

Abstract
Glanzmann’s thrombasthenia (GT) is a congenital qualitative platelet disorders due to the deficiency or defect of platelet membrane GPIIb/IIIa (integrin αIIbβ3). The standard treatment for bleeding is platelet transfusion but repeated transfusion may result in the development of anti-platelet antibodies (to HLA and/or GPIIbIIIa) rendering future platelet transfusion ineffective. Alternative effective agent(s) are needed. There are increasing reports documenting efficacy of high dose rFVIIa in GT patients with adverse events uncommon. The efficacy is supported by evidence that high concentration FVIIa binds to activated platelet surface and improves thrombin generation to enhance deposition (adhesion) and aggregation of platelets lacking GPIIb/IIIa. While there are increasing clinical experiences, evidence-based clinical data are not available. There is a need for more clinical studies, particularly clinical trials, to further assess the efficacy, safety (particularly thrombotic events) and optimal regimen of rFVIIa in GT patients, either singly or in combination with other hemostatic agents such as platelet transfusion. In the absence of this data, for treatment of severe bleeding in GT patients with platelet antibodies and platelet refractoriness, rFVIIa at dose 90 μg/kg every 2 h for 3 or more doses could be considered. This more “optimal regimen” derived from a recent International Survey needs confirmation with larger studies. What the optimal regimen for surgical coverage is remains unresolved.
Keywords: Glanzmann’s thrombasthenia, recombinant human activated factor VII (rFVIIa), bleeding, surgery, platelet transfusion, GPIIb/IIIa
Introduction
Glanzmann’s thrombasthenia
Glanzmann’s thrombasthenia (GT) is a congenital bleeding disorder of platelet dysfunction due to the deficiency or dysfunction of platelet membrane glycoprotein (GP) IIb/IIIa complex (integrin αIIbβ3). (George et al 1990; Nurden 1999) GT is a rare autosomal recessive disorder with an incidence of about 1:1 million, although in areas where marriage between close family relatives are prevalent, the incidence is much higher. The genetic defect can be on the GPIIb or GPIIIa gene and homozygosity of the same mutation are more likely a result of consanguineous marriage, while many other patients may be compound heterozygous for different mutations from each parent. Bleeding symptoms are confined to GT patients with homozygous or compound heterozygous for GPIIb/IIIa mutations. Heterozygotes are asymptomatic, so that family history of bleeding may be absent.
GPIIb/IIIa complex is involved with platelet aggregation mediated primarily by the binding of fibrinogen to this glycoprotein complex on activated platelets (Peerschke et al 1980). Thus absence of platelet aggregation in response to physiologic agonists such as adenosine-5’-phosphate (ADP), epinephrine, collagen and thrombin is the hallmark laboratory abnormality. The diagnosis can be confirmed by flow cytometry using monoclonal antibodies to GPIIb, GPIIIa or intact GPIIa/IIIa complex. For convenience, GT can be classified according to the platelet membrane GPIIb/IIIa levels, being <5% for type I disease, 5%–15% for type II disease and higher for variant disease which is a result of dysfunction rather than a deficiency of the GPIIb/IIIa complex. Asymptomatic heterozygotes usually have about 50% platelet membrane GPIIb/IIIa.
Bleeding symptoms has been reviewed by George et al in 1990 (George et al 1990) on 113 patients. The most common symptoms are menorrhagia (98%), easy bruising and purpura (86%), epistaxis (73%) and gingival bleeding (55%). Less common are gastrointestinal bleeding (12%), hematuria (6%) while hemarthrosis (3%), intracranial bleeding (2%) and deep visceral bleeding (1%) are rare. Although bleeding manifestations are variable, and occasional patients may have very mild symptoms, overall, GT is a severe bleeding disorder, as majority of patients would have a history of red cell and/or platelet transfusion. Epistaxis is common particularly in children and can be severe requiring transfusion. Menorrhagia could be a critical bleeding problem with a particularly high risk of severe and prolonged bleeding requiring transfusion at menarche.
Management options for the treatment of GT bleeding include conservative measures for mild bleeding. These include the use of local pressure including nasal packing with gelatin sponge for epistaxis, local hemostatics such as fibrin glue and topical thrombin, as well as the use of antifibrinolytics. The standard treatment for serious bleeding is platelet transfusion. There are also anecdotal reports of success with DDAVP, but DDAVP is often not effective.
The use of platelets however can be complicated by the development of allo-antibodies to GPIIb/IIIa and/or antigens of the HLA system. For this reason, single-donor HLA-matched platelet transfusion is preferred over random donor platelets so as to at least delay or prevent HLA allo-immunization, even though antibodies to GPIIb/IIIa may still develop. Patients with platelet antibodies can become refractory to future platelet transfusion treatments. Furthermore, anti-GPIIb/IIIa antibodies may cross the placenta in pregnant women resulting in neonatal thrombocytopenia and/or bleeding in the fetuses or newborns, including intracranial bleeding. Platelet concentrates, as a blood product that must be stored at room temperature can also be associated with blood-borne infections. The residual risk of blood-borne virus infection is now minimal (Dodd et al 2002). Bacterial contamination of platelet concentrates, on the other hand, could be as high as 1 in 2000 to 3000 units and one out of six of the contaminated units transfused may result in severe septic reactions. (Jacobs et al 2001) An alternative effective agent is therefore needed for management of GT patients particularly those who are refractory to platelet transfusion and in patients who live in areas where platelets are not readily available.
Recombinant human activated factor VIIa (rFVIIa)
Recombinant Human activated Factor VIIa (rFVIIa, NovoSeven/Niastase, Novo Nordisk A/S, Bagsvaerd, Denmark) is produced by recombinant DNA technology in baby hamster kidney (BHK) cells cultured in a medium containing fetal calf serum, but no human proteins or derivatives (Jurlander et al 2001) so that human pathogen transmission is not a risk. The final product has a specific activity of approximately 50,000 IU/mg. As a reference, average normal plasma contains 1 U FVII activity (plasma concentrations of FVII and FVIIa are respectively 10 nM and 0.1 nM) (Morrissey and Mutch 2006). The median in vivo recovery of rFVIIa after infusion at doses 17.5–70 μg/kg in the bleeding and nonbleeding state was determined to be approximately 46% and 44%, respectively (Lindley et al 1994).
rFVIIa exerts its hemostatic effect only after complexing with tissue-factor (TF). TF is normally not exposed to flowing blood, but is found in various cells in the deeper layer of the blood vessel wall. (Morrissey and Mutch 2006) Clotting is initiated at the wound site after tissue injury and complexing of FVIIa with the exposed TF. Under normal physiological circumstances, rFVIIa is otherwise proteolytically inert in the circulation and infusion of rFVIIa does not appear to cause systemic activation of coagulation except in unusual circumstances. Other cells that express TF include some tumor cells as well as monocytes and neutrophils that have been stimulated by bacterial endotoxin and certain other inflammatory mediators. (Morrissey and Mutch 2006) Studies in rabbit stasis model at rFVIIa doses 100–1000 μg/kg showed no significant clot formation over 10 minutes of stasis. (Diness et al 1992) Significant platelet and fibrinogen changes were not observed 3 hours after infusion. Turacek et al (1997) confirmed in a similar Rabbit stasis model rFVIIa to have low thrombogenic potential, which was enhanced by the addition of soluble TF.
Pharmacokinetic studies in adult hemophilia patients with or without inhibitors show this agent, when given at doses 17.5–70 μg/kg, to have a relatively short half-life (T1/2), at 2.3 and 2.9 hours respectively in the bleeding and non-bleeding state.(Lindley et al 1994) Similar T1/2 (3.0 h) was obtained in patients with factor VII deficiency (Berrettini et al 2001). The T1/2 in children at a dose of 90–180 μg/kg was similar at 2.6 hr.(Villar et al 2004) The short T1/2 makes it necessary to give this agent at frequent short-intervals.
rFVIIa is currently approved for the treatment of hemophilia A or B patients with inhibitors in many countries including New Zealand, Australia, Japan and those in North America, and Europe. It is also approved in the European Union (EU) for treatment of patients with acquired hemophilia, Factor VII deficiency and GT with history of platelet antibodies (to GPIIb/IIIa and/or HLA) and past or present history of platelet refractoriness and in the US for bleeding in Factor VII deficiency.
Mechanism of action of rFVIIa in patients with Glanzmann’s thrombasthenia
Physiologically, FVIIa exerts its hemostatic effect after complexing with TF. In normal persons, FVIIa-TF complex on TF-bearing cells at the site of vascular injury activates FX and FIX resulting in initial thrombin generation that activates a number of clotting factors (eg, FV, FVIII, FXI) as well as platelets. Activated platelets are recruited to the wound site where they aggregate, mediated in part by binding of soluble fibrinogen to platelet surface GPIIb/IIIa (Phillips et al 1988), for primary hemostatic plug formation. Activated platelets at the wound site support further coagulation activation resulting in sufficient thrombin generation (thrombin burst) for fibrin formation and hemostasis (Hoffman et al 1998). In GT, this TF mechanism is important in generating the initial thrombin on TF-bearing cells at the site of tissue injury for initiating platelet activation. However, this small amount of thrombin generation is not sufficient in GT to result in platelet aggregation to support sufficient thrombin burst at the wound site, because of the lack of GPIIbIIIa receptors for fibrinogen binding. GT platelets have thus been shown to have impaired thrombin generation capacity (Reverter et al 1996; Dargaud et al 2006). Experimental evidence suggests that high dose rFVIIa effect hemostasis in GT via a TF independent mechanism. High dose rFVIIa can bind to activated platelet surface with a low affinity (Kd ~ 100 nM).(Monroe et al 1997) At high concentration, the bound rFVIIa could directly activate FX to FXa resulting in bursts of thrombin generation sufficient to convert fibrinogen to fibrin. In experimental models, this thrombin burst mediated by rFVIIa bound to activated GT platelet surface was shown to allow for further platelet activation, and enhancement of adhesion and aggregation of the Glanzmann’s thrombasthenia platelets (Lisman et al 2003, 2004) (Figure 1). The enhanced adhesion of Glanzmann’s thrombasthenia platelets appear to require the participation of von Willebrand factor-GPIb interaction, mediated also by enhanced thrombin generation.(Lisman et al 2003) Despite the lack of surface GPIIb/IIIa, and therefore unable to bind soluble fibrinogen, GT platelets have been shown to agglutinate in the presence of fibrin, particularly polymeric fibrin (Niewiarowski et al 1981; McGregor et al 1989; Osdoit and Rosa 2001). Lisman and his colleague (Lisman et al 2004) showed that GT platelets aggregated in parallel with fibrin conversion related to thrombin generation mediated by rFVIIa bound to activated GT platelet surface. This fibrin mediated GT platelet aggregation was partially dependent on binding of thrombin to GPIb (Lisman et al 2004) shown in normal platelets to mediate binding of fibrin to an as yet unidentified receptor. Fibrin appeared an active participant in mediating platelet aggregation partly in a receptor-mediated manner, as opposed to being passively trapped during platelet aggregation, since aggregation was less efficient if viable platelets were replaced by fixed platelets.
Figure 1.
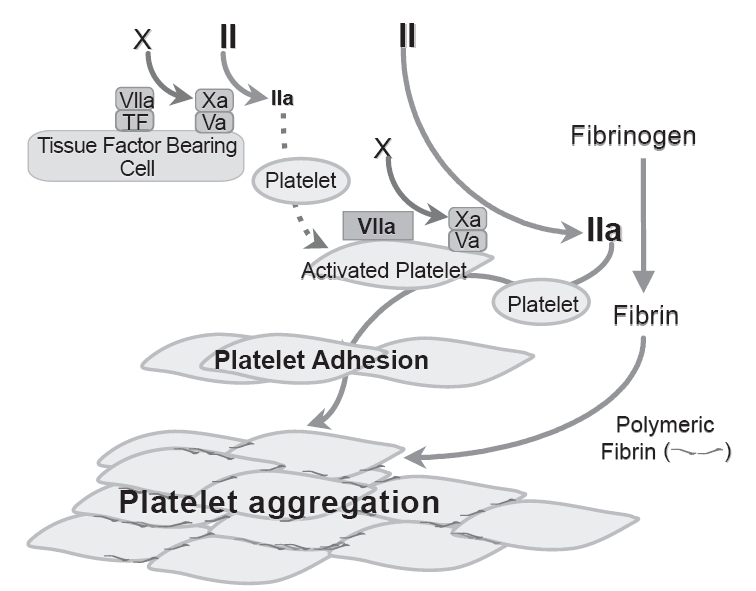
Schematic tissue factor-independent, platelet-dependent model of primary hemostatic plug formation in Glanzmann’s thrombasthenia (GT) platelets deficient in membrane glycoprotein (GP) IIb-IIIa (integrin aIIbβ3). FVIIa-tissue factor (TF) complex on TF-bearing cells at the site of vascular injury activates FX to FXa, which in turn complexes with FVa on the TF-bearing cells to initiate generation of a small amount of thrombin (FIIa) from prothrombin (FII). This initially generated thrombin is not sufficient to allow fibrin formation, but is sufficient to activate the GT platelets, causing degranulation and release of FV. FVIIa binds to activated platelets weakly. At high concentration (eg, high dose rFVIIa therapy), the bound FVIIa can directly activate FX to FXa to mediate generation of high concentration of thrombin (thrombin burst). The augmented thrombin generation results in increased number of activated platelets deposited (adhesion) to the wound site, and increased available platelet procoagulant surface to facilitate more thrombin generation and more platelet activation. The augmented thrombin generated also converts fibrinogen to fibrin. Activated GT platelets cannot utilize fibrinogen for aggregation reaction as they lack the fibrinogen receptor integrin aIIbβ3. However, binding of fibrin/polymeric fibrin to an as yet unidentified platelet surface receptor can mediate aggregation of the GT platelets at the wound site (even though less potent than fibrinogen mediated aggregation of normal platelets) resulting in primary hemostatic plug formation. Adapted from Poon (2006).
rFVIIa in the treatment and prevention of bleeding in Glanzmann’s thrombasthenia patients
Tengborn and Petruson (1996) were the first to use rFVIIa to successfully treat a hereditary platelet disorder in a child with GT and severe epistaxis. Since then there have been a large number of reports on efficacy of rFVIIa in GT (Poon et al 1999; Chuansumrit et al 1999, 2003; Robinson et al 2000; d’Oiron et al 2000; Monte and Lyons 2002; van Buuren and Wielenga 2002; Almeida et al 2003; Caglar et al 2003; Bell and Savidge 2003; Poon et al 2004; Kaleelrahman et al 2004; Kale et al 2004; Uzunlar et al 2004; Coppola et al 2004; Yilmaz et al 2005; Dargaud et al 2006; Inagaki et al 2006; Lombardo and Sottilotta 2006) as well as other platelet function disorders including Bernard Soulier Syndrome (Peters and Heijboer 1998; Almeida et al 2003; Kaleelrahman et al 2004; Ozelo et al 2005); platelet storage pool defect including Hermansky-Pudlak syndrome (Pozo Pozo et al 2002; Almeida et al 2003; Langendonck and Appel 2005), and platelet-type (pseudo) von Willebrand disease (Fressinaud et al 1998).
In 1999, we reported a pilot prospective open-label Canadian study on 4 children treated for 24 bleeding episodes (3 GI, 13 nose, 7 oropharynx and 1 traumatic facial hematoma) and one herniorrhaphy treated with rFVIIa at 89–116 μg/kg every two hours. (Poon et al 1999). The study was initiated because one of the participating children had required frequent admission to intensive care unit for severe epistaxes with hemoglobin dropping to as low as 40 g/L and had become refractory to platelet transfusion because of the development of antibodies against GPIIb/IIIa. In this study, the severity of some of the bleedings was indicated by the need for red cell transfusion in all 3 GI bleeds and in 4 epistaxes. Bleeding stopped promptly or within 6 hr of starting rFVIIa in 16 bleeding episodes (67%) and in less than 24 h in another 7 episodes. The remaining one GI bleed failed to respond to 24 every-two-hour doses and required rescue with platelet transfusion. Two bleeds, one GI and one frenulum/lip cut, each initially stopped after 3 rFVIIa doses at 4 and 5 hours respectively, recurred at 36 and 63 hours later. In both cases, bleeding stopped after additional doses without rebleeding. The herniorrhaphy was managed with one dose preoperatively and one dose 2 hours later without perioperative or postoperative bleeding and the patient was discharged 36 hours postoperatively with minor ecchymoses at the wound site. Antifibrinolytic drugs were given concurrently with rFVIIa in all but two minor bleeding episodes, one with oropharyngeal bleed and one with facial hematoma. No thrombotic adverse event was noted.
A British study of five GT patients (Almeida et al 2003) showed more variable results with good/excellent results in only 12 of the 25 bleeding episodes, but all three surgical procedures were successfully covered. Further analysis of the data showed an important observation. The poor results could be related to delayed treatment, as good/excellent responses were observed in 10 of their 14 episodes (71%) treated within 12 hours of bleeding onset, but in only two of 11 (18%) treated after 12 h. This study underlines the importance of early treatment as has also been observed when rFVIIa were used to treat hemophilia patients with inhibitors for muscle, joint and mucocutaneous bleeds. (Lusher 1998; Santagostino et al 1999; Lusher 2000).
International survey on the use of rFVIIa in the treatment and prevention of bleeding in GT
Randomized trials would have been helpful to evaluate the effectiveness and risk-benefit of rFVIIa in GT. However, such studies are hindered by the rarity of GT. Moreover, many bleeds in GT can be treated with local measures and/or antifibrinolytic drugs and the number of bleeds requirimg the use of systemic hemostatic treatment is not high. We (Coordinators: M-C Poon and R d’Oiron) began an international survey in 1999 to better assess the efficacy and safety of rFVIIa in GT patients. There was wide participation from 49 centers in 17 countries, providing data that include some previously published cases.(Tengborn and Petruson 1996; Musso et al 1999; Poon et al 1999; d’Oiron et al 2000; Robinson et al 2000; Almeida et al 2003; Devecioglu et al 2003) The survey data analyzed for reporting in 2004 (Poon et al 2004) include 59 patients treated for 108 bleeding episodes and 34 invasive procedures. There are limitations on this kind of survey that need to take into account in the interpretation of the data. These include the heterogeneity of treatment regimens used since management was at the discretion of the investigators, the different guidelines for the minimum rFVIIa doses to be used before declaring failure and switching to other available treatment product(s), and the lack of data on the time between bleeding onset and treatment initiation. Also, although investigators were asked to report all positive and negative experiences, the possibility for under-reporting of negative experience still could not be excluded. Nonetheless, useful information and observations were obtained.
The 59 patients treated include 24 males and 35 females, aged 1–72 years (median 22) including 27 children (age 15 years or younger). The majority of patients had type I disease (39 of 49 known) and 35 patients had platelet antibodies (to GPIIb/IIIa and/or HLA) and/or platelet refractoriness.
Data on bleeding episodes
Of the 108 bleeding episodes, 76 were severe and 32 were moderate. Bleeding was considered severe if it was intracranial, resulted from severe trauma, compressed a vital organ, or led to a fall in hemoglobin level of 20 g/L or more within 4 days. Bleeds that require systemic hemostatic treatment other than antifibrinolytic drugs, but did not fit at least one of these criteria were considered moderate. These 108 bleeds include 17 from the gastrointestinal tract (GI, 16 severe, 1 mild), 45 nose (32 severe, 13 moderate), 29 oropharynx (19 severe, 10 moderate), and 17 miscellaneous sites (9 severe, 8 moderate). As indicated in Table 1, 5 successfully treated episodes (2 nose bleeds and 3 bleeding in miscellaneous sites) were not evaluable due to confounding by concurrent platelet transfusion. The response (cessation of bleeding) rate among the evaluable patients was 53%, 74%, 79%, and 93% respectively for bleeding from the GI tract, nose, oropharynx and miscellaneous sites (Table 1). The overall response rate for intention to treat and response rate for evaluable episodes were respectively 71% and 75%, However, 8 of 77 episodes that stopped bleeding had a recurrence, so that the overall success rate when the recurrences were excluded was 64% for intention-to-tread and 67% for evaluable episodes, and was much lower than that observed in the pilot Canadian study (Poon et al 1999). Here, several clinically relevant observations were made:
Table 1.
Outcome of rFVIIa treatment for 34 surgical procedures and 108 bleeding episodes in patients with Glanzmann’s thrombasthenia (data from Poon et al (2004))
| Category | N | Responsea (rate %)b |
Recurancec | Failured | Not evaluablea |
|---|---|---|---|---|---|
| Bleeding episodes (all) | 108 | 77(71/75) | 8 | 26 | 5 |
| GI | 17 | 9(53/53) | 1 | 8 | 0 |
| Epistaxes | 45 | 32(71/74) | 4 | 11 | 2 |
| Oropharynx | 29 | 23(79/79) | 2 | 6 | 0 |
| Miscellaneous sites | 17 | 13(76/93) | 1 | 1 | 3 |
| Surgical procedures (all) | 34 | 29(85/94) | 2 | 3 | |
| Major | 9 | 6(67/86) | 1 | 2 | |
| Minor – dental extraction | 9 | 9(100/100) | 0 | 0 | |
| Minor – others | 16 | 14(88/93) | 1 | 1 |
Response = cessation of bleeding within 48 h of initiation of rFVIIa treatment.
(intention-to-treat response rate in % / response rate of evaluable episodes in %).
Recurrence = bleeding that stopped in ≤48 h but with recurrence of bleeding in ≤48 h following cessation of initial bleeding.
Failure = Bleeding that stopped more than 48 h after the initiation of rFVIIa treatment and/or if another treatment (other than antifibrinolytic drugs) was needed.
Not evaluable = platelet transfusion(s) were given concurrently with rFVIIa.
Some rFVIIa treatment regimens are better than others
Of note is that 10 of the 26 failures were declared after a one or two bolus dose of rFVIIa (n = 5 each), so that the rFVIIa treatment may not have been sufficient. Indeed, when evaluable bleeds were analyzed according to the treatment regimen, the overall success (without recurrences) rate in the 41 severe and moderate bleeds treated with bolus rFVIIa dose of at least 80 μg/kg, at interval of 2.5 h or less for at least 3 doses was 78% (32/41), compared with 60% (37/62) with other rFVIIa regimens including rFVIIa given by continuous infusion. For severe bleeds, the success rate using this arbitrary but more “optimal” regimen was 77% (24/31) and was significantly higher than the 55% (18/33) (p = 0.01) for regimens using lower dose or longer dosing intervals or less number of doses or when continuous infusion was used. It is therefore not surprising that Inagaki et al (Inagaki et al 2006) found that a single dose of rFVIIa, while effective in stopping a mild epistaxis was not effective for a severe prolonged epistaxis. Thus, for severe bleeds when rFVIIa is used, doses at about 90 μg/kg or higher, every 2 h for a minimum of 3 doses until bleeding stops should therefore be considered. In the treatment of hemophilia with inhibitors, Kenet et al (2003) showed that megadose rFVIIa at 300 μg/kg may be more effective than the standard rFVIIa dose of 90 μg/kg. It is therefore reasonable that a higher dose rFVIIa up to 300 μg/kg can be tried in patients who fail after several standard doses, but there is at this time no trial study data to support this contention. In clinical practice, how a patient is managed and when treatment should be switched will obviously depend on the ready availability of various treatment options and knowledge of relative efficacy, risk and cost of each.
Continuous infusion is not effective in stopping bleeding
Continuous infusion appears ineffective to stop bleeding, as 6 of 7 CI treatments of evaluable bleeding episodes failed. The failures include 4 GI bleeds, one nose and one post-intubation pharyngolarygeal bleed using doses up to 32 μg/kg/h (after initial bolus) for up to 22 days. One GI bleed did respond to 40 μg/kg/h CI (after initial bolus) carried out for 60 hours with bleeding stopped at 24 h. The number of episode treated was too small for analysis of CI dose effect.
Maintenance doses are useful particularly for severe bleeds
The recurrence rate could apparently be decreased by the use of maintenance doses after bleeding stopped. Only one of the 8 episodes (13%) that stopped but with a recurrence received a maintenance dose. On the contrary, in the 62 episodes that bleeding stopped without a recurrence (and with the timing of bleeding cessation and rFVIIa discontinuation known) 36 (58%) received maintenance doses (p = 0.022). One to three (or more) maintenance doses at 90 μg/kg or higher (similar to the treatment dosage), every 2–3 hourly should therefore be considered for severe bleeding episodes, bleeding related to severe trauma and GI bleeds even though the bleeding may stop after one or two doses.
GI bleeding is particularly difficult to treat
Only 9 of 17 GI bleeding episodes stopped, of which one recurred 36 hr later. The 8 failures may be related to the severity of the GI lesion as there were 3 angiodysplasia, one Mallory-Weiss syndrome and one radiation gut injury. The treatment may also be inadequate in some episodes; one episode was declared failure after received a single dose rFVIIa, while 4 were treated by continuous infusion. One additional patient was given 3 doses then stopped despite continuing bleeding.
Use of antifibrinolytic drugs
Antifibrinolytic drugs were given concurrently with rFVIIa in 12 of 17 GI bleeds and 70 of 91 non-GI bleeds. The success rate for those receiving concurrent antifibrinolytic drugs were similar to those not receiving antifibrinolytic drugs. On general principle, the use of antifibrinolytic agents in conjunction with rFVIIa should be considered at least for muscosal bleeds when there is no contraindication such as bleeding in the urinary tract. In the survey, one episodes of post-intubation pharyngolaryngeal bleed stopped only when antifibrinolytic drug was added after failing 9 days of continuous rFVIIa infusion.
Possibility of using rFVIIa as first-line therapy
A high proportion (58/80 or 83%) of the successfully treated bleeds stopped within 6 hours (0.1–6 hours) after the first rFVIIa injection, suggesting that rFVIIa could be used as first-line therapy while waiting for the availability of adequate apheresis platelet concentrates. On the contrary, in patients with anti-platelet antibodies and refractory to platelet transfusion, high-dose HLA-compatible platelets transfusions, with or without antibody removal therapy (Ito et al 1991; Martin et al 2002), should not be delayed in case of life-threatening bleeding or when rFVIIa therapy fails.
Data on invasive procedures
Of the 34 surgical procedures covered with rFVIIa, 9 were major, 25 were minor. Two major and one minor successful procedures were not evaluable due to confounding by concurrent platelet transfusion. Of the 7 evaluable major procedures covered with rFVIIa, 6 were successful, 3 by bolus rFVIIa injection and 3 by continuous infusions (Tables 1 and 2). Of the 24 evaluable minor procedures treated, 23 were successful, 20 by bolus injection and 3 by continuous infusion (Tables 1 and 2). Thus the intention-to-treat success rate for the 34 procedures was 85%, and the success rate for the 31 evaluable episodes was 94%. The following observations are made:
Table 2.
Invasive procedures (N = 29) successfully treated with rFVIIa as first-line therapy (3 non-evaluablea episodes and 2 failuresb not included) (data from Poon (2004))
| Bolus injections | Median (range) | Continuous infusion | Median (range) |
|---|---|---|---|
| Major surgery: all 3 episodesc | Major Surgery: all 3 episodesd | ||
| Dosage (μg/kg per injection) | 92(80–92) | Initial bolus dosage (μg/kg) | 49(28–70) |
| Doses used (N) | 14(4–33) | CI dosage (μg/kg/h) | 5(5–30) |
| Duration of treatment (h) | 47(11–141) | Duration of treatment (h) | 282(225–336) |
| Minor surgery: all 20 epidsodese | Minor surgery: all 3 episodesf | ||
| Dosage (μg/kg per injection) | 109(74–150) | Initial bolus dosage (μg/kg) | 88(72–110) |
| Doses used (N) | 3(1–19) | CI dosage (μg/kg/h) | 12(9–20) |
| Duration of treatment (h) | 7(0.1–152) | Duration of treatment (h) | 67(60–267) |
| Minor surgery: 9 dental extraction | |||
| Dosage (μg/kg per injection) | 100(78–134) | ||
| Doses used (N) | 3(1–19) | ||
| Duration of treatment (h) | 6(0.1–152) |
Nonevaluables: two major (pyelonephrectomy, skin grafting) and 1 minor (central catheter insertion).
Failures: persistent oozing after endoscopic uterine myometomy; recurrent hematuria following ureteric stent removal.
Major by bolus injection: laparotomy, laparoscopic bilateral oophorectomy, hysterectomy.
Major by CI: colostomy, colostomy revision, intestinal resection.
Minor by bolus injection: Nine dental extractions, 4 other dental procedures, 2 colonoscopy/polypectomy, one each of cystoscopy/ureteric stent insertion, hernia repair, tracheostomy, knee injection, otolaryngological cryosurgery.
Minor by CI: central cathetre removal, colonoscopy/polypectomy, cystoscopy/ureteric stent insertion.
Dental extractions
Dental extractions are frequent minor procedures. In this survey, all 9 dental extractions were successfully covered by bolus injections using, in median (range), rFVIIa dose of 100 μg/kg (78–134) for 3 doses (1–19) and treatment duration of 6 h (0.1–152) (Table 2). The first dose was obviously given immediately or shortly before the procedure with subsequent doses given mostly at 2 h interval. Although some episodes were covered with only the initial dose with no further injection because there was no bleeding, in general 1 or more maintenance dose(s) were given following the procedure. Antifibrinolytic agents were given concurrently with rFVIIa in all the 9 dental extractions.
Continuous Infusion (CI) is effective but not necessarily advantageous compared to bolus injections
Unlike treatment of bleeding episodes where continuous infusion appears ineffective, CI was effective in preventing bleeding in all 6 surgical procedures. However, although CI was used with the intention to avoid the valley and peak rFVIIa concentrations that would theoretically save on factor concentrates, in fact, CI resulted in longer treatment duration (Table 2). Objective data on reasons for the longer treatment are lacking. It is tempting to speculate that perhaps once CI has been initiated and became routine, it became more likely to let the routine continued in the absence of bleeding, but again supporting data is lacking. Thus, the total rFVIIa usage was higher in CI than in bolus injection treatment with the median (range) for major procedures 1779 μg (1182–10080) by CI vs. 1288 μg (368–2640) by bolus; and for minor procedures 948 μg (715–2376) by CI vs 300 μg (112–2042) by bolus. As indicated in the Adverse Event section, the only adverse events in this study were in patients receiving high-dose CI for a prolonged period following major surgery.
Use of antifibrinolytic drugs
Antifibrinolytic drugs were used concurrently with rFVIIa in 27 of the 34 procedures. The drug was omitted in 4 minor and 3 major procedures including 3 that would have been contraindicated because the surgical site involved the urinary tract and one procedure that was done on a patient with preexisting coronary disease. The data is insufficient to analyze the contribution of antifibrinolytics in preventing surgical bleeding. None-the-less, in the absence of contraindications, antifibrinolytics should be considered for surgery involving mucosal sites and in dental extractions/procedures where there are no contra-indications. In this survey, as indicted earlier, antifibrinolytics were used in all the 9 successful dental extractions.
Is there an optimal regimen?
The number of surgical procedures treated with rFVIIa remains relatively small such that an optimal regimen could not be derived from the available data.
Unresolved issues
There are unresolved issues raised by this survey. One already stated is the inability to determine the optimal rFVIIa regimen for surgical coverage in GT patients. In addition, although some preliminary suggestions for the optimal rFVIIa regimen in bleeding episodes can be derived from the analysis, confirmation from larger studies will be required. There are also insufficient data to evaluate the safety of rFVIIa in GT patients, particularly with regard to the incidence of thromboembolic events.
In 2004, the European Medical Evaluation Agency (EMEA) approved the use of rFVIIa for European Union patients with GT with platelet antibodies and past and/or present history of platelet refractoriness. As required by the EMEA, an international post-marketing pharmacovigilance study (http://www.glanzmann-reg.org) has recently been launched to assess the efficacy and safety of rFVIIa and other systemic hemostatic agents (including platelet transfusion) on a larger number of GT patients with or without platelet antibodies or platelet refractoriness (Poon 2006). The study is open to any interested investigator both within and outside Europe.
Adverse events
rFVIIa when used in patients with Glanzmann’s thrombas-thenia appears to be safe. In the International survey (Poon et al 2004), a 72-year old woman with GT developed deep vein thrombosis and pulmonary embolism after bowel resection surgery covered with rFVIIa (d’Oiron et al 2000). She received a bolus of 90 μg/kg rFVIIa followed by high dose continuous infusion at 30 μg/kg/h for 16 hours. The thrombotic event occurred 6 days after rFVIIa was discontinued. Another 23-year old woman with GT developed clots in the right renal pelvis and ureter after gynecological surgery covered with rFVIIa by continuous infusion (25 μg/kg/h tapered gradually to 12 μg/kg/h for 4 days, and antifibrinolytics (Robinson et al 2000). This patient likely had inadvertent trauma to the kidney during surgery, with bleeding and clotting in the renal pelvis and ureter that did not lyse. This does not represent an intravascular clotting complication. Among patients with other platelet disorders receiving rFVIIa, thrombotic events in one patient with Bernard-Soulier syndrome and one with uremic dysfunction have been reported to the US Food and Drug Administration (FDA) MedWatch Pharmacovigilance Program (Aledort 2004).
rFVIIa has been used more extensively in hemophilia patients with inhibitors and thrombotic complications are rare except in unusual circumstances (Abshire and Kenet 2004; Aledort 2004). Abshire and Kenet (2004) reviewed 20 thrombotic and 5 DIC events reported spontaneously to the manufacturers, reported in clinical trials or reported in the literature. The amount of rFVIIa administered between 1996 and April 2003 to hemophilia patients (number not specified) was more than 700,000 standard doses of rFVIIa (each equivalent to 90 μg/kg × 40 kg). Most of these patients apparently had comorbid or predisposing factor, such as diabetes mellitus, coronary artery disease, atherosclerosis, hypertension, obesity, advanced age or indwelling catheter or other DIC risk. rFVIIa has also been used on an investigational basis (off-label) for diverse bleeding situation in patients without hemophilia or congenital bleeding disorders, including GI, intracranial and postoperative bleeds; bleeding after trauma or after bone marrow/stem cell or organ transplantation. Thirty-eight thrombotic events were reported to the FDA MedWatch Pharmacovigilance Program or as published case reports between April 1999 and June 2000 (Aledort 2004); Sallah et al 2005). O’Connell et al (2006) reviewed adverse events related to rFVIIa use reported to the US FDA Adverse Event Reporting system from March 25, 1999 to December 31, 2005. They found 220 reports on 246 thromboembolic events (23 reports in hemophilia and 197 reports in off-label indications) that included 129(52%) arterial events (non hemorrhagic intracranial cerebrovascular accident, acute myocardial infarction, other arterial thromboembolism), 100(41%) venous events (including deep vein thrombosis and pulmonary embolism), 15(6%) devise occlusion and 2(1%) with sites not stated. Forty-three of the 67 deaths were probably related to thromboembolic events. In all series, the contribution of patient factors as well as other hemostatic agents that might have been used, relative to rFVIIa use, in these adverse events was not clear, so that the true incidence of rFVII-related thrombosis remains unknown. Finally, in a clinical trial of 399 patients with intracranial bleeds, arterial thrombosis (primarily myocardial ischemia and cerebral infarct) was significantly higher in the rFVIIa group (16 of 303, 5%) compared to the controls (0 of 96, 0%) while venous thrombosis were the same.(Mayer et al 2005) The incidence of arterial thrombosis at rFVIIa dose of 40, 80 and 160 μg/kg in this study were respectively 6%, 2% and 8%.
Irrespective, it is expected that correction of the hemostatic defect may predispose the patient (with bleeding disorders) to thrombosis if predisposing factors are present. Thus, caution should be exercised when using rFVIIa in patients with underlying conditions that may predispose them to arterial or venous thrombosis, or DIC and in patients with advanced age.
Conclusion
High dose rFVIIa has been shown to have documented efficacy for the treatment and prevention of bleeding in patients with Glanzmann’s thrombasthenia. Experimental evidences are emerging suggesting that high dose rFVIIa improves thrombin generation, though not necessarily to normal levels, resulting in enhanced adhesion and aggregation of GT platelets that do not respond to physiologic aggregating agents because of the lack of the fibrinogen receptor GPIIbIIIa. At this time, high dose rFVIIa is indicated for GT patients who have development anti-platelet antibodies (to HLA and/or GPIIbIIIa) and history of past or present refractoriness to platelet transfusion. High dose rFVIIa could also be considered for situations where avoidance of anti-platelet antibodies development is important or desirable. One such example is female patients approaching and during reproductive age when development of anti-GPIIbIIIa antibodies capable of passing through the placenta during pregnancy may result in fetal/neonatal thrombocytopenia and serious bleeding (including intracranial bleeding). As indicated earlier, the International Survey data on Glanzmann’s thrombasthenia allow a preliminary suggestion of a more optimal regimen for treatment of bleeding episodes, but the appropriate regimen for surgical prophylaxis remains unknown. The experience on the use of rFVIIa in GT patients is also too limited to allow proper assessment of safety of rFVIIa. There is, therefore, a major need for clinical studies, particularly clinical trials, to assess efficacy, safety (particularly thrombotic events) and to provide evidence-based optimal treatment regimens of rFVIIa in GT patients with normal plasma coagulation pathways. We need data on rFVIIa as an independent treatment modality especially in patients refractory to platelet transfusion, or as an adjunct to other hemostatic treatment including platelet transfusion. The data also need to be stratified according to mild/moderate and severe bleeding episodes, as well as minor and major surgical procedures. At this time the efficacy of high dose rFVIIa can only be followed by clinical observations. There is therefore a need to find laboratory parameters that will predict clinical efficacy.
References
- Abshire T, Kenet G. Recombinant factor VIIa: review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost. 2004;2:899–909. doi: 10.1111/j.1538-7836.2004.00759.x. [DOI] [PubMed] [Google Scholar]
- Aledort LM. Comparative thrombotic event incidence after infusion of recombinant factor VIIa versus factor VIII inhibitor bypass activity. J Thromb Haemost. 2004;2:1700–08. doi: 10.1111/j.1538-7836.2004.00944.x. [DOI] [PubMed] [Google Scholar]
- Almeida AM, Khair K, Hann I, et al. The use of recombinant factor VIIa in children with inherited platelet function disorders. Br J Haematol. 2003;121:477–81. doi: 10.1046/j.1365-2141.2003.04286.x. [DOI] [PubMed] [Google Scholar]
- Bell JA, Savidge GF. Glanzmann’s thrombasthenia proposed optimal management during surgery and delivery. Clin Appl Thromb Hemost. 2003;9:167–70. doi: 10.1177/107602960300900213. [DOI] [PubMed] [Google Scholar]
- Berrettini M, Mariani G, Schiavoni M, et al. Pharmacokinetic evaluation of recombinant, activated factor VII in patients with inherited factor VII deficiency. Haematologica. 2001;86:640–5. [PubMed] [Google Scholar]
- Caglar K, Cetinkaya A, Aytac S, et al. Use of recombinant factor VIIa for bleeding in children with Glanzmann thrombasthenia. Pediatr Hematol Oncol. 2003;20:435–8. [PubMed] [Google Scholar]
- Chuansumrit A, Sangkapreecha C, Hathirat P. Successful epistaxis control in a patient with Glanzmann thrombasthenia by increased bolus injection dose of recombinant factor VIIa. Thromb Haemost. 1999;82:1778. [PubMed] [Google Scholar]
- Chuansumrit A, Suwannuraks M, Sri-Udomporn N, et al. Recombinant activated factor VII combined with local measures in preventing bleeding from invasive dental procedures in patients with Glanzmann thrombasthenia. Blood Coagul Fibrinolysis. 2003;14:187–90. doi: 10.1097/00001721-200302000-00011. [DOI] [PubMed] [Google Scholar]
- Coppola A, Tufano A, Cimino E, et al. Recombinant factor VIIa in a patient with Glanzmann’s thrombasthenia undergoing gynecological surgery: open issues in light of successful treatment. Thromb Haemost. 2004;92:1450–2. [PubMed] [Google Scholar]
- d’Oiron R, Menart C, Trzeciak MC, et al. Use of recombinant factor VIIa in 3 patients with inherited type I Glanzmann’s thrombasthenia undergoing invasive procedures. Thromb Haemost. 2000;83:644–7. [PubMed] [Google Scholar]
- Dargaud Y, Bordet JC, Trzeciak MC, et al. A case of Glanzmann’s thrombasthenia successfully treated with recombinant factor VIIa during a surgical procedure: observations on the monitoring and the mechanism of action of this drug. Haematologica. 2006;91(6 Suppl):17–20. [PubMed] [Google Scholar]
- Devecioglu O, Unuvar A, Anak S, et al. Pyelolithotomy in a patient with Glanzmann thrombasthenia and antiglycoprotein IIb/IIIa antibodies: the shortest possible duration of treatment with recombinant activated factor VII and platelet transfusions. Turk J Pediatr. 2003;45:64–6. [PubMed] [Google Scholar]
- Diness V, Bregengaard C, Erhardtsen E, et al. Recombinant human factor VIIa (rFVIIa) in a rabbit stasis model. Thromb Res. 1992;67:233–41. doi: 10.1016/0049-3848(92)90142-w. [DOI] [PubMed] [Google Scholar]
- Dodd RY, Notari EP, Stramer SL. Current prevalence and incidence of infectious disease markers and estimated window-period risk in the American Red Cross blood donor population. Transfusion. 2002;42:975–9. doi: 10.1046/j.1537-2995.2002.00174.x. [DOI] [PubMed] [Google Scholar]
- Fressinaud E, Sigaud-Fiks M, Le Boterff C, et al. Use of recombinant factor VIIa (NovoSeven®) for dental extraction in a patient affected by platelet-type (pseudo-) von Willebrand disease (Abstract) Haemophilia. 1998;4:299. [Google Scholar]
- George JN, Caen JP, Nurden AT. Glanzmann’s thrombasthenia: the spectrum of clinical disease. Blood. 1990;75:1383–95. [PubMed] [Google Scholar]
- Hoffman M, Monroe DM, III, Roberts HR. Activated factor VII activates factors IX and X on the surface of activated platelets: thoughts on the mechanism of action of high-dose activated factor VII. Blood Coagul Fibrinolysis. 1998;9(Suppl 1):S61–5. [PubMed] [Google Scholar]
- Inagaki M, Mori T, Tsunematsu Y, et al. Use of recombinant activated factor VII to control bleeding in a young child with qualitative platelet disorder: a case report. Blood Coagul Fibrinolysis. 2006;17:317–22. doi: 10.1097/01.mbc.0000224853.50248.6f. [DOI] [PubMed] [Google Scholar]
- Ito K, Yoshida H, Hatoyama H, et al. Antibody removal therapy used successfully at delivery of a pregnant patient with Glanzmann’s thrombasthenia and multiple anti-platelet antibodies. Vox Sang. 1991;61:40–6. doi: 10.1111/j.1423-0410.1991.tb00925.x. [DOI] [PubMed] [Google Scholar]
- Jacobs MR, Palavecino E, Yomtovian R. Don’t bug me: the problem of bacterial contamination of blood components – challenges and solutions. Transfusion. 2001;41:1331–4. doi: 10.1046/j.1537-2995.2001.41111331.x. [DOI] [PubMed] [Google Scholar]
- Jurlander B, Thim L, Klausen NK, et al. Recombinant activated factor VII (rFVIIa): characterization, manufacturing, and clinical development. Semin Thromb Hemost. 2001;27:373–84. doi: 10.1055/s-2001-16971. [DOI] [PubMed] [Google Scholar]
- Kale A, Bayhan G, Yalinkaya A, et al. The use of recombinant factor VIla in a primigravida with Glanzmann’s thrombasthenia during delivery. J Perinat Med. 2004;32:456–8. doi: 10.1515/JPM.2004.147. [DOI] [PubMed] [Google Scholar]
- Kaleelrahman M, Minford A, Parapia LA. Use of recombinant factor VIIa in inherited platelet disorders. Br J Haematol. 2004;125:95–6. doi: 10.1111/j.1365-2141.2004.04878.x. [DOI] [PubMed] [Google Scholar]
- Kenet G, Lubetskyb A, Luboshitz J, et al. A new approach to treatment of bleeding episodes in young hemophilia patients: a single bolus megadose of recombinant activated factor VII (NovoSeven) J Thromb Haemost. 2003;1:450–5. doi: 10.1046/j.1538-7836.2003.00059.x. [DOI] [PubMed] [Google Scholar]
- Langendonck L, Appel IM. Modification of biological parameters after treatment with recombinant factor VIIa in a patient with thrombocytopathy due to storage pool disease. Pediatr Blood Cancer. 2005;44:676–8. doi: 10.1002/pbc.20199. [DOI] [PubMed] [Google Scholar]
- Lindley CM, Sawyer WT, Macik BG, et al. Pharmacokinetics and pharmacodynamics of recombinant factor VIIa. Clin Pharmacol Ther. 1994;55:638–48. doi: 10.1038/clpt.1994.80. [DOI] [PubMed] [Google Scholar]
- Lisman T, Adelmeijer J, Heijnen F, et al. Recombinant factor VIIa restores aggregation of alphaIIbbeta3-deficient platelets via tissue factor-independent fibrin generation. Blood. 2004;103:1720–7. doi: 10.1182/blood-2003-07-2287. [DOI] [PubMed] [Google Scholar]
- Lisman T, Moschatsis S, Adelmeijer J, et al. Recombinant factor VIIa enhances deposition of platelets with congenital or acquired alpha IIb beta 3 deficiency to endothelial cell matrix and collagen under conditions of flow via tissue factor-independent thrombin generation. Blood. 2003;101:1864–70. doi: 10.1182/blood-2002-09-2761. [DOI] [PubMed] [Google Scholar]
- Lombardo VT, Sottilotta G. Recombinant activated factor VII combined with desmopressin in preventing bleeding from dental extraction in a patient with Glanzmann’s thrombasthenia. Clin Appl Thromb Hemost. 2006;12:115–16. doi: 10.1177/107602960601200120. [DOI] [PubMed] [Google Scholar]
- Lusher JM. Early treatment with recombinant factor VIIa results in greater efficacy with less product. Eur J Haematol Suppl. 1998;63:7–10. doi: 10.1111/j.1600-0609.1998.tb01103.x. [DOI] [PubMed] [Google Scholar]
- Lusher JM. Acute hemarthroses: the benefits of early versus late treatment with recombinant activated factor VII. Blood Coagul Fibrinolysis. 2000;11(Suppl 1):S45–9. doi: 10.1097/00001721-200004001-00010. [DOI] [PubMed] [Google Scholar]
- Martin I, Kriaa F, Proulle V, et al. Protein A Sepharose immunoadsorption can restore the efficacy of platelet concentrates in patients with Glanzmann’s thrombasthenia and anti-glycoprotein IIb-IIIa antibodies. Br J Haematol. 2002;119:991–7. doi: 10.1046/j.1365-2141.2002.03936.x. [DOI] [PubMed] [Google Scholar]
- Mayer SA, Brun NC, Begtrup K, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2005;352:777–85. doi: 10.1056/NEJMoa042991. [DOI] [PubMed] [Google Scholar]
- McGregor L, Hanss M, Sayegh A, et al. Aggregation to thrombin and collagen of platelets from a Glanzmann thrombasthenic patient lacking glycoproteins IIb and IIIa. Thromb Haemost. 1989;62:962–7. [PubMed] [Google Scholar]
- Monroe DM, Hoffman M, Oliver JA, et al. Platelet activity of high-dose factor VIIa is independent of tissue factor. Br J Haematol. 1997;99:542–7. doi: 10.1046/j.1365-2141.1997.4463256.x. [DOI] [PubMed] [Google Scholar]
- Monte S, Lyons G. Peripartum management of a patient with Glanzmann’s thrombasthenia using Thrombelastograph. Br J Anaesth. 2002;88:734–8. doi: 10.1093/bja/88.5.734. [DOI] [PubMed] [Google Scholar]
- Morrissey JH, Mutch NJ. Tissue factor structure and function. Hemostasis and Thrombosis. In: Colman RE, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Basic Principles and Clinical Practice. 5. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 91–106. [Google Scholar]
- Musso R, Cultrera D, Russo M, et al. Recombinant activated factor VII as haemostatic agent in Glanzmann’s thrombasthenia (Abstract) Thromb Haemost. 1999;82(Suppl):621. [Google Scholar]
- Niewiarowski S, Levy-Toledano S, Caen JP. Platelet interaction with polymerizing fibrin in Glanzmann’s thrombasthenia. Thromb Res. 1981;23:457–63. doi: 10.1016/0049-3848(81)90207-3. [DOI] [PubMed] [Google Scholar]
- Nurden AT. Inherited abnormalities of platelets. Thromb Haemost. 1999;82:468–80. [PubMed] [Google Scholar]
- O’Connell KA, Wood JJ, Wise RP, et al. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 2006;295:293–8. doi: 10.1001/jama.295.3.293. [DOI] [PubMed] [Google Scholar]
- Osdoit S, Rosa J-P. Polymeric fibrin interacts with platelets independently from integrin αIIβ3 (Abstract) Blood. 2001;98:518a. [Google Scholar]
- Ozelo MC, Svirin P, Larina L. Use of recombinant factor VIIa in the management of severe bleeding episodes in patients with Bernard-Soulier syndrome. Ann Hematol. 2005;84:816–22. doi: 10.1007/s00277-005-1080-y. [DOI] [PubMed] [Google Scholar]
- Peerschke EI, Zucker MB, Grant RA, et al. Correlation between fibrinogen binding to human platelets and platelet aggregability. Blood. 1980;55:841–7. [PubMed] [Google Scholar]
- Peters M, Heijboer H. Treatment of a patient with Bernard-Soulier, syndrome and recurrent nosebleeds with recombinant factor VIIa [letter] Thromb Haemost. 1998;80:352. [PubMed] [Google Scholar]
- Phillips DR, Charo IF, Parise LV, et al. The platelet membrane glycoprotein IIb-IIIa complex. Blood. 1988;71:831–43. [PubMed] [Google Scholar]
- Poon M-C. Factor VIIa. In: Michelson AA, editor. Platelets. 2. Boston: Elsevier Science; 2006. pp. 1251–61. [Google Scholar]
- Poon M-C, d’Oiron R, von Depka M, et al. Prophylactic and therapeutic recombinant factor VIIa administration to patients with Glanzmann’s thrombasthenia: results of an international survey. J Thromb Haemost. 2004;2:1096–103. doi: 10.1111/j.1538-7836.2004.00767.x. [DOI] [PubMed] [Google Scholar]
- Poon M-C, Demers C, Jobin F, et al. Recombinant factor VIIa is effective for bleeding and surgery in patients with Glanzmann thrombasthenia. Blood. 1999;94:3951–3. [PubMed] [Google Scholar]
- Poon M-C, Zotz R, DiMinno G, et al. Glanzmann’s thrombasthenia treatment: a prospective observational registry on the use of recombinant human activated factor VII and other hemostatic agents. Semin Hematol. 2006;43:S33–6. doi: 10.1053/j.seminhematol.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Pozo Pozo AI, Jimenez-Yuste V, Villar A, et al. Successful thyroidectomy in a patient with Hermansky-Pudlak syndrome treated with recombinant activated factor VII and platelet concentrates. Blood Coagul Fibrinolysis. 2002;13:551–3. doi: 10.1097/00001721-200209000-00010. [DOI] [PubMed] [Google Scholar]
- Reverter JC, Beguin S, Kessels H, et al. Inhibition of platelet-mediated, tissue factor-induced thrombin generation by the mouse/human chimeric 7E3 antibody. Potential implications for the effect of c7E3 Fab treatment on acute thrombosis and “clinical restenosis”. J Clin Invest. 1996;98:863–74. doi: 10.1172/JCI118859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KL, Savoia H, Street AM. Thrombotic complications in two patients receiving NovoSeven® (Abstract) Haemophilia. 2000;6:349. [Google Scholar]
- Sallah S, Isaksen M, Seremetis S, et al. Comparative thrombotic event incidence after infusion of recombinant factor VIIa vs. factor VIII inhibitor bypass activity – a rebuttal. J Thromb Haemost. 2005;3:820–2. doi: 10.1111/j.1538-7836.2005.01254.x. [DOI] [PubMed] [Google Scholar]
- Santagostino E, Gringeri A, Mannucci PM. Home treatment with recombinant activated factor VII in patients with factor VIII inhibitors: the advantages of early intervention. Br J Haematol. 1999;104:22–6. doi: 10.1046/j.1365-2141.1999.01128.x. [DOI] [PubMed] [Google Scholar]
- Tengborn L, Petruson B. A patient with Glanzmann thrombasthenia and epistaxis successfully treated with recombinant factor VIIa [letter] Thromb Haemost. 1996;75:981–2. [PubMed] [Google Scholar]
- Turecek PL, Richter G, Muchitsch EM, et al. Thrombogenicity of recombinant factor VIIa and recombinant soluble tissue factor in an in vivo rabbit model (Abstract) Thromb Haemost. 1997;78(Suppl):222. [Google Scholar]
- Uzunlar HI, Eroglu A, Senel AC, et al. A patient with Glanzmann’s thrombasthenia for emergent abdominal surgery. Anesth Analg. 2004;99:1258–60. doi: 10.1213/01.ANE.0000131726.09685.BF. [DOI] [PubMed] [Google Scholar]
- van Buuren HR, Wielenga JJ. Successful surgery using recombinant factor VIIa for recurrent, idiopathic nonulcer duodenal bleeding in a patient with Glanzmann’s thrombasthenia. Dig Dis Sci. 2002;47:2134–6. doi: 10.1023/a:1019605803467. [DOI] [PubMed] [Google Scholar]
- Villar A, Aronis S, Morfini M, et al. Pharmacokinetics of activated recombinant coagulation factor VII (NovoSeven) in children vs. adults with haemophilia A. Haemophilia. 2004;10:352–9. doi: 10.1111/j.1365-2516.2004.00925.x. [DOI] [PubMed] [Google Scholar]
- Yilmaz BT, Alioglu B, Ozyurek E, et al. Successful use of recombinant factor VIIa (NovoSeven) during cardiac surgery in a pediatric patient with Glanzmann thrombasthenia. Pediatr Cardiol. 2005;26:843–5. doi: 10.1007/s00246-004-0919-7. [DOI] [PubMed] [Google Scholar]