily, which includes Fas, TNFR1, and DR3–DR6. The binding of death ligands, such as Fas ligand or TNF-α, to their cognate receptors at the plasma membrane causes homotrimerization of the receptor and recruitment of specific adaptor proteins, such as Fas-associated death domain and procaspase-8, into a death-inducing signaling complex (DISC). This, in turn, leads to activation of initiator caspase-8, which subsequently activates effector caspases 3, 6, and 7 (Hirata et al 1998; Danial and Korshmeyer 2004). In contrast, in the intrinsic pathway (Figure 3), the mitochondria detect and respond to unfavorable changes in the internal environment and play a central role in the integration and execution of a wide variety of apoptotic signals. The mitochondria provide the energy required for execution of the apoptotic program and release of pro-apoptotic proteins such as cytochrome c, endonuclease G, and apoptosis-inducing factor. Release of these substances occurs during opening of the mitochondrial permeability transition (MPT) pore, a large non-selective ion channel in the outer mitochondrial membrane. Gating of the MPT pore is controlled by a combination of calcium, redox potential, pH and transmembrane voltage; when the MPT is opened, the normally impervious mitochondrial membrane undergoes a reversible transition to permeability (Zoratti and Szab’o 1995; Chelli et al 2001). Either brief or sustained MPT opening can lead to apoptosis (Bishopric et al 2001). On release from mitochondria, cytochrome c forms a complex with procaspase-9 and a cytosolic cofactor, Apaf-1 (Li et al 1997; Zou et al 1999). In the presence of sufficient ATP, caspase-9 undergoes autocatalytic cleavage to create an active ‘apoptosome’ that cleaves caspase-3 and initiates the apoptotic program. Thus, the extrinsic and intrinsic pathways have different initiator caspases but converge at the level of the effector caspases.
Figure 2.
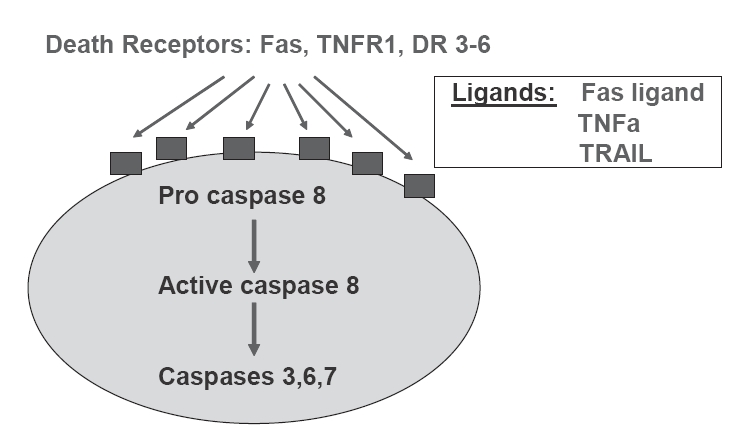
Schematic representation of the death receptor (extrinsic) pathway of caspase activation to provoke the mechanism of apoptosis.
Figure 3.
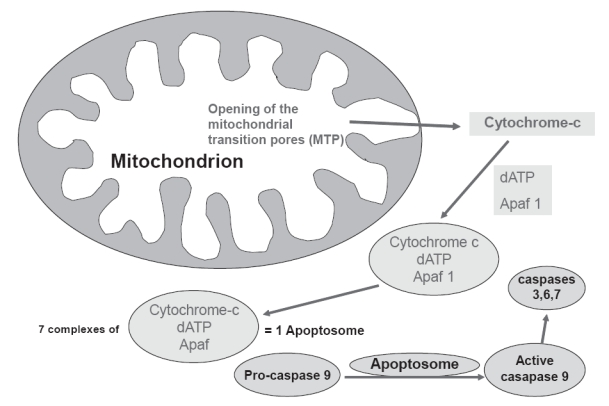
Schematic representation of the mitochondrial death (intrinsic) pathway leading to the formation of apoptosome, activation of caspases and apoptosis.
The intrinsic pathway of apoptosis is regulated by members of the Bcl-2 family. This family is composed of anti-apoptotic eg, Bcl2, Bcl-xL, Bcl-w, A1, and Mcl-1 and pro-apoptotic proteins eg, Bax, Bad, Bak, Bid, Bim, Bnip3, Bnip3L (Nix), and Bik. All of these proteins possess up to four conserved regions known as Bcl-homology (BH) domains (Cory et al 2003). The principal site of action of the Bcl-2 family proteins is at the mitochondria, where they regulate cytochrome c release and caspase activation. The exact mechanisms by which Bcl-2 proteins modulate apoptosis are still subject to much debate and controversy. One hypothesis is that both pro-apoptotic and anti-apoptotic Bcl-2 proteins bind directly to components of the mitochondrial pore, leading to either its opening or closure, respectively (Gross et al 1999; Mattson and Kroemer 2003). A second hypothesis is that upon activation, pro-apoptotic members such as Bax and Bak are inserted into the outer mitochondrial membrane where they oligomerize to form a protein-permeant pore of their own (Mattson and Kroemer 2003).
The most clinical relevant stimulants, which initiate the process of apoptosis apart from ischemia/reperfusion, include pressure or volume overload, cellular calcium overload, oxygen derived free radicals, increased levels of catecholamines and angiotensin II, Fas ligand, TNF-α, as well as decreased coronary flow reserve (Khoynezhad et al 2004; Scarabelli et al 2006). It is obvious that there is a complex interrelation between all the above factors. Decreased coronary flow reserve, for example, results in periods of ischemia and reperfusion and in generation of oxygen-derived free radicals. Angiotensin II may cause pressure overload and heart failure results in increased circulating catecholamine levels. All the above mentioned conditions may lead to contractile dysfunction of the heart, to dysrhythmias and to cell death because of apoptosis or necrosis. The rationale of current therapy is mainly to modify myocyte dysfunction and to minimize the intensity of the lethal injury acting on the heart. Well documented clinical interventions such as restoration of ischemia by pharmacological or interventional means, valve replacement or surgical correction of any anatomic abnormalities which may be considered responsible for pressure or volume overload, use of beta blockers, ACE inhibitors or angiotensin II receptor antagonists may prevent, suppress or restore cardiac dysfunction. However, preservation of cells subjected to lethal injury remains an attractive goal and inhibition of cardiac myocyte apoptosis may represent a novel approach for treatment of cardiac disease. For this reason, a better understanding of the pathways of apoptosis and their regulation is required. Therapeutic approaches such as the use of inhibitors of TNF-α, caspases and sodium-hydrogen exchanger, upregulation of insulin like growth factor-1, antioxidants, ischemic preconditioning, which will be discussed in the next section, and others are currently under investigation (Gill et al 2002; Khoynezhad et al 2004).
Preconditioning and apoptosis
Accumulating evidence from in vivo and in vitro studies suggests that ischemic preconditioning significantly reduces DNA fragmentation (Figure 4) and apoptotic myocyte death that is associated with ischemia/reperfusion in isolated rabbit and rat heart (Gottlieb et al 1996; Maulik et al 1999; Nakamura et al 2000; Zhao and Vinten-Johansen 2002; Lazou et al 2006). In addition, activation of receptors that are involved in the initiation of the mechanism of preconditioning, such as δ-opioid and bradykinin, also results in reduced apoptosis after ischemia/reperfusion (Okubo et al 2004; Feng et al 2005). It is clear from these studies that attenuation of apoptosis by ischemic preconditioning is associated with a reduction in infarct size analysed early at reperfusion. It is not known, however, whether a reduction in infarct size measured days after reperfusion is achieved by inhibiting apoptosis since apoptosis progresses after reperfusion over a number of days. The potential mechanisms underlying preconditioning attenuated myocardial apoptosis have not been fully clarified and they are under intense investigation. However, a number of mediators have been implicated.
Figure 4.
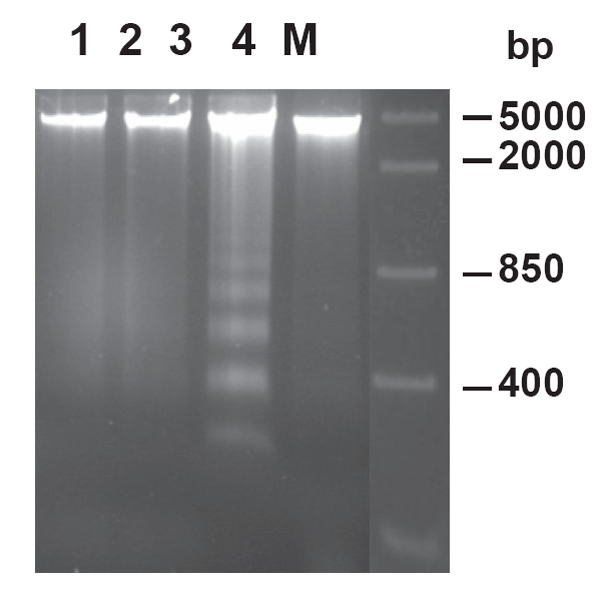
Effect of ischemic preconditioning on DNA fragmentation in rabbit hearts subjected to ischemia/reperfusion Lane 1 represents control non-ischemic tissue; lane 2 represents ischemic tissue after ischemia without reperfusion; lanes 3 and 4 represent ischemic tissue after ischemia/reperfusion in nonpreconditioned and preconditioned hearts, respectively. M, marker lane (from Lazou et al 2006).
Several studies have shown that preconditioning inhibits apoptosis by reducing the burst of reactive oxygen species generated from inflammatory cells (ie, PMNs and macrophages) and noninflammatory cell types (ie, endothelial cells and myocytes). In this regard, antioxidants and free radical scavengers have been reported to inhibit the appearance of apoptosis (Karzelewski et al 1999; Zhao and Vinten-Johansen 2002). Other studies have implicated that preconditioning reduces myocardial apoptosis through altering the balance between anti-apoptotic and pro-apoptotic proteins, the release of cytochrome c from mitochondria, caspase activation, and activation of protein kinase C isozymes. Preconditioning causes a decrease in pro-apoptotic Bax and increase in the anti-apoptotic Bcl-2 proteins resulting in an altered Bcl-2/Bax ratio and attenuation of cytochrome c release from mitochondria (Maulik et al 1999, 2000; Nakamura et al 2000). Inhibition of caspase activity, a key event in the development of apoptosis, has also been demonstrated in preconditioning (Piot et al 1999). A reduction in ceramide production during sustained ischemia has been suggested to reduce apoptosis in rabbit hearts (Argaud et al 2004). However, an interrelationship between changes in the expression of Bcl-2 family proteins, mitochondrial activity and reduction of apoptosis after ischemia/reperfusion by ischemic preconditioning needs to be further investigated in in vivo models.
As discussed above, PKC has a central role in ischemic preconditioning. In particular, activation of PKCε is important in the preservation of cell viability. Although the effect of ischemic preconditioning on necrosis through activation of PKC isoforms has been intensively investigated (Yellon and Downey 2003), only a few studies have shown that preconditioning reduces apoptosis by a PKC-dependent pathway. Okamura et al (1999) showed that blockade of PKC may interrupt the protective effect of preconditioning and simultaneously it may promote the mechanism of apoptosis. Furthermore, PKCε but not PKCδ is involved in the inhibition of apoptosis by preconditioning after simulated ischemia and reoxygenation (Liu et al 2001).
The mitochondrial KATP channels significantly contribute to the mechanism of protection in ischemic preconditioning by the generation of the oxygen-derived free radicals. Opening of these channels by pharmacological means like nicorandil or diazoxide, trigger the mechanism of protection while the addition of 5HD blunts this effect (Pain et al 2000; Sato et al 2000; Iliodromitis et al 2003; O’Rourke 2004). It is of interest that diazoxide is also capable in preventing apoptosis and again the simultaneous use of 5HD abolishes this benefit (Ardehali et al 2005). In addition to direct effects on mitochondrial function, opening of mitochondrial KATP channels may have secondary effects on cell signaling such as activation of PKCε (Liu et al 2002).
As mentioned above, activation of kinases of the RISC pathway has been implicated in mediating the cardioprotection associated with ischemic preconditioning (Housenloy et al 2005; Hausenloy and Yellon 2007). Recent studies have shown that pharmacological activation of these kinases is associated with recruitment of anti-apoptotic signaling components such as the phosphorylation and inhibition of the proapoptotic proteins Bax and Bad, the inhibition of caspase 3 activation, the phosphorylation and activation of p70S6K (which acts to inhibit Bad) and the phosphorylation and activation of the antiapoptotic protein Bcl-2 (Harada et al 2001; Hausenloy and Yellon 2007). In addition, the inhibition of the mitochondrial permeability transition pore (MTP), a mitochondrial channel which mediates cell death at the time of myocardial reperfusion by uncoupling oxidative phosphorylation and inducing mitochondrial swelling (Hausenloy and Yellon 2003), has been identified as a downstream target of the RISK pathway (Davidson et al 2006; Bopassa et al 2006). Although it has been shown that ischemic preconditioning prevents the increased permeability of the transition pore by the activation of kinases, Akt, PI3 kinase, and ERK1/2, the mechanism through which the RISK pathway inhibits the opening of the MTP is unclear.
In conclusion, ischemic preconditioning is a protective mechanism in limiting the infarct size and there is a great body of evidence that it reduces both necrosis and apoptosis. We expect that in the future the acquired experience and the increased knowledge of the underlying mechanisms in preconditioning and apoptosis would allow a more appropriate use of the proper pharmacological agents that would mimic preconditioning and that confer reduction of both necrosis and apoptosis. However, the translation of the laboratory findings in the clinical practice should be performed very cautiously and with prudence.
References
- Andreotti F, Pasceri V, Hackett DR, et al. Preinfarction angina as a predictor of more rapid coronary thrombolysis in patients with acute myocardial infarction. N Engl J Med. 1996;334:7–12. doi: 10.1056/NEJM199601043340102. [DOI] [PubMed] [Google Scholar]
- Ardehali H, O’Rourke B. Mitochondrial KATP channels in cell survival and death. J Mol Cell Cardiol. 2005;39:7–16. doi: 10.1016/j.yjmcc.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaud L, Prigent AF, Chalabreysse L, et al. Ceramide in the antiapoptotic effect of ischemic preconditioning. Am J Physiol. 2004;286:H246–51. doi: 10.1152/ajpheart.00638.2003. [DOI] [PubMed] [Google Scholar]
- Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:427–36. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Baines CP, Wang L, Cohen MV, et al. Protein tyrosine kinase is downstream of protein kinase C for ischemic preconditioning’s anti-infarct effect in the rabbit heart. J Mol Cell Cardiol. 1998;30:383–92. doi: 10.1006/jmcc.1997.0601. [DOI] [PubMed] [Google Scholar]
- Bishopric NH, Andreka P, Slepak TI, et al. Molecular mechanisms of apoptosis in the cardiac myocyte. Curr Opin Pharmacol. 2001;1:141–50. doi: 10.1016/s1471-4892(01)00032-7. [DOI] [PubMed] [Google Scholar]
- Bopassa JC, Ferrera R, Gateau-Roesch O, et al. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc Res. 2006;69:178–85. doi: 10.1016/j.cardiores.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Buja LM. Myocardial ischemia and reperfusion. Cardiovasc Pathol. 2005;14:170–5. doi: 10.1016/j.carpath.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Chelli B, Falleni A, Salvetti F, et al. Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem Pharmacol. 2001;61:695–705. doi: 10.1016/s0006-2952(00)00588-8. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. Conscious rabbits become tolerant to multiple episodes of ischemic preconditioning. Circ Res. 1994;74:998–1004. doi: 10.1161/01.res.74.5.998. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Liu GS, et al. Acetylcholine, bradykinin, opioids and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circ Res. 2001;89:273–8. doi: 10.1161/hh1501.094266. [DOI] [PubMed] [Google Scholar]
- Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Davidson SM, Hausenloy D, Duchen MR, et al. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006;38:414–19. doi: 10.1016/j.biocel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Eefting F, Rensing B, Wigman J, et al. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004;61:414–26. doi: 10.1016/j.cardiores.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Elsasser A, Suzuki K, Lorenz-Meyer S, et al. The role of apoptosis in myocardial ischemia: a critical appraisal. Basic Res Cardiol. 2001;96:219–26. doi: 10.1007/s003950170052. [DOI] [PubMed] [Google Scholar]
- Feng J, Bianchi C, Sandmeyer JL, et al. Bradykinin preconditioning improves the profile of cell survival proteins and limits apoptosis after cardioplegic arrest. Circulation. 2005;112(Suppl I):I190–5. doi: 10.1161/CIRCULATIONAHA.104.524454. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Young PR. Apoptosis in cardiac diseases: Stress- and mitogen-activated signaling pathways. Cardiovasc Res. 2000;45:560–9. doi: 10.1016/s0008-6363(99)00372-7. [DOI] [PubMed] [Google Scholar]
- Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–56. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- Fryer RM, Eells JT, Hsu AK, et al. Ischemic preconditioning in rats: role of mitochondrial KATP channel in preservation of mitochondrial function. Am J Physiol. 2000;278:H305–12. doi: 10.1152/ajpheart.2000.278.1.H305. [DOI] [PubMed] [Google Scholar]
- Gheeraert PJ, Henriques JPS, De Buyzere ML, et al. Preinfarction angina protects against out-of-hospital ventricular fibrillation in patients with acute occlusion of the left coronary artery. J Am Coll Cardiol. 2001;38:1369–76. doi: 10.1016/s0735-1097(01)01561-3. [DOI] [PubMed] [Google Scholar]
- Gill C, Mestril R, Samali A. Losing heart: the role of apoptosis in heart disease-a novel therapeutic target? FASEB J. 2002;16:135–46. doi: 10.1096/fj.01-0629com. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, Burleson KO, Kloner RA, et al. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–28. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Gruol DL, Zhu JY. Preconditioning rabbit cardiomyocytes: role of pH, vacuolar proton ATPase, and apoptosis. J Clin Invest. 1996;97:2391–8. doi: 10.1172/JCI118683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Engler RL. Apoptosis in myocardial ischemia-reperfusion. Ann NY Acad Sci. 1999;874:412–26. doi: 10.1111/j.1749-6632.1999.tb09255.x. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;3:1899–911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Harada H, Andersen JS, Mann M, et al. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA. 2001;98:9666–70. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Miura T, Dairaku Y, et al. NO donor-activated PKC delta plays a pivotal role in ischemic myocardial protection through accelerated opening of K-ATP mitochondrial channels. J Cardiovasc Pharmacol. 2004;44:35–41. doi: 10.1097/00005344-200407000-00005. [DOI] [PubMed] [Google Scholar]
- Haunstetter A, Izumo S. Apoptosis: basic mechanisms and implications for cardiovascular disease. Circ Res. 1998;82:1111–29. doi: 10.1161/01.res.82.11.1111. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003;35:339–41. doi: 10.1016/s0022-2828(03)00043-9. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Tsang A, Mocanu M, et al. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol. 2005;288:H971–H976. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- Housenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling:taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–34. doi: 10.1007/s10741-007-9026-1. [DOI] [PubMed] [Google Scholar]
- Hirata H, Takahashi A, Kobayashi S, et al. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J Exp Med. 1998;187:587–600. doi: 10.1084/jem.187.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliodromitis EK, Papadopoulos C, Paraskevaidis IA, et al. Protection from preconditioning can be reinstated at various reperfusion intervals. Cardiovasc Drugs Ther. 1996;10:341–6. doi: 10.1007/BF02627958. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Kremastinos DT, Katritsis DG, et al. Multiple cycles of preconditioning cause loss of protection in open-chest rabbits. J Mol Cell Cardiol. 1997;29:915–20. doi: 10.1006/jmcc.1996.0328. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Miki T, Liu GS, et al. The PKC activator PMA preconditions rabbit heart in the presence of adenosine receptor blockade: is 5’-nucleotidase important? J Mol Cell Cardiol. 1998;30:2201–11. doi: 10.1006/jmcc.1998.0780. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Cokkinos P, Zoga A, et al. Oral nicorandil recaptures the waned protection from preconditioning in vivo. Br J Pharmacol. 2003;38:1101–6. doi: 10.1038/sj.bjp.0705149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliodromitis EK, Tasouli A, Andreadou I, et al. Intravenous atenolol and esmolol maintain the protective effect of ischemic preconditioning in vivo. Eur J Pharmacol. 2004;499:163–9. doi: 10.1016/j.ejphar.2004.07.093. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Caitanaki C, Lazou A, et al. Differential activation of mitogen-activated kinases in ischemic and nitroglycerin-induced preconditioning. Basic Res Cardiol. 2006;101:327–35. doi: 10.1007/s00395-006-0594-3. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Sato H, Tateishi H, et al. Implications of prodromal angina pectoris in anterior wall acute myocardial infarction: acute angiographic findings and long-term prognosis. J Am Coll Cardiol. 1997;30:970–5. doi: 10.1016/s0735-1097(97)00238-6. [DOI] [PubMed] [Google Scholar]
- Jenkins DP, Baxter GF, Yellon DM. The pathophysiology of ischemic preconditioning. Pharmacol Res. 1995;84:350–6. doi: 10.1016/1043-6618(95)80022-0. [DOI] [PubMed] [Google Scholar]
- Jenkins DP, Pugsley WB, Alkhulaifi AM, et al. Ischemic preconditioning reduces troponin T release in patients undergoing coronary artery bypass surgery. Heart. 1997;77:314–18. doi: 10.1136/hrt.77.4.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzelewski M, Czarnowska E, Maczewski M, et al. Effect of ischemic preconditioning on endothelial dysfunction and granulocyte adhesion in isolated guinea-pig hearts subjected to ischemia / reperfusion. J Physiol Pharmacol. 1999;50:617–28. [PubMed] [Google Scholar]
- Kharbanda RK, Peters M, Walton B, et al. Ishemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia reperfusion in humans in vivo. Circulation. 2001;103:1624–30. doi: 10.1161/01.cir.103.12.1624. [DOI] [PubMed] [Google Scholar]
- Khoynezhad A, Jalali Z, Tortolani AJ. Apoptosis: Pathophysiology and therapeutic implications for the cardiac surgeon. Ann Thorac Surg. 2004;78:1109–18. doi: 10.1016/j.athoracsur.2003.06.034. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Muller J, Davis V, et al. Effects of previous angina pectoris in patients with first acute myocardial infarction not receiving thrombolytics. Am J Cardiol. 1995a;75:615–17. doi: 10.1016/s0002-9149(99)80628-6. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Shook T, Przyklenk K, et al. Previous angina alters in hospital outcome in TIMI 4. A clinical correlate to preconditioning? Circulation. 1995b;91:37–45. doi: 10.1161/01.cir.91.1.37. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Miyazaki S, Miyao Y, et al. Effect on survival of previous angina pectoris after acute myocardial infarction. Am J Cardiol. 1997;79:1534–8. doi: 10.1016/s0002-9149(97)00188-4. [DOI] [PubMed] [Google Scholar]
- Lazou A, Iliodromitis EK, Cieslak D, et al. Ischemic but not mechanical preconditioning attenuates ischemia/reperfusion induced myocardial apoptosis in anaesthetized rabbits: The role of Bcl-2 family proteins and ERK1/2. Apoptosis. 2006;11:2195–204. doi: 10.1007/s10495-006-0292-5. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Lundergan CF, Hodgson J, et al. for the IONA study group. Effect of nicorandil on coronary events in patients with stable angina: the impact of nicorandil in angina (IONA) randomized trial. Lancet. 2002;359:1269–75. doi: 10.1016/S0140-6736(02)08265-X. [DOI] [PubMed] [Google Scholar]
- Li GC, Vasquez JA, Gallagher KP, et al. Myocardial protection with preconditioning. Circulation. 1990;82:609–19. doi: 10.1161/01.cir.82.2.609. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liu GS, Thornton J, Van Winkle DM, et al. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–6. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- Liu H, McPherson BC, Yao Z. Preconditioning attenuates apoptosis and necrosis: role of protein kinase Cε and -δ isoforms. Am J Physiol. 2001;281:H404–10. doi: 10.1152/ajpheart.2001.281.1.H404. [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang HY, Zhu X, et al. Preconditioning blocks cardiocyte apoptosis: role of K(ATP) channels and PKC-epsilon. Am J Physiol. 2002;282:H380–6. doi: 10.1152/ajpheart.00348.2001. [DOI] [PubMed] [Google Scholar]
- Liu Y, Downey JM. Ischemic preconditioning protects against infarction in rat heart. Am J Physiol. 1992;263:H1107–12. doi: 10.1152/ajpheart.1992.263.4.H1107. [DOI] [PubMed] [Google Scholar]
- Mahaffey KW, Puma JA, Bardagelata NA, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of multicenter, randomized, placebo-controlled trial: the AMISTAD trial. J Am Coll Cardiol. 1999;34:1711–20. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- Majno G, Joris I. Apoptosis, oncosis and necrosis: an overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- Mangano DT, Miao Y, Tudor IC, et al. Post-reperfusion myocardial infarction. Long-term survival improvement using adenosine regulation with acadesine. J Am Coll Cardiol. 2006;48:206–14. doi: 10.1016/j.jacc.2006.04.044. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med. 2003;9:196–205. doi: 10.1016/s1471-4914(03)00046-7. [DOI] [PubMed] [Google Scholar]
- Maulik N, Engelman RM, Rousou JA, et al. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation. 1999;100:II369–75. doi: 10.1161/01.cir.100.suppl_2.ii-369. [DOI] [PubMed] [Google Scholar]
- Maulik N, Sasaki H, Addya S, et al. Regulation of cardiomyocyte apoptosis by redox-sensitive transcription factors. FEBS Lett. 2000;485:7–12. doi: 10.1016/s0014-5793(00)02174-8. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nakanura M, Wang NP, Zhao ZQ, et al. Preconditioning decreases Bax expression, PMN accumulation and apoptosis in reperfused rat heart. Cardiovasc Res. 2000;45:661–70. doi: 10.1016/s0008-6363(99)00393-4. [DOI] [PubMed] [Google Scholar]
- O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–32. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura T, Miura T, Iwamoto H, et al. Ischemic preconditioning attenuates apoptosis through protein kinas C in rat hearts. Am J Physiol. 1999;277:H1997–2001. doi: 10.1152/ajpheart.1999.277.5.H1997. [DOI] [PubMed] [Google Scholar]
- Okubo S, Tanabe Y, Takeda K, et al. Ischemic preconditioning and morphine attenuate myocardial apoptosis and infarction after ischemia-reperfusion in rabbits: role of δ-opioid receptor. Am J Physiol. 2003;287:H1786–91. doi: 10.1152/ajpheart.01143.2003. [DOI] [PubMed] [Google Scholar]
- Pain T, Yang X, Critz SD, et al. Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–6. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- Paraskevaidis IA, Iliodromitis EK, Mavrogeni S, et al. Repeated exercise test identifies early and late preconditioning. Int J Cardiol. 2005;98:221–6. doi: 10.1016/j.ijcard.2003.10.040. [DOI] [PubMed] [Google Scholar]
- Patel DJ, Pucell HJ, Fox KM, et al. Cardioprotection by opening of the KATP channel inn unsteable angina. Is this a clinical manifestation of myocardial preconditioning? Results of a randomized study with nicorandil. Eur Heart J. 1999;20:51–7. doi: 10.1053/euhj.1998.1354. [DOI] [PubMed] [Google Scholar]
- Piot CA, Martini JF, Bui SK, et al. Ischemic preconditioning attenuates ischemia/reperfusion-induced activation of caspases and subsequent cleavage of poly(ADP-ribose) polymerase in rat hearts in vivo. Cardiovasc Res. 1999;44:536–42. doi: 10.1016/s0008-6363(99)00227-8. [DOI] [PubMed] [Google Scholar]
- Przyklenk K, Kloner RA. Ischemic preconditioning: exploring the paradox. Progress Cardiovasc Dis. 1999;40:517–47. doi: 10.1016/s0033-0620(98)80002-9. [DOI] [PubMed] [Google Scholar]
- Przyklenk K. Ischemic preconditioning. J Thromb Thrombolysis. 2000;9:99–103. doi: 10.1023/a:1018629727410. [DOI] [PubMed] [Google Scholar]
- Ross AM, Gibbons RJ, Stone GW, et al. A randomized, double-blind, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45:1775–80. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res. 2000;45:528–37. doi: 10.1016/s0008-6363(99)00384-3. [DOI] [PubMed] [Google Scholar]
- Sato T, Sasaki N, O’Rourke B, et al. Nicorandil, a potent cardioprotective agent, acts by opening mitochondrial ATP-dependent potassium channels. J Am Coll Cardiol. 2000;35:514–8. doi: 10.1016/s0735-1097(99)00552-5. [DOI] [PubMed] [Google Scholar]
- Scarabelli TM, Knight R, Stephanou A, et al. Clinical implications of apoptosis in ischemic myocardium. Curr Probl Cardiol. 2006;31:181–264. doi: 10.1016/j.cpcardiol.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Schott RJ, Rohmann S, Braun ER, et al. Ischemic preconditioning reduces infarct size in swine myocardium. Circ Res. 1990;66:1133–42. doi: 10.1161/01.res.66.4.1133. [DOI] [PubMed] [Google Scholar]
- Schulz R, Cohen MV, Behrens M, et al. Signal transduction of ischemic preconditioning. Cardiovasc Res. 2001;52:181–98. doi: 10.1016/s0008-6363(01)00384-4. [DOI] [PubMed] [Google Scholar]
- Schwarz ER, Reffelmann T, Kloner RA. Clinical effects of ischemic preconditioning. Curr Opin Cardiol. 1999;14:340–8. doi: 10.1097/00001573-199907000-00010. [DOI] [PubMed] [Google Scholar]
- Searle J, Kerr JF, Bishop CJ. Necrosis and apoptosis: Distinct modes of cell death with fundamentally different significance. Pathol Annu. 1982;17:229–59. [PubMed] [Google Scholar]
- Shiki K, Hearse DJ. Preconditioning of ischemic myocardium: reperfusion-induced arrhythmias. Am J Physiol. 1987;253:H1470–6. doi: 10.1152/ajpheart.1987.253.6.H1470. [DOI] [PubMed] [Google Scholar]
- Singal PK, Li T, Kumar D, et al. Adriamycin induced heart failure: mechanism and modulation. Mol Cell Biochem. 2000;207:77–86. doi: 10.1023/a:1007094214460. [DOI] [PubMed] [Google Scholar]
- Tomai F, Crea F, Gaspardone A, et al. Effects of A1 adenosine receptor blockade by by bamiphylline on ischemic preconditioning during coronary angioplasty. Eur Heart J. 1996;17:846–53. doi: 10.1093/oxfordjournals.eurheartj.a014965. [DOI] [PubMed] [Google Scholar]
- Tomai F, Crea F, Gaspardone A, et al. Phentolamine prevents adaptation to ischemia during coronary angioplasty. Role of a-adrenergic receptors in ischemic preconditioning. Circulation. 1997;96:2171–7. doi: 10.1161/01.cir.96.7.2171. [DOI] [PubMed] [Google Scholar]
- Tsang A, Hausenloy DJ, Mocanu MM, et al. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res. 2004;95:230–2. doi: 10.1161/01.RES.0000138303.76488.fe. [DOI] [PubMed] [Google Scholar]
- Valen G. The basic biology of apoptosis and its implications for cardiac function and viability. Ann Thorac Surg. 2003;75:S656–60. doi: 10.1016/s0003-4975(02)04687-8. [DOI] [PubMed] [Google Scholar]
- Van Winkle DM, Thornton JD, Downey DM, et al. The natural history of preconditioning: Cardioprotection depends on duration of transient ischemia and time to subsequent ischemia. Coron Artery Dis. 1991;2:613–19. [Google Scholar]
- Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci. 2000;113:753–7. doi: 10.1242/jcs.113.5.753. [DOI] [PubMed] [Google Scholar]
- Yang XM, Baxter GF, Heads RJ, et al. Infarct limitation of the second window of protection in conscious rabbit model. Cardiovasc Res. 1996;31:777–83. doi: 10.1016/0008-6363(96)00026-0. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Alkulaifi AM, Pugsley WB. Preconditioning the human myocardium. Lancet. 1993;342:276–7. doi: 10.1016/0140-6736(93)91819-8. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Baxter GF. A “second window of protection” or delayed preconditioning phenomenon: future horizons for myocardial protection? J Mol Cell Cardiol. 1995;27:1023–34. doi: 10.1016/0022-2828(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1133–51. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Vinten-Johansen J. Myocardial apoptosis and ischemic preconditioning. Cardiovasc Res. 2002;55:438–55. doi: 10.1016/s0008-6363(02)00442-x. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Szab’o I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–76. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, et al. An APAF-1. cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]