

Abstract
Streptokinase (SK) activates human fibrinolysis by inducing non-proteolytic activation of the serine proteinase zymogen, plasminogen (Pg), in the SK·Pg* catalytic complex. SK·Pg* proteolytically activates Pg to plasmin (Pm). SK-induced Pg activation is enhanced by lysine-binding site (LBS) interactions with kringles on Pg and Pm, as evidenced by inhibition of the reactions by the lysine analogue, 6-aminohexanoic acid. Equilibrium binding analysis and [Lys]Pg activation kinetics with wild-type SK, carboxypeptidase B-treated SK, and a COOH-terminal Lys414 deletion mutant (SKΔK414) demonstrated a critical role for Lys414 in the enhancement of [Lys]Pg and [Lys]Pm binding and conformational [Lys]Pg activation. The LBS-independent affinity of SK for [Glu]Pg was unaffected by deletion of Lys414. By contrast, removal of SK Lys414 caused 19- and 14-fold decreases in SK affinity for [Lys]Pg and [Lys]Pm binding in the catalytic mode, respectively. In kinetic studies of the coupled conformational and proteolytic activation of [Lys]Pg, SKΔK414 exhibited a corresponding 17-fold affinity decrease for formation of the SKΔK414·[Lys]Pg* complex. SKΔK414 binding to [Lys]Pg and [Lys]Pm and conformational [Lys]Pg activation were LBS-independent, whereas [Lys]Pg substrate binding and proteolytic [Lys]Pm generation remained LBS-dependent. We conclude that binding of SK Lys414 to [Lys]Pg and [Lys]Pm kringles enhances SK·[Lys]Pg* and SK·[Lys]Pm catalytic complex formation. This interaction is distinct structurally and functionally from LBS-dependent Pg substrate recognition by these complexes.
Streptokinase (SK)2 activates the human fibrinolytic system by activating the zymogen, plasminogen (Pg) to form the fibrin-degrading proteinase, plasmin (Pm) (1). The mechanism of SK-activated Pm formation is unique in that it is initiated by formation of an SK·Pg* complex in which the zymogen catalytic site is activated non-proteolytically (2–5). SK·Pg* binds free Pg and converts it into Pm by intermolecular proteolytic cleavage (6). Pm binds tightly to SK in the catalytic mode (7, 8) and SK·Pm propagates proteolytic Pg activation (6, 9, 10) through expression of a Pg substrate binding exosite (7). The mechanism is regulated by intrinsic differences in affinity of SK for [Glu]Pg, [Lys]Pg, and [Lys]Pm (5–8,11). [Glu]Pg consists of an NH2-terminal 77-residue peptide, five kringle domains, and a serine proteinase catalytic domain (12). [Glu]Pg is maintained in a compact conformation through intramolecular interaction of the NH2-terminal peptide with kringles 4 and 5 (13–15). Pm cleavage of the NH2-terminal peptide of [Glu]Pg generates the more reactive [Lys]Pg, which assumes an extended conformation with expression of enhanced lysine-binding site (LBS) interactions (12, 14–16).
Formation of SK·[Lys]Pg* and SK· [Lys]Pm catalytic complexes and subsequent [Lys]Pg substrate recognition are enhanced by interactions of SK with LBS of Pg and Pm kringle domains (5–8, 11, 17–20). Recent studies of the Pg activation mechanism demonstrate that LBS interactions enhance SK· [Lys]Pg* catalytic complex formation and Pg substrate binding but are not absolutely required for these interactions (5, 6). SK binding to the compact conformation of [Glu]Pg in the catalytic mode is LBS-independent (5, 6, 8, 11).
Several studies have sought to define SK lysine residues that mediate its interactions with Pg and Pm kringles. The crystal structure of SK bound to the isolated Pm catalytic domain (micro-Pm) shows that SK consists of three homologous, independently folded β-grasp domains connected by flexible linking sequences (21). SK forms a “crater” around the Pm catalytic site which provides a surface for Pg substrate binding (21). Studies of SK domain truncation and deletion mutants, isolated domains, and point mutants have led to diverse interpretations, indicating that each of the SK domains may participate in kringle interactions (18–20, 22, 23). Some studies support a role for the flexible 250-loop of the SK β-domain in LBS-dependent Pg substrate binding (19, 20). By contrast, no studies have demonstrated the structural basis for the LBS dependence of catalytic complex formation.
Kringles of Pg and Pm bind zwitterionic COOH-terminal lysine residues and lysine analogues specifically, notably 6-aminohexanoic acid (6-AHA) (13, 24). This is the basis for Pg binding by several proteins, including fibrin (25, 26), antiplasmin (27), histidine-rich glycoprotein (28), and tetranectin (29). In fibrinolysis, LBS interactions with fibrin mediated by COOH-terminal lysine residues localize and accelerate Pg activation and fibrin degradation by Pm (25, 26, 30) and protect fibrin-bound Pm from inactivation by antiplasmin (31). Surprisingly, the fact that the COOH-terminal residue of SK is Lys414 (32) has been overlooked in previous studies. Here, we show that Lys414 is responsible for the LBS-dependent enhancement in affinity of SK· [Lys]Pg* and SK·[Lys]Pm catalytic complex formation. This interaction is shown to be structurally and functionally distinct from the LBS-dependent binding of [Lys]Pg as a substrate of the catalytic complexes.
EXPERIMENTAL PROCEDURES
Wild-type SK and an SK Mutant Lacking the COOH-terminal Lys
Wild-type SK (wtSK) was prepared by methods described previously (33, 34) or was expressed as a fusion protein with a tobacco etch virus (TEV) proteinase cleavage site (underlined) encoded before the wtSK protein, Met-His6-Ser-Ala-Gly-Gly-Ser-Pro-Trp-Asn-Glu-Asn-Leu-Try-Phe-Gln-SKIle1-SKAla2-SKGly3 … (His6-wtSK). From a pET30a(+) vector backbone, a single nucleotide substitution mutated the P13 residue of a thrombin cleavage site (from Arg to Trp), which eliminated this unnecessary site and generated a NcoI restriction site. Flanking NcoI and XhoI restriction sites (underlined) where incorporated into the 5′- and 3′-PCR primers, respectively, with the sense primer, 5′-TCACTCCGCGGGTGGTAGTCCATGGAACGAGAACCTGTATTTTCAGATTGCTGGACCTGAGTGGCTG-3′, the same for wtSK and SKΔK414 constructs, and the antisense primer, 5′-ATAATGGTGCTCGAGTTATTTGTCGTTAGGGTTATCAGG-3′, only differed by the Lys414 codon (bold). A construct that encoded a His6-tagged TEV proteinase was kindly provided by Dr. Laura Mizoue of the Vanderbilt University Center for Structural Biology and used to remove the His6-tag from the NH2 terminus of wtSK.
His6-wtSK was expressed from Rosetta(DE3) pLysS cells induced with 20 g/liter lactose for 12–16 h at 37 °C. Cells were harvested by centrifugation, resuspended in 50 mM Hepes, 125 mM NaCl, 1 mg/ml polyethylene glycol 8000, pH 7.4 (Buffer A) with 1 mM EDTA and 0.2% sodium azide, lysed by three cycles of sonication (~45 s cycles) on ice, and centrifuged to clarify lysates. The pellet was resuspended in Buffer A containing 3 M NaSCN. The solubilized wtSK was dialyzed into 50 mM Hepes, 400 mM NaCl, 50 mM imidazole, pH 7.4 (Buffer B) and purified by Ni2+-iminodiacetic acid-Sepharose chromatography with a 50–500 mM imidazole gradient in Buffer B. TEV proteinase was added to the eluted protein in a 1 to 5 molar ratio of enzyme to substrate. The reaction mixture was first dialyzed overnight into 50 mM Hepes, 300 mM NaCl, 1 mM dithiothreitol, 5% glycerol, pH 7.8 at 4 °C, and subsequently dialyzed back into Buffer B. Uncleaved fusion protein, cleaved His6-tag, and the TEV proteinase bound to Ni2+-iminodiacetic acid-Sepharose, and wtSK was obtained from the column flow-through. wtSK was dialyzed against Buffer A without polyethylene glycol, quick-frozen, and stored at −80 °C. SKΔ414K was prepared following an identical procedure. The correct NH2-terminal sequence for wtSK and SKΔK414 was confirmed.
Native and Carboxypeptidase B (CpB)-treated SK
Native SK purchased from Diapharma and purified as described previously (7, 8, 11) was treated with porcine pancreatic CpB (Sigma Type-1, diisopropylfluorophosphate-treated, 4.7 mg/ml in 0.1 M NaCl) in 50 mM Hepes, 125 mM NaCl, pH 7.4. SK (2.5 mg/ml) was incubated with CpB (35 μg/ml) for 30 min at 25 °C, and the reaction was stopped by addition of 10 mM EDTA. Titrations of [5F]FFR-[Lys]Pg were performed in 50 mM Hepes, 125 mM NaCl, 1 mM EDTA, 10 μM Val-Phe-Arg-CH2Cl, 1 mg/ml bovine serum albumin, ±100 mM 6-AHA, pH 7.4.
Fluorescence Equilibrium Binding
[Glu]Pg, [Lys]Pg, [Lys]Pm, and the active site-labeled fluorescein analogues were prepared as described (5, 6, 11). Fluorescence titrations were performed in Buffer A containing 1 mM EDTA and 1 μM D-Phe-Phe-Arg-CH2Cl ± 10 mM 6-AHA as described previously (5, 6, 33). Fluorescence changes ((Fobs − Fo)/Fo = ΔF/Fo) as a function of total SK concentration, were fit by the quadratic binding equation to determine the maximum fluorescence change (ΔFmax/Fo) and dissociation constant (KD), with the stoichiometric factor (n) fixed at 1. Competitive binding titrations of native [Lys]Pg were performed by addition of wtSK or SKΔ414 to mixtures of [5F]FFR-[Lys]Pg as a function of native [Lys]Pg concentration, as described previously (5). Results were analyzed by fitting of the cubic binding equation to determine the KD of wtSK and SKΔ414 for native [Lys]Pg.
Plasminogen Activation Kinetics
Coupled conformational and proteolytic activation of [Lys]Pg by wtSK and SKΔK414 were quantitated as described previously (5, 6). Fitting of parabolic progress curves of D-Val-Leu-Lys-pNA (VLK-pNA) hydrolysis at 200 μM, in the presence of 15 nM [Lys]Pg and increasing wtSK and SKΔK414 concentrations gave the initial rates (v1) of VLK-pNA hydrolysis, reflecting conformational activation of the SK·Pg* complex, and the rates of activity increase (v2), reflecting Pm generation. The SK dependences of v1 and v2 were analyzed using the simplified equations described previously (5, 6).
The data were also analyzed by fitting of the family of progress curves as a function of SK concentration with a more complete mechanism including both SK·Pg*- and SK·Pm-catalyzed Pg activation pathways under bimolecular reaction conditions (Scheme 1). For this analysis, the Km values for chromogenic substrate hydrolysis by Pm, SK·Pg*, and SK·Pm were fixed at the previously determined values, whereas the corresponding kcat values were allowed to vary within the experimental error of their determination to optimize the fit. for SK·Pm binding was fixed at 12 pM (7). The fitted parameters were KA for SK·Pg* formation and the bimolecular rate constants for Pm generation by SK·Pg* (kPg) and SK·Pm (kPm) (Scheme 1) (5, 6). Non-linear least squares fitting was performed with SCIENTIST (MicroMath) or DYNAFIT (35). Error estimates represent the 95% confidence interval.
SCHEME 1.
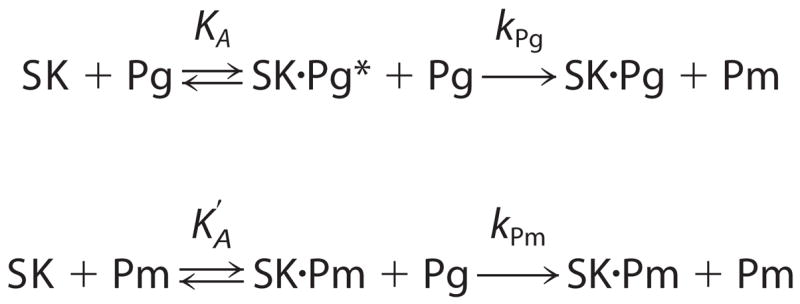
RESULTS AND DISCUSSION
Binding of Native SK and CpB-treated SK to [Lys]Pg
Native SK was treated with CpB to remove the COOH-terminal lysine residue, under conditions where there was no detectable degradation of SK observable by SDS-gel electrophoresis (not shown). Titrations of [5F]FFR-[Lys]Pg with native SK and CpB-SK were performed in the absence and presence of saturating 6-AHA to evaluate the effect of CpB treatment on the LBS dependence of SK affinity (Fig. 1A). In the absence and presence of 6-AHA, native SK bound fluorescein-labeled [Lys]Pg with KD 20 ± 5 nM and 380 ± 40 nM, respectively, and with ΔFmax/Fo of −15 ± 1% and −19 ± 1%. By contrast, CpB-SK bound labeled [Lys]Pg with KD 130 ± 20 nM and ΔFmax/Fo −17 ± 1% in the absence of 6-AHA, and with KD 300 ± 50 nM and ΔFmax/Fo −19 ± 1% in the presence of 6-AHA. The affinities for native and CpB-treated SK in the presence of 6-AHA were indistinguishable. CpB treatment of SK decreased the effect of 6-AHA on [Lys]Pg affinity from 19-fold to 2.3-fold. This demonstrated that CpB treatment of SK resulted in a selective loss of LBS-dependent affinity for labeled [Lys]Pg.
FIGURE 1. Effect of removal of the COOH-terminal Lys414 of SK on binding to [Glu]Pg, [Lys]Pg, and [Lys]Pm.
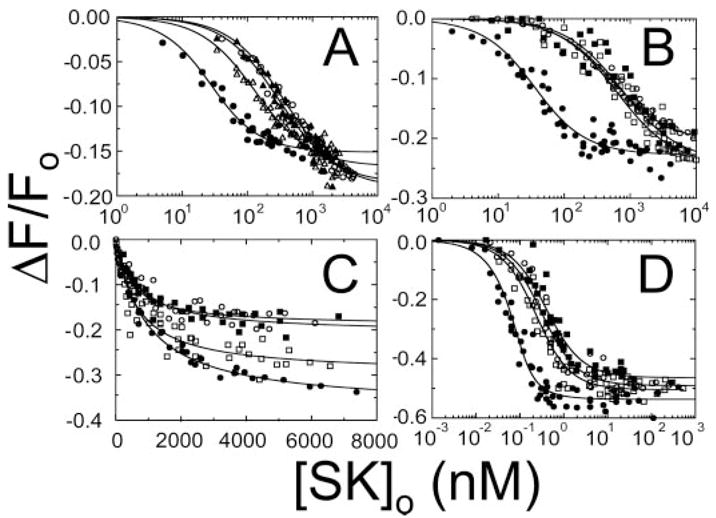
A, effect of CpB treatment of native SK on affinity for [5F]FFR-[Lys]Pg in the absence and presence of 6-AHA. Titrations of the decrease in fluorescence (ΔF/Fo) of 15 nM [5F]FFR-[Lys]Pg as a function of total SK concentration ([SK]o) for native SK in the absence (●) and presence (○) of 100 mM 6-AHA and CpB-treated SK in the absence (△) and presence (▲) of 6-AHA. B, titrations of 15 nM fluorescein-labeled [Lys]Pg with wtSK in the absence (●) and presence (○) of 10 mM 6-AHA and with SKΔK414 in the absence (□) and presence (■) of 10 mM 6-AHA. C and D, analogous titrations of 15 nM fluorescein-labeled [Glu]Pg (C) and 75 pM fluorescein-labeled [Lys]Pm (D) using the same symbols as in B. Lines represent the least squares fits with the parameters given in the “Results and Discussion.” Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
Binding of wtSK and SKΔK414 to [Lys]Pg
Titrations of [5F]FFR-[Lys]Pg with wtSK and SKΔK414 were performed in the absence and presence of 10 mM 6-AHA (Fig. 1B). In the absence and presence of 6-AHA, wtSK bound labeled [Lys]Pg with KD 28 ± 6 nM and 520 ± 70 nM, respectively, and with ΔFmax/Fo of −23 ± 1% and −21 ± 1%. Like CpB-treated SK, SKΔK414 bound to labeled [Lys]Pg with weaker affinity than wtSK, with KD 610 ± 200 nM and 750 ± 350 nM in the absence and presence of 6-AHA, respectively, and ΔFmax/Fo of −24 ± 2% and −24 ± 3%. Native and wtSK bound with indistinguishable affinity to labeled Pg, and their LBS-dependent losses of affinity in the presence of 6-AHA were the same (Fig. 1, A and B). Deletion of the COOH-terminal lysine decreased the weakening effect of 6-AHA on affinity from 19- to 1.2-fold. This demonstrated that the SK mutant lacking the COOH-terminal lysine exhibited a selective loss of LBS-dependent affinity for labeled [Lys]Pg.
Binding of wtSK and SKΔK414 to [Glu]Pg
Titrations of [5F]FFR-[Glu]Pg with wtSK and SKΔK414 were performed in the absence and presence of 10 mM 6-AHA (Fig. 1C). In the absence and presence of 6-AHA, wtSK bound labeled [Glu]Pg with KD 930 ± 120 nM and 471 ± 140 nM, respectively, and with ΔFmax/Fo −37 ± 1% and −19 ± 1%. SKΔK414 bound labeled [Glu]Pg with KD 634 ± 160 nM and 560 ± 150 nM in the absence and presence of 6-AHA, respectively, and with ΔFmax/Fo −30 ± 2% and −19 ± 1%. Although the amplitudes of the fluorescence changes were decreased by 6-AHA, the affinities of wtSK and SKΔK414 for labeled [Glu]Pg in the presence or absence of 6-AHA were indistinguishable and LBS-independent. This was consistent with the LBS independence of SK binding to [Glu]Pg in the compact conformation (5, 8, 11) and an independence of the affinity on deletion of Lys414.
Binding of wtSK and SKΔK414 to [Lys]Pm
To determine whether the COOH-terminal lysine of SK also interacted with [Lys]Pm kringles, titrations of [5F]FFR-Pm with wtSK and SKΔK414 were performed in the absence and presence of 10 mM 6-AHA (Fig. 1D). wtSK bound labeled [Lys]Pm with KD 19 ± 7 pM, consistent with the previously reported affinity (7) and ΔFmax/Fo of −54 ± 2% in the absence of 6-AHA, compared with a 14-fold higher KD of 260 ± 70 pM and ΔFmax/Fo of −49 ± 2% in the presence of 6-AHA. SKΔK414 bound labeled [Lys]Pm with impaired affinity represented by KD 190 ± 40 pM and 350 ± 80 pM in the absence and presence of 6-AHA, respectively, and with ΔFmax/Fo of −50 ± 1% and −47 ± 2%. The affinities of wtSK for [5F]FFR-Pm in the presence of 6-AHA and that of SKΔK414 in the absence and presence of 6-AHA were indistinguishable within the experimental error. The results indicated that similar to [Lys]Pg, the affinity for complex formation between wtSK and [Lys]Pm was enhanced by interaction of SK Lys414 with kringles on [Lys]Pm.
Binding of SKΔK414 and wtSK to Native Pg
The affinities of active site-labeled [Glu]Pg and [Lys]Pg for SK are consistently ~5-fold lower than those of the native proteins, as determined in previous studies, but maintain the same magnitude of the effects of 6-AHA on binding (5, 11). To characterize binding of wtSK and SKΔK414 to native [Lys]Pg, [5F]FFR-[Lys]Pg was used as a probe of the native and labeled [Lys]Pg competitive binding equilibria in the absence of proteinase inhibitors. To resolve the rapid binding equilibrium from slower proteolytic cleavage, individual measurements were made as a function of time after addition of wtSK or SKΔK414 to mixtures of [5F]FFR-[Lys]Pg and various concentrations of native [Lys]Pg at fixed wtSK and SKΔK414 concentrations as described previously (5, 6). Simultaneous fits of the direct titration and the competitive titration data in Fig. 2 by the cubic binding equation gave a KD of 8.8 ± 4.3 nM for wtSK (Fig. 2A), which was increased to 124 ± 71 nM by 6-AHA (Fig. 2B). SKΔK414 bound to native [Lys]Pg with indistinguishable values of 157 ± 66 nM and 106 ± 40 nM in the absence and presence of 6-AHA (Fig. 2C). The values for wtSK were in good agreement with the previously determined dissociation constants of 10 ± 3 nM and 115 ± 32 nM for native SK in the absence and presence of 6-AHA, respectively (5).
FIGURE 2. Competitive binding of native [Lys]Pg and [5F]FFR-[Lys]Pg to wtSK and SKΔK414.
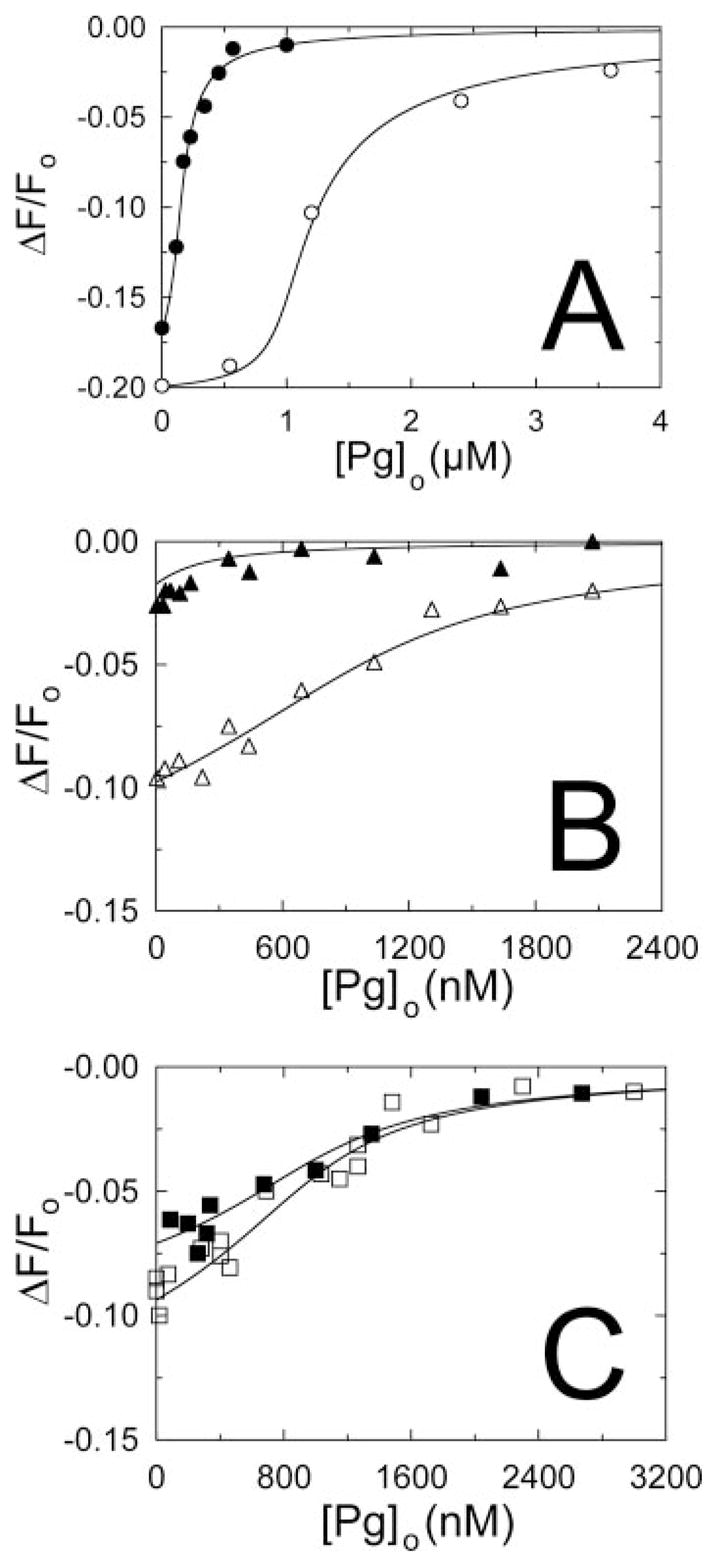
A, the fractional change in fluorescence (ΔF/Fo) of 15 nM [5F]FFR-[Lys]Pg plotted against the total concentration of native [Lys]Pg ([Pg]o) for 150 nM (●) and 1 μM (○) wtSK. B, similar titrations are shown in the presence of 10 mM 6-AHA for 100 nM (▲) and 1 μM (△) wtSK. C, titrations at 1 μM SKΔK414 in the absence (■) and presence (□) of 10 mM 6-AHA. The lines represent the fit by the cubic competitive binding equation with the parameters given in the “Results and Discussion” and n fixed at 1. Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
Plasminogen Activation Kinetics
Determination of the affinities of wtSK and SKΔK414 for native [Lys]Pg was necessary to interpret the kinetics of native [Lys]Pg activation by the SK mutant. The kinetics of [Lys]Pg activation were examined by analysis of reaction progress curves in the presence of VLK-pNA, monitored by hydrolysis of the chromogenic substrate. As previously detailed (5, 6), the parabolic progress curves were resolved into an initial rate of substrate hydrolysis (v1) representing the activity of the conformationally activated SK·Pg* complex and the rate of acceleration (v2) representing the subsequent proteolytic generation of Pm. The dependences of v1 and v2 on SK concentration were analyzed as described under “Experimental Procedures.” A second method was also used for the analysis in which families of progress curves collected as a function of SK concentration were fit by numerical integration of the rate equations for the complete mechanism, including both SK·Pg*- and SK·Pm-catalyzed sequential reactions (Scheme 1 and see “Experimental Procedures”).
Analysis of the hyperbolic v1 dependence on SK concentration in the absence of 6-AHA gave apparent KA of 2 ± 1 nM and 68 ± 27 nM for wtSK and SKΔK414, respectively (Fig. 3A). In the presence of 6-AHA, the formation of the wtSK·[Lys]Pg* complex was weakened to 43 ± 12 nM (22-fold), whereas the affinity of SKΔK414 was indistinguishable at 87 ± 28 nM, indicating that formation of the SKΔK414· [Lys]Pg* catalytic complex was LBS-independent. Analysis of the bimodal v2 dependence on wtSK concentration in the absence of 6-AHA gave a similar affinity of 1.5 ± 0.1 nM for SK·[Lys]Pg* formation. The v2 dependence of SKΔK414 was shifted to higher SK concentration, due to coupling between the formation of SKΔK414·[Lys]Pg* and Pm generation, corresponding to KA 63 ± 54 nM for SKΔK414·[Lys]Pg* formation. Values of KA determined kinetically in the absence and presence of 6-AHA were in good agreement with the dissociation constants determined above by competitive binding. The bimolecular rate constants (kPg), representing Pg substrate binding and Pm formation, were indistinguishable for wtSK and SKΔK414, as indicated by the same v2 maxima. In the presence of 6-AHA, v2 was decreased 20-fold for wtSK and an indistinguishable ~25-fold for SKΔK414. Analysis of reaction progress curves by numerical integration of the rate equations gave KA 3.2 ± 0.1 nM and 88 ± 7 nM for wtSK and SKΔK414, respectively, in the absence of 6-AHA, and indistinguishable kPg of 0.0006 ± 0.0001 nM−1 s−1, whereas in the presence of 6-AHA, KA for wtSK and SKΔK414 increased to 39 ± 1 nM (12-fold) and 131 ± 8 nM (1.5-fold), respectively.
FIGURE 3. Kinetics of [Lys]Pg activation by wtSK and SKΔK414.
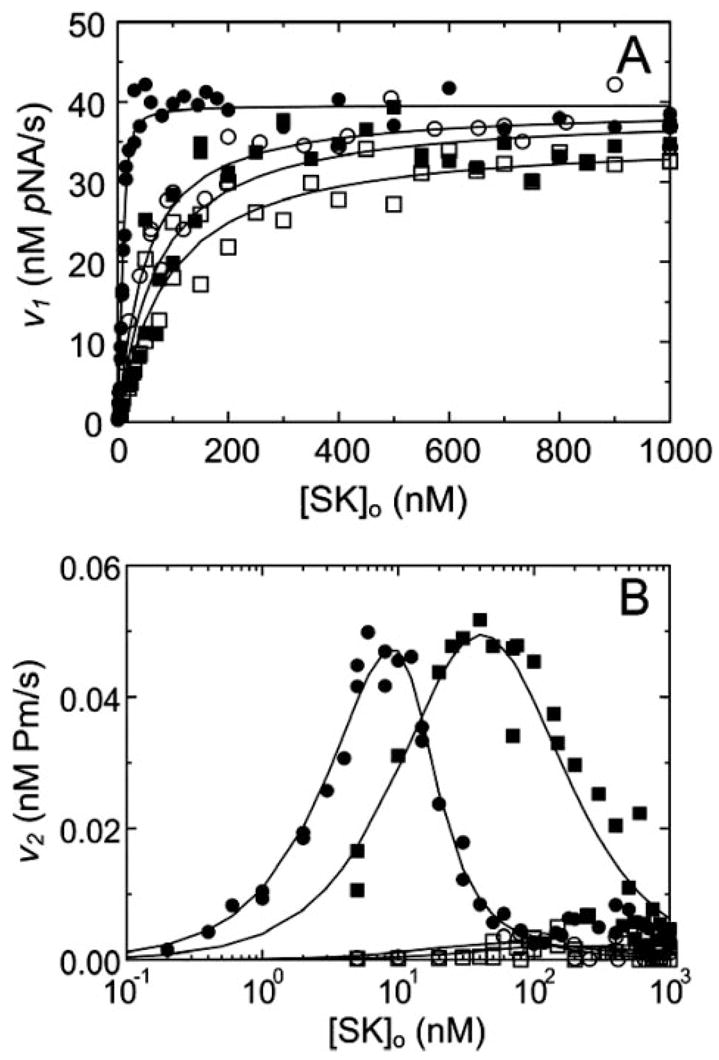
A, dependence of v1 on the total SK species concentration ([SK]o) obtained from reactions of 15 nM [Lys]Pg in the presence of 200 μM VLK-pNA and in the absence (●) and presence (○) of 10 mM 6-AHA and with SKΔK414 in the absence (■) and presence (□) of 10 mM 6-AHA. B, SK concentration dependencies of v2 rates for wtSK in the absence (●) and presence (○) of 10 mM 6-AHA and SKΔK414 in the absence (■) and presence (□) of 10 mM 6-AHA. Lines represent the fits by the equations described previously (5, 6) with the parameters given in the “Results and Discussion.” Activation reactions were performed and analyzed as described under “Experimental Procedures.”
In conclusion, results of quantitative equilibrium binding and [Lys]Pg activation kinetic studies with wtSK, CpB-treated SK, and a COOH-terminal SKΔK414 deletion mutant demonstrated a critical role for the COOH-terminal lysine of SK in the enhancement of [Lys]Pg and [Lys]Pm binding and conformational [Lys]Pg activation. Removal of SK Lys414 resulted in complete loss of the LBS dependence of SK binding to [Lys]Pg and [Lys]Pm in the catalytic mode and a corresponding loss of LBS-dependent affinity for formation of the conformationally activated SK·[Lys]Pg* complex measured kinetically.
SKΔK414 binding to [Lys]Pg and [Lys]Pm and conformational [Lys]Pg activation were LBS-independent, as shown by the absence of a significant effect of 6-AHA, whereas [Lys]Pg substrate binding remained LBS-dependent to a comparable extent as that of wtSK. We conclude that binding of SK Lys414 to [Lys]Pg and [Lys]Pm kringles enhances SK·Pg* and SK·Pm catalytic complex formation. The Pg substrate interaction is concluded to be mediated by a distinct SK structure responsible for Pg substrate recognition by the SK·Pg* and SK·Pm complexes. Further studies will be required to clarify the SK structure responsible for the LBS dependence of Pg substrate recognition.
Acknowledgments
We thank Malabika Laha and Ronald R. Bean for excellent technical assistance.
Footnotes
This work was supported by National Institutes of Health/NHLBI Grant HL056181 (to P. E. B.).
The abbreviations used are: SK, streptokinase; wtSK, recombinant wild-type SK; SKΔK414, streptokinase mutant lacking the COOH-terminal lysine; CpB, carboxypeptidase B; 6-AHA; 6-aminohexanoic acid; pNA, p-nitroaniline; Pg, plasminogen; [Glu]Pg, compact form of Pg; [Lys]Pg, [Glu]Pg lacking the 77-residue NH2-terminal peptide; [Lys]Pm, Pm, plasmin; fluorescein-labeled analogues of Pg or Pm prepared with Nα-[(acetylthio)acetyl]-(D-Phe)-Phe-Arg-CH2Cl and 5-(iodoacetamido)fluorescein are represented by [5F]FFR-Pg or –Pm; LBS, lysine-binding site; TEV, tobacco etch virus.
Schechter-Berger (36) notation referring to the residues of a substrate (from the NH2-terminal end) as …P4-P3-P2-P1-P1′-P2′ … with the scissile bond at P1-P1′.
References
- 1.Collen D, Lijnen HR. Blood. 1991;78:3114–3124. [PubMed] [Google Scholar]
- 2.McClintock DK, Bell PH. Biochem Biophys Res Commun. 1971;43:694–702. doi: 10.1016/0006-291x(71)90670-x. [DOI] [PubMed] [Google Scholar]
- 3.Reddy KN, Markus G. J Biol Chem. 1972;247:1683–1691. [PubMed] [Google Scholar]
- 4.Schick LA, Castellino FJ. Biochem Biophys Res Commun. 1974;57:47–54. doi: 10.1016/s0006-291x(74)80355-4. [DOI] [PubMed] [Google Scholar]
- 5.Boxrud PD, Verhamme IM, Bock PE. J Biol Chem. 2004;279:36633–36641. doi: 10.1074/jbc.M405264200. [DOI] [PubMed] [Google Scholar]
- 6.Boxrud PD, Bock PE. J Biol Chem. 2004;279:36642–36649. doi: 10.1074/jbc.M405265200. [DOI] [PubMed] [Google Scholar]
- 7.Boxrud PD, Fay WP, Bock PE. J Biol Chem. 2000;275:14579–14589. doi: 10.1074/jbc.275.19.14579. [DOI] [PubMed] [Google Scholar]
- 8.Boxrud PD, Bock PE. Biochemistry. 2000;39:13974–13981. doi: 10.1021/bi000594i. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Gronow M, Siefring GE, Jr, Castellino FJ. J Biol Chem. 1978;253:1090–1094. [PubMed] [Google Scholar]
- 10.Wohl RC, Summaria L, Robbins KC. J Biol Chem. 1980;255:2005–2013. [PubMed] [Google Scholar]
- 11.Bock PE, Day DE, Verhamme IM, Bernardo MM, Olson ST, Shore JD. J Biol Chem. 1996;271:1072–1080. doi: 10.1074/jbc.271.2.1072. [DOI] [PubMed] [Google Scholar]
- 12.Ponting CP, Marshall JM, Cederholm-Williams SA. Blood Coagul Fibrinolysis. 1992;3:605–614. [PubMed] [Google Scholar]
- 13.Ponting CP, Holland SK, Cederholm-Williams SA, Marshall JM, Brown AJ, Spraggon G, Blake CC. Biochim Biophys Acta. 1992;1159:155–161. doi: 10.1016/0167-4838(92)90020-e. [DOI] [PubMed] [Google Scholar]
- 14.Marshall JM, Brown AJ, Ponting CP. Biochemistry. 1994;33:3599–3606. doi: 10.1021/bi00178a017. [DOI] [PubMed] [Google Scholar]
- 15.McCance SG, Castellino FJ. Biochemistry. 1995;34:9581–9586. doi: 10.1021/bi00029a035. [DOI] [PubMed] [Google Scholar]
- 16.Violand BN, Byrne R, Castellino FJ. J Biol Chem. 1978;253:5395–5401. [PubMed] [Google Scholar]
- 17.Lin LF, Houng A, Reed GL. Biochemistry. 2000;39:4740–4745. doi: 10.1021/bi992028x. [DOI] [PubMed] [Google Scholar]
- 18.Conejero-Lara F, Parrado J, Azuaga AI, Dobson CM, Ponting CP. Protein Sci. 1998;7:2190–2199. doi: 10.1002/pro.5560071017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhar J, Pande AH, Sundram V, Nanda JS, Mande SC, Sahni G. J Biol Chem. 2002;277:13257–13267. doi: 10.1074/jbc.M108422200. [DOI] [PubMed] [Google Scholar]
- 20.Chaudhary A, Vasudha S, Rajagopal K, Komath SS, Garg N, Yadav M, Mande SC, Sahni G. Protein Sci. 1999;8:2791–2805. doi: 10.1110/ps.8.12.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Lin X, Loy JA, Tang J, Zhang XC. Science. 1998;281:1662–1665. doi: 10.1126/science.281.5383.1662. [DOI] [PubMed] [Google Scholar]
- 22.Loy JA, Lin X, Schenone M, Castellino FJ, Zhang XC, Tang J. Biochemistry. 2001;40:14686–14695. doi: 10.1021/bi011309d. [DOI] [PubMed] [Google Scholar]
- 23.Sazonova IY, Robinson BR, Gladysheva IP, Castellino FJ, Reed GL. J Biol Chem. 2004;279:24994–25001. doi: 10.1074/jbc.M400253200. [DOI] [PubMed] [Google Scholar]
- 24.Castellino FJ, McCance SG. Ciba Found Symp. 1997;212:46–60. doi: 10.1002/9780470515457.ch4. [DOI] [PubMed] [Google Scholar]
- 25.Lucas MA, Fretto LJ, McKee PA. J Biol Chem. 1983;258:4249–4256. [PubMed] [Google Scholar]
- 26.Bok RA, Mangel WF. Biochemistry. 1985;24:3279–3286. doi: 10.1021/bi00334a031. [DOI] [PubMed] [Google Scholar]
- 27.Hortin GL, Gibson BL, Fok KF. Biochem Biophys Res Commun. 1988;155:591–596. doi: 10.1016/s0006-291x(88)80535-7. [DOI] [PubMed] [Google Scholar]
- 28.Lijnen HR, Hoylaerts M, Collen D. J Biol Chem. 1980;255:10214–10222. [PubMed] [Google Scholar]
- 29.Clemmensen I, Petersen LC, Kluft C. Eur J Biochem. 1986;156:327–333. doi: 10.1111/j.1432-1033.1986.tb09586.x. [DOI] [PubMed] [Google Scholar]
- 30.Wiman B, Collen D. Nature. 1978;272:549–550. doi: 10.1038/272549a0. [DOI] [PubMed] [Google Scholar]
- 31.Wiman B, Collen D. Eur J Biochem. 1978;84:573–578. doi: 10.1111/j.1432-1033.1978.tb12200.x. [DOI] [PubMed] [Google Scholar]
- 32.Jackson KW, Tang J. Biochemistry. 1982;21:6620–6625. doi: 10.1021/bi00269a001. [DOI] [PubMed] [Google Scholar]
- 33.Bean RR, Verhamme IM, Bock PE. J Biol Chem. 2005;280:7504–7510. doi: 10.1074/jbc.M411637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boxrud PD, Verhamme IM, Fay WP, Bock PE. J Biol Chem. 2001;276:26084–26089. doi: 10.1074/jbc.M101966200. [DOI] [PubMed] [Google Scholar]
- 35.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 36.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]