

Abstract
Activation of the capsaicin receptor (VR1 or TRPV1) in bronchial epithelial cells by capsaicinoids and other vanilloids promotes pro-inflammatory cytokine production and cell death. The purpose of this study was to investigate the role of TRPV1-mediated calcium flux from extracellular sources as an initiator of these responses and to define additional cellular pathways that control cell death. TRPV1 antagonists and reduction of calcium concentrations in treatment solutions attenuated calcium flux, induction of interleukin-6 and 8 gene expression, and IL-6 secretion by cells treated with capsaicin or resiniferatoxin. Most TRPV1 antagonists also attenuated cell death, but the relative potency and extent of protection did not directly correlate with inhibition of total calcium flux. Treatment solutions with reduced calcium content or chelators had no effect on cytotoxicity. Inhibitors of arachidonic acid metabolism and cyclo-oxygenases also prevented cell death indicating that TRPV1 agonists disrupted basal arachidonic acid metabolism and altered cyclo-oxygenase function via a TRPV1-dependent mechanism in order to produce toxicity. These data confirm previous results demonstrating calcium flux through TRPV1 acts as a trigger for cytokine production by vanilloids, and provides new mechanistic insights on mechanisms of cell death produced by TRPV1 agonists in respiratory epithelial cells.
Keywords: Capsaicin, Resiniferatoxin, TRPV1, Calcium, Cyclo-oxygenase (COX), Cytotoxicity, Inflammation
INTRODUCTION
The capsaicin receptor (VR1 or TRPV1) has been described as a temperature- (>42°C), pH- (<6.4 at 37°C), and vanilloid-sensitive homo-tetrameric cation channel exhibiting moderate selectivity for calcium (pCa++:pNa+ = 9.6) [1–3]. TRPV1 expression was initially demonstrated in peripheral afferent sensory nerve fibers (C- and Aδ-) originating from dorsal root ganglia [1], but ongoing studies have also revealed expression and function in a variety of nonneuronal tissues and cell types [4] including keratinocytes [5] and lung epithelial cells [6–8]. TRPV1 subunits consist of six transmembrane domains, a putative pore loop region, and cytosolic N- and C-terminal domains that possess a variety of regulatory features including multiple phosphorylation sites [9–12], subcellular localization sequences [13], a calcium/calmodulin binding site [14], and a phosphatidylinositol diphosphate (PiP2) binding site [15]. Differences in binding of regulatory constituents at these sites negatively and positively regulate channel gating thresholds and calcium flux. It has been proposed that these features play a definitive role in “fine-tuning” receptor responses to agonists under diverse physiological states. The relevance of these structural and functional aspects of TRPV1 to vanilloid-induced cell death and cytokine production is essentially unknown.
In human airway epithelial cells, TRPV1 has been shown to regulate inflammatory cytokine production following exposure to concentrated ambient particulate pollutants and TRPV1 agonists [8, 16–19]. A direct relationship was shown for TRPV1 activation by negatively charged particles and the production of cytokines and apoptotic cell death in several respiratory epithelial cell lines including BEAS-2B, A549, NHBE, and a small alveolar epithelial cell (SAEC) line.
Our laboratory has investigated mechanisms by which pepper sprays produce toxicity in respiratory tissues; the active ingredients in pepper spray products are the capsaicinoids [7]. Rats exposed to capsaicin by nose-only inhalation exhibited marked inflammation, the appearance of neutrophils and proliferating macrophages, and extensive damage to tracheal epithelial, bronchial epithelial, and alveolar cells [7]. Complimentary in vitro studies demonstrated that TRPV1 could mediate both pro-inflammatory cytokine production and cell death. It was found that the level of TRPV1 expression in multiple cell types (i.e., BEAS-2B, A549, and HepG2) correlated to their relative sensitivity to TRPV1 agonist-induced toxicities. Furthermore, selective overexpression of TRPV1 in BEAS-2B cells conferred greater (~100–200-fold) sensitivity to agonists using IL-6 production and cell death as endpoints. However, a confounding outcome of these studies was that capsazepine, the prototypical TRPV1 antagonist, and EGTA, a calcium chelator, did not prevent the cytotoxicity of capsaicin or resiniferatoxin (RTX) in either BEAS-2B or TRPV1-overexpressing cells, despite inhibiting cytokine production. Inhibition of alternate TRPV1-independent mechanisms of cell death (e.g., capsaicin-dependent inhibition of protein synthesis, ROS production, etc.) [20–23] was investigated, but was not involved in this unique cell death process.
In this study, we used a number of novel, selective, and potent antagonists of TRPV1 [24–27] and overt manipulations to extracellular calcium content to define the precise role of TRPV1-mediated calcium flux from extracellular sources in the production of pro-inflammatory cytokines and cell death in response to treatment with prototypical TRPV1 agonists. Many of these antagonists have been shown to exhibit greater potency, affinity, and selectivity for TRPV1 than capsazepine and, thus, may provide additional insights into the biochemical basis of TRPV1-mediated toxicities. Additional studies to identify specific components of the cell death process following TRPV1 activation were also performed.
MATERIALS AND METHODS
Chemicals
Capsaicin (n-vanillylnonanamide) (97%), capsazepine (CPZ), ionomycin, 5,8,11,14-eicosatetraynoic acid (ETYA), indomethacin, acetylsalicylic acid, etodolac, diclofenac, ABTS (2,2’-azino-bis-[ethylbenzo-thiazoline-6-sulfonic acid]-diammonium salt), sulfinpyrazone, Tween-20, thapsigargin, EGTA, ruthenium red, and 30% hydrogen peroxide (H2O2) were purchased from Sigma Chemical Corporation (St. Louis, MO). RTX and 5-iodo-RTX were purchased from Alexis Biochemicals (San Diego, CA). Fluo-4-AM was purchased from Molecular Probes (Eugene, OR). The synthesis and characterization of SC0030 (N-(4-tert-butylbenzyl)-N’-[3-fluoro-4-(methylsulfonylamino)-benzyl]thiourea), JYL-1433, and KMJ-642 have been previously described [26,27]. Synthesis of LJO-328 (N-(4-tert-butylbenzyl)-N’-(1-[3-fluoro-4-(methylsulfonylamino)phenyl]ethyl)thiourea), an α-substituted SC0030 analogue, is presented in the following patent document (WO 2005/003084). Two proprietary competitive TRPV1 antagonists having structures similar to KMJ-642, LJO-328, JYL-1433, and SC0030 (antagonists A and B) were also obtained from Dr. Jeewoo Lee. For reference, the chemical structures of LJO-328, SC0030, JYL-1433, KMJ-642, 5-iodo-RTX, and capsazepine are shown in Figure 1; structures for antagonists A and B are proprietary. All other chemicals were purchased from established chemical suppliers.
FIGURE 1.
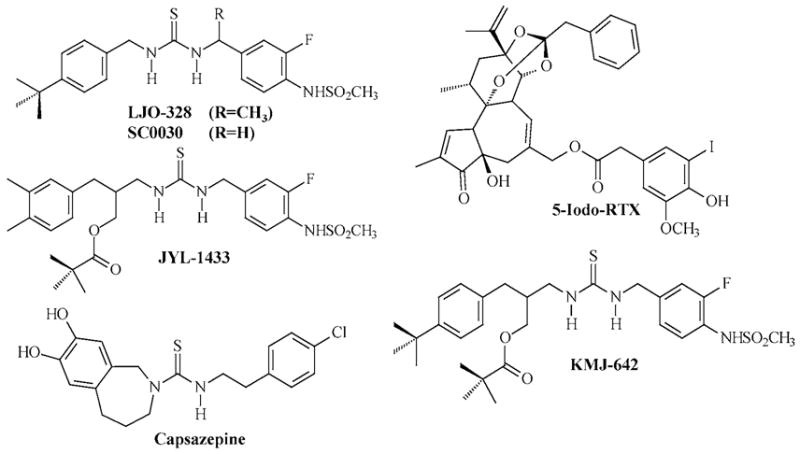
Chemical structures of LJO-328, SC0030, JYL-1433, KMJ-642, 5-iodo-RTX, and capsazepine.
Cell Culture
BEAS-2B bronchial epithelial cells (CRL-9609) were purchased from ATCC (Rockville, MD). TRPV1-overexpressing cells were generated by transfecting BEAS-2B with the human TRPV1 cDNA cloned into the pcDNA 3.1D-V5/His6 mammalian expression vector (InVitrogen, Carlsbad, CA) and selecting for stably transformed cells, as previously described [7]. Cells were cultured in LHC-9 media (BioSource, Camarillo, CA) in coated polystyrene cell culture flasks. The plate coat consisted of LHC basal media fortified with collagen (30 μg/mL), fibronectin (10 μg/mL), and bovine serum albumin (10 μg/mL). Cells were maintained between 30–90% confluence and were passaged every 2–4 days. Some experiments assessing the effects of calcium on cytotoxicity and cytokine responses were performed in keratinocyte growth medium (KGM) complete with calcium and deficient in calcium (KGM 2) (Cambrex Bioscience, Walkersville, MD). Cells were cultured for 2 h in complete or calcium-free KGM prior to treatments.
Fluorometric Calcium Assays
Cells were subcultured into coated 96 well cell culture plates and grown to ~90% confluence over 48 h. Cells were loaded with Fluo-4-AM (2.5 μM), a membrane permeable fluorogenic calcium indicator, for 90 min at room temperature (~22°C) in LHC-9 media containing 200 μM sulfinpyrazone, washed with LHC-9, and incubated at room temperature for an additional 20–30 min to permit methyl ester hydrolysis and activation of Fluo 4 within the cells. All loading steps were performed in the dark. Changes in cellular fluorescence in response to agonist and antagonist treatments were assessed microscopically at 10× magnification on cell populations (~500 cells in a field) using a Nikon Diaphot inverted microscope, equipped with a filter set designed to visualize green fluorescent protein. Fluoromicrographs were captured at high resolution using a SPOT Insight QE digital camera interfaced with the SPOT data system software (Diagnostic Instruments, Inc., Sterling Heights, MI). Images were collected immediately prior to the addition of the various substances and 1 min after treatment. All agonist and antagonist solutions were prepared in LHC-9 (or KMG and KGM 2) and were added to the cells in 50–100 μL volumes at room temperature. Antagonists were added to cells 1 min prior to agonist exposure. Data are presented as the mean fluorescence intensity, and standard deviation for cell populations is determined using the NIH Image J software package.
Cytotoxicity Assays
Cells were subcultured into coated multiwell cell culture plates and allowed to reach ~80% confluence over 24 h. The cells were washed once with sterile phosphate-buffered saline and treated for 24 h with the various agonists and antagonists. Treatment solutions were prepared in LHC-9 (or KGM and KGM 2). Cells were treated with antagonists for 30 min prior to agonist treatments and were included in the agonist treatment solutions. Cell viability was assessed using the Dojindo Cell Counting Kit-8 (Dojindo Laboratories, Gaithersburg, MD), according to the supplier recommendations. Cell viability was determined spectrophotometrically assaying for the formation of a water-soluble formazan dye product produced by active mitochondrial dehydrogenase enzymes in viable cells. Data are expressed as the percentage of viable cells relative to untreated control cells.
RT-PCR Analysis of Cytokine Gene Expression
Cells were subcultured into coated 25 cm2 cell culture flasks and grown to a density of ~80% over 48 h. Cells were treated with capsaicin in the presence and absence of various antagonists for 4 h at 37°C. Treatment solutions were prepared in LHC-9. Cells were treated with antagonists for 30 min prior to agonist exposure and were also included in the treatment solutions at the specified concentrations. Total RNA was extracted from the cells using the RNeasy mini RNA isolation kit (Qiagen, Valencia, CA), quantified using the UV absorbance ratio at 260/280 nm, and 5 μg of total RNA transcribed into cDNA using Superscript II (InVitrogen, Carlsbad, CA). IL-6, IL-8, and β-actin cDNA was selectively amplified by PCR from 2.5 μL of the cDNA synthesis reaction and the following primers: IL-6 sense 5’-CTTCTCCACAAGCGCCTTC-3’ and antisense 5’-GGCAAGTCTCCTCATTGAATC-3’ (325 nt), IL-8 sense 5’-GTGGCTCTCTTGGCAGCCTTC-3’ and antisense 5’-CAGGAATCTTGTATTGCATCTG-3’ (410 nt), and β-actin sense 5’-GACAACGGCTCCGGCATGTGCA-3’ and antisense 5’-TGAGGATGCCTCTCTTGCTCTG-3’ (183 nt). The PCR program consisted of an initial 2 min incubation at 94°C and 28 cycles of 94°C (30 s), 55°C (30 s), and 72°C (30 s). A final extension period of 10 min at 72°C was also included. PCR products were resolved on a 1% sodium borate agarose gel, and the images were analyzed using a Bio-Rad Gel-Doc imaging system. Relative band intensities for each PCR product relative to the internal PCR control (β-actin) are reported.
IL-6 ELISA
Cells were subcultured into 24 or 48-well coated cell culture dishes at ~40% confluence and maintained for 48 h until a confluence of ~80% was achieved. Treatments were performed in LHC-9 (or KGM, KGM 2) fortified with capsaicin and the various modulators of TRPV1 function for 24 h. Cells were treated with antagonists for 30 min prior to agonist treatment. After 24 h, the media was collected, clarified by centrifugation, and stored at −20°C until assayed for IL-6 content.
IL-6 production was quantified using a validated ELISA assay developed in our laboratory. Briefly, Nunc MaxiSorb 96-well plates (Fischer Scientific) were coated for 12 h at 4°C using a rat IgG1 monoclonal anti-human IL-6 antibody (eBioscience, San Diego, CA) at a concentration of 1 μg/mL in 100 mM sodium carbonate buffer, pH 9.5. The coating solution was removed, washed three times with phosphate-buffered saline (PBS) containing 0.05% Tween-20, and incubated for 1 h at room temperature with PBS containing 10% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT). Samples and standards (100 μL) were aliquoted into the wells and incubated at room temperature for 2 h. The samples were removed; the plate was washed five times and incubated for an additional 2 h at room temperature with PBS + FBS containing 1 μg/mL of an affinity purified biotinylated rat IgG2A monoclonal anti-human IL-6 antibody (eBioscience). The plate was washed five times and incubated for 30 min at room temperature with a horseradish peroxidase-avidin conjugate (eBioscience) diluted 1/1000 in PBS + FBS. The wells were washed five times and developed for 30–60 min at room temperature by incubating with 0.03% H2O2 and 0.55 mM ABTS in 100 mM citrate buffer, pH 4.4. Reactions were terminated by addition of 50 μL of 20% SDS: 50% dimethyformamide and the concentration of IL-6 calculated using the absorbance at 405 nm and a semilog calibration curve constructed using recombinant human IL-6 as the standard (R&D Systems, Minneapolis, MN). The limit of quantitation (LOQ) for this assay was 20 pg/mL. All experiments were performed in triplicate.
Statistical Analysis
Data were analyzed for statistical significance using ANOVA, paired t-tests, and posttesting using Dunnett’s test. Statistically significant differences in responses are represented within the figures and are described in the figure legends.
RESULTS
TRPV1-overexpressing BEAS-2B cells treated with the prototypical TRPV1 agonists, RTX, and capsaicin, exhibited dose-dependent increases in cellular fluorescence relative to untreated control cells (Figure 2A). EC50 values for the induction of calcium flux were 0.4 ± 0.1 and 1.0 ± 0.4 μM for RTX and capsaicin, respectively. Agonist induced calcium flux was decreased by ~45% in reduced calcium media (open bars), and further decreased by ~6% when EGTA (100 μM) and the polar, nonselective calcium channel blocker, ruthenium red (10 μM), were included in the treatment solutions (solid bars) (Figure 2B). Depletion of endoplasmic reticulum stores of calcium with thapsigargin (gray bars) decreased calcium flux by ~65 and 85% in calcium deficient and complete media, respectively (Figure 2B). These data indicated that ~45–50% of the total calcium flux observed was attributable to uptake from the treatment solutions. All TRPV1 selective antagonists inhibited calcium flux in a dose-dependent manner. IC50 values for the antagonists are presented in Table 1. The rank order for inhibition of calcium flux was SC0030, capsazepine and 5-iodo-RTX, antagonist A, JYL-1433, KMJ-642, antagonist B, and LJO-328.
FIGURE 2.
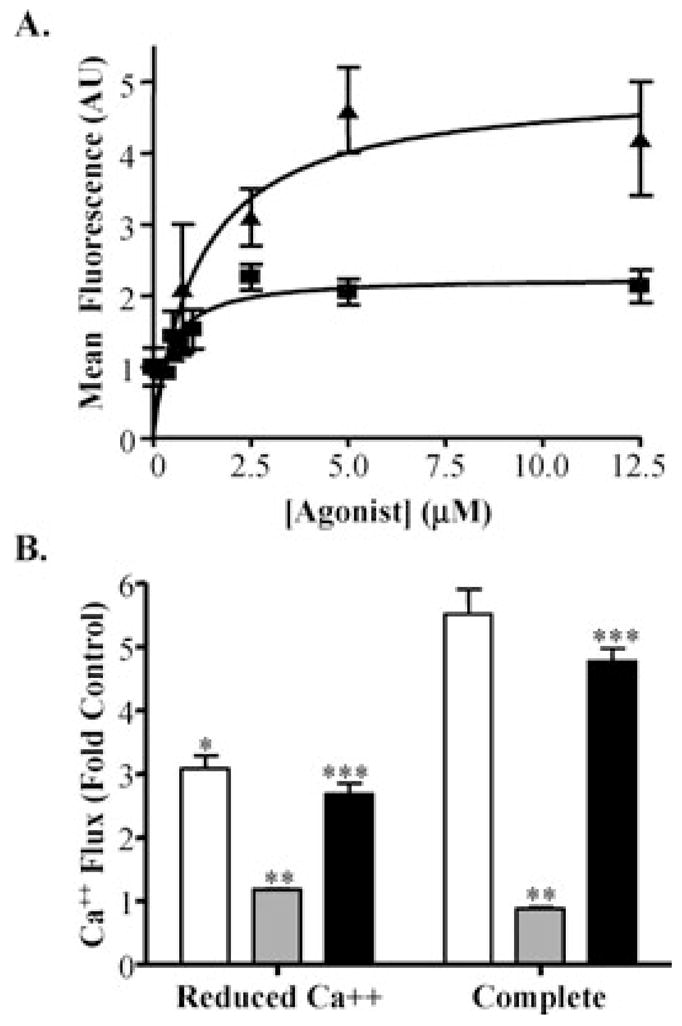
(A) Dose-response data for the induction of intracellular calcium flux in TRPV1-overexpressing cells by RTX (squares) and capsaicin (triangles). Data represent the mean fluorescence values for cell populations and standard deviation (n = 4). EC50 values were obtained by nonlinear regression analysis (Prism 4, GraphPad Software, Inc., San Diego, CA) using the one-site binding model. (B) Attenuated capsaicin-induced (20 μM) calcium flux (open bars) in TRPV1-overexpressing cells using reduced calcium solutions (left group), depletion of ER-calcium stores with thapsigargin (1.5 μM, 5 min) (gray bars), and treating with 100 μM EGTA and 10 μM ruthenium red (black bars). Data represent the mean fluorescence values for cell populations and standard deviation (n = 4). *Statistically significant decreases relative to complete media, **significant decreases due to depletion of ER calcium stores, and ***additional decreases afforded by EGTA and ruthenium red (p ≤ 0.05) are identified.
TABLE 1.
IC50 Values for the Inhibition of RTX-and Capsaicin-Induced Calcium Flux Using Various TRPV1 Antagonists
| TRPV1 Inhibitor | IC50 (μM)a |
|---|---|
| SC0030 | 0.3 ± 0.1 |
| Capsazepine | 0.5 ± 0.3 |
| 5-Iodo-RTX | 0.6 ± 0.3 |
| Antagonist A | 0.8 ± 0.2 |
| JYL-1433 | 3 ± 2 |
| KMJ-642 | 3.0 ± 0.5 |
| Antagonist B | 5 ± 2 |
| LJO-328 | 7 ± 4 |
| LJO-328 (Capsaicin) | 0.8 ± 0.1 |
IC50 values were determined from semilog plots using the one-site competition model provided in the GraphPad Software package. Data represent the mean and standard deviation (n = 4).
Inhibition of cell death by various TRPV1-selective antagonists was also assessed. Figures 3A and 3B present dose-response data for the inhibition of cell death by TRPV1 antagonists. 5-Iodo-RTX was the most potent inhibitor of capsaicin toxicity followed by SC0030, KMJ-642, antagonist A, JYL-1433, LJO-328, and antagonist B. The rank order for the degree of protection provided by the effective antagonists was 5-iodo-RTX, LJO-328, antagonist A, SC0030, antagonist B, JYL-1433, KMJ-642, and capsazepine; decreases in cell viability at high antagonist concentrations were due to the toxicity of the antagonists themselves. Interestingly, capsazepine did not prevent cell death while KMJ-642 provided only minimal protection, despite the ability of both antagonists to prevent calcium flux. Figure 3C compares the inhibition of capsaicin- and RTX-induced cell death by 5-iodo-RTX and LJO-328. Threshold concentrations of LJO-328 that prevented cell death were >5–7.5 μM for capsaicin and >10 μM for RTX, consistent with RTX being a more potent and selective TRPV1 agonist with a lower Kd than capsaicin [28,29]. 5-Iodo-RTX was the most potent inhibitor of cell death induced by RTX, but also required a minimum ratio of ~5:1 to be effective despite having a Kd similar to RTX itself (Figure 3C). An approximate 25-fold increase in the LD50 for capsaicin was observed when LJO-328 was included in treatment solutions (Figure 3D), confirming results from Figure 3B that a minimum ratio of ~5–10:1 LJO-328:capsaicin was required to compete for TRPV1 binding and to mitigate toxicity by this antagonist. A ratio >5–10:1 was also required for all of the other antagonists tested (Figures 3A and 3B).
FIGURE 3.
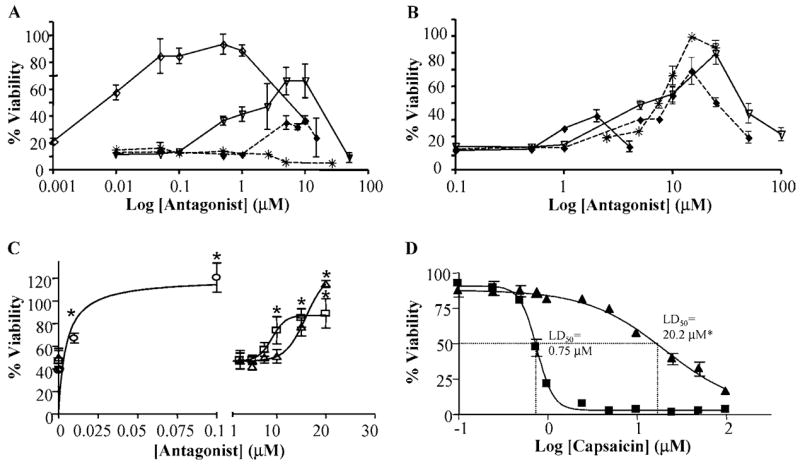
(A) Inhibition of cell death (1 μM capsaicin) in TRPV1-overexpressing cells by various TRPV1 selective antagonists. SC0030 (upside-down open triangles, solid line), JYL-1433 (filled diamonds, dashed line), capsazepine (stars, dashed line), and 5-iodo-RTX (open diamonds, solid line). (B) Inhibition of cell death by LJO-328 (stars, dashed line), KMJ-642 (filled diamonds, solid line), antagonist A (upside-down open triangles, solid line), and antagonist B (filled diamonds, dashed line). Data are representative of the mean viability and standard deviation (n = 3). For clarity, statistical significance has not been noted in the figures. (C) The effects of LJO-328 and 5-iodo-RTX on cell death induced by vanilloid treatment. TRPV1-overexpressing cells were treated with 1 μM capsaicin or 10 nM RTX with increasing concentrations of LJO-328 or 5-iodo-RTX for 24 h. Data represent the mean and standard deviation (n = 3). Data are as follows: 10 nM RTX plus 5-iodo-RTX (circles), 10 nM RTX plus LJO-328 (triangles), and 1 μM capsaicin plus LJO-328 (squares). Statistically significant changes in cell viability relative to capsaicin- or RTX-treated controls (p ≤ 0.05) are identified with an asterisk. (D) Dose-response cytotoxicity data for TRPV1-overexpressing cells treated with increasing concentrations of capsaicin in the presence (triangles) and absence of 20 μM LJO-328 (squares). Data represent the mean and standard deviation (n = 4).
Several TRPV1 antagonists were also assessed for modulation of agonist-induced cytokine responses. IL-6 and 8 are common biomarkers of cellular injury and the induction of acute pro-inflammatory processes. Cells treated with capsaicin exhibited significant (2.5-and 8-fold) increases in the relative abundance of IL-6 and IL-8 mRNA transcripts (Figure 4A) in response to capsaicin treatment. IL-6 and IL-8 gene induction was markedly suppressed by LJO-328 (Figure 4A), as well as by capsazepine, SC0030, EGTA, and ruthenium red (Figure 4B). Differences in the inhibition patterns for IL-6 and IL-8 were observed with EGTA and ruthenium red. These effects appeared to be a direct result of antagonist/chelator treatment rather than influences on TRPV1-mediated functions since increases in IL-6 occurred in control samples (data not shown). Addition of LJO-328 to treatment solutions completely blocked the induction of IL-6 secretion by cells treated with capsaicin and RTX in a concentration-dependent manner (Figure 4C). Concentrations of LJO-328 that blocked IL-6 production were approximately twofold lower for capsaicin than RTX, similar to the trends observed for inhibition of cell death.
FIGURE 4.
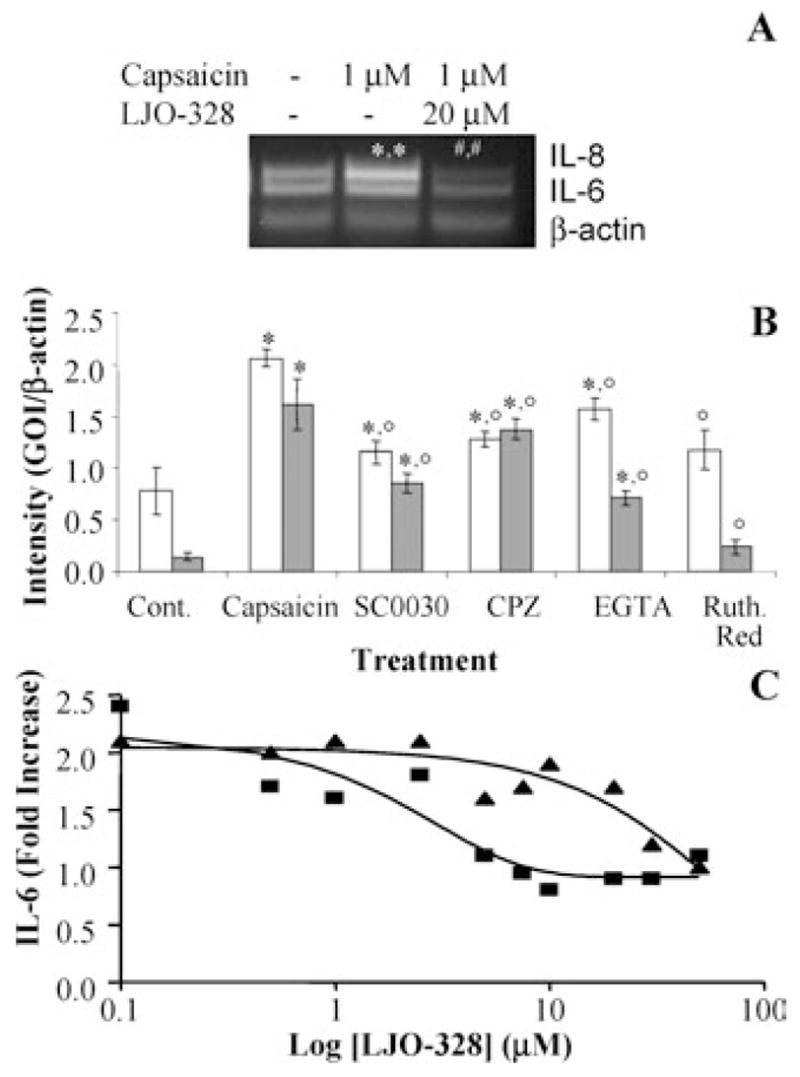
(A) Induction of IL-6 and IL-8 gene expression in TRPV1-overexpressing cells by capsaicin and inhibition by LJO-328. Cells were treated as shown in the figure for 4 h, harvested, and changes in gene expression assessed by RT-PCR, as described under the materials and methods section. (*) Represents a statistical increase over untreated control cells while (#) represents significant differences from treated and control cells. (B) The effects of multiple TRPV1 antagonists and modulators of TRPV1 function or calcium concentration on the induction of IL-6 (open bars) and IL-8 (shaded bars) genes in TRPV1-overexpressing cells treated with capsaicin (1 μM) for 4 h at 37°C. Concentrations of antagonist were SC0030 (100 nM), capsazepine (15 μM), EGTA (75 μM), and ruthenium red (150 μM). Points at which statistically greater levels of gene expression were observed versus untreated control cells are indicated by an asterisk (*), while lower levels of expression relative to capsaicin-treated cells are represented with an open circle (°). Data represent the mean and standard deviation (n = 5). (C) Inhibition of capsaicin- and RTX-induced IL-6 secretion by TRPV1-overexpressing cells with LJO-328. Cells were treated with increasing concentrations of LJO-328 and capsaicin (1 μM) (squares) or RTX (10 nM) (triangles) for 24 h at 37°C. IL-6 concentration in media was determined by ELISA using pooled samples (n = 3). The concentration of IL-6 in untreated control cells was ~265 pg/mL.
Additional experiments to fully elucidate the significance of extracellular to intracellular calcium flux in TRPV1 agonist-induced cell death and cytokine production were performed by treating TRPV1-overexpressing cells with increasing concentrations of capsaicin in either complete (KGM) or calcium-deficient (KGM 2) cell culture media. Inhibition of cell death was not observed in reduced calcium media (Figure 5A), while IL-6 production was completely prevented (Figure 5B).
FIGURE 5.
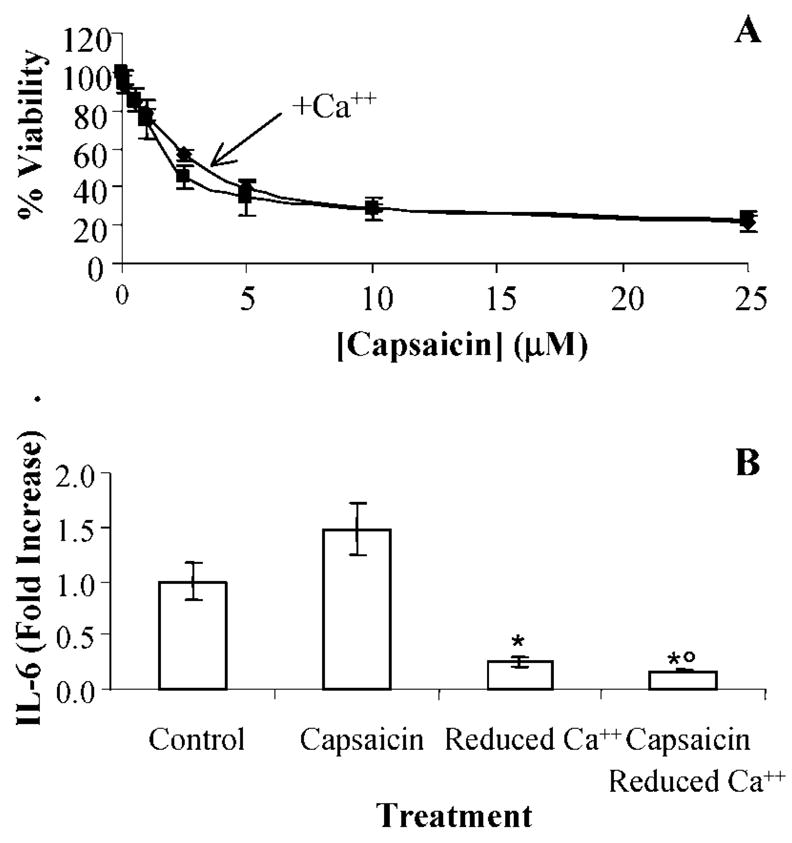
(A) Dose-response cytotoxicity data for TRPV1-overexpressing cells treated with increasing concentrations of capsaicin in complete (diamonds) and calcium deficient (squares) cell culture media. Data represent the mean and standard deviation (n = 4). Statistical differences (p ≤ 0.05) were not observed. (B) Inhibition of IL-6 production by cells treated with capsaicin (1 μM) in complete and calcium-deficient cell culture media. The concentration of IL-6 in media collected from untreated cells was 270 ± 50 pg/mL. IL-6 was lower than complete cell culture media (*) and in cells treated with capsaicin (°) (p ≤ 0.05). Data represent the mean and standard deviation (n = 3).
Co-treatment of cells with capsaicin and inhibitors of arachidonic acid metabolism (ETYA) and cyclo-oxygenase (COX) enzymes (indomethacin, etodolac, aspirin, and diclofenac) also showed significant inhibition of cell death (Figure 6). LJO-328 and capsazepine were evaluated for inhibition of recombinant human COX-1 and COX-2 in vitro; however, neither TRPV1 antagonist was active (data not shown) demonstrating that LJO-328 and the other TRPV1 antagonists exerted their protective effects through TRPV1 inhibition and not by altering associated cell death pathways.
FIGURE 6.
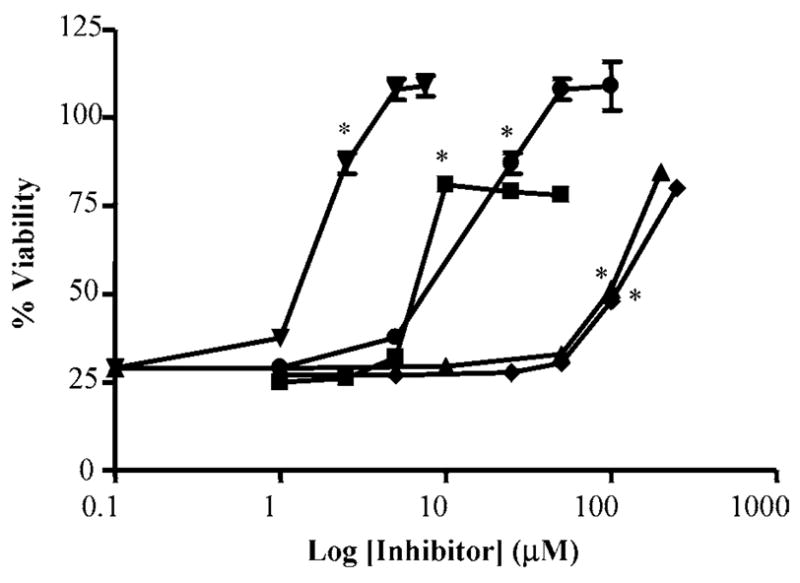
Inhibition of cell death by inhibitors of arachidonic acid metabolism and COX activity. Inhibition of cell death induced by capsaicin (1 μM) in TRPV1-overexpressing cells using ETYA (upside down triangles), indomethacin (squares), etodolac (triangles), acetyl-salicylic acid (diamonds), and diclofenac (circles). Error bars less than 5% are not shown. Data represent the mean and standard deviation (n = 3). The lowest concentrations of inhibitor at which statistical significance (p ≤ 0.05) was observed are indicated with an asterisk (*).
DISCUSSION
TRPV1 is a vital component of mammalian sensory function. However, definitive physiological functions of TRPV1 and the consequences of activation in respiratory epithelial cells have not been fully established. It has been suggested that TRPV1 serves as a molecular sensor of potentially noxious inhaled environmental substances, whose activation acts, in concert with neuronal TRPV1, to initiate protective defense mechanisms including cough and immune responses. Data presented here support this idea and confirm an essential role for TRPV1 in promoting pro-inflammatory cytokine production by bronchial epithelial cells via a mechanism that was dependent upon the ability of TRPV1 to mediate the transfer of calcium ions from the extracellular matrix into the cytosol.
Data are also presented that highlight the existence of ancillary, deleterious TRPV1-mediated processes that ultimately lead to cell death. These processes were independent of extracellular calcium content and cellular uptake through TRPV1. These results contrasted with the neurotoxic mechanism of capsaicinoids, which involves agonist-induced influx and prolonged accumulation of calcium (termed excitotoxicity) [30]. Excitotoxic mechanisms have also been shown for Jurkat, HEK293, and CHO cells that transiently overexpressed recombinant rat TRPV1 [31–33]. In all instances, toxicity was inhibited by capsazepine and treatments that interfered with TRPV1-mediated calcium flux and accumulation in cells. Here, we clearly demonstrate the existence of an alternate mechanism for cell death by TRPV1 agonists, one that was not influenced by changes in extracellular calcium content or inhibited by capsazepine. Rather cell death involved disruption of normal COX function following TRPV1 activation.
Our previous studies [7] provided preliminary evidence that the cytotoxicity of capsaicinoids and other TRPV1 agonists to BEAS-2B and TRPV1-overexpressing cells occurred via a mechanism that was independent of calcium. These studies provide additional mechanistic insights and support this conclusion. Most TRPV1 antagonists prevented cell death to some degree (Figures 3A and 3B). However, inhibition of cell death by TRPV1-selective antagonists did not directly correlate with their ability to attenuate calcium flux (Table 1 and Figures 3A and 3B). For example, capsazepine and KMJ-642 were potent inhibitors of calcium flux, yet little to no protection against cytotoxicity was observed using these two antagonists (Figure 3A). Similarly, removal of calcium from the treatment solutions had no effect on cell death by TRPV1 agonists (Figure 5), despite having significant impact on overall agonist-induced calcium flux and cytokine responses.
Three hypotheses to describe the mechanism(s) of cell death were considered. A concomitant increase in intracellular sodium, as a result of TRPV1 activation, has been suggested as a possible mechanism for cell death. However, this hypothesis was dismissed because TRPV1 inhibitors (e.g., capsazepine) have been shown to concomitantly inhibit both calcium and sodium flux [33]. If the influx of sodium ions through TRPV1 caused cell death, then inhibition of cell death would correlate directly with the relative IC50 values of the antagonists. This correlation was not observed. A second potential calcium independent mechanism of cytotoxicity that also account for the level of TRPV1 expression as a determinant of sensitivity was that agonist-induced tetramerization of TRPV1 subunits [2] served as an intrinsic cell death signal. However, tetramer formation has also been shown to be blocked by TRPV1 antagonists, such as capsazepine, and calcium chelators such as EGTA [2]. Unfortunately, neither of these substances prevented cell death. A third hypothesis to explain the data presented in this study stated that cell death occurred via the activation of intracellular, ER-bound TRPV1 to promote toxicity. This mechanism explains data that shows a strong correlation between TRPV1 expression and the inability of extracellular modifications to calcium content to alter cell death processes. Data presented in Figure 2B support the existence of two distinct populations of TRPV1 in these cells and provide significant support for this proposed mechanism.
Additional studies to identify prodeath pathways associated with TRPV1 signaling demonstrated a role for altered arachidonic acid homeostasis and COX metabolism (Figure 6). Previous research has demonstrated the selective upregulation of COX-2 and PGE2 by BEAS-2B cells following treatment with residual oil-fly ash (ROFA) [17,34,35], an activator of TRPV1 [17], and in keratinocytes treated with capsaicin [5]. Non-specific inhibition of arachidonic acid metabolism using ETYA and COX inhibition using various NSAIDs prevented cell death by capsaicin (Figure 6). These data indicate that changes in COX-mediated metabolism via a TRPV1 mediated process contribute to the cell death cascade.
Collectively, these studies expand our current understanding of the mechanism(s) by which TRPV1 mediates pro-inflammatory and cell death processes in BEAS-2B cells exposed to capsaicin, RTX, and, likely, other TRPV1 agonists. The presence of multiple functionally distinct subcellular pools of TRPV1 has been shown and processes integrally related to TRPV1 function in cells have been identified. The proposed mechanism(s) of cytokine induction and cell death are presented graphically in Figure 7. Based on our results, it may be reasonable to predict that variations in the regulation of TRPV1 function (e.g., PIP2 or calmodulin binding, phosphorylation, etc.), that can be influenced by genetic and/or environmental factors, may have a significant impact on the toxicities of various TRPV1 agonists in vivo, depending upon which pool of TRPV1 that becomes activated. Likewise, differences in TRPV1 expression and location, rather than differences in channel gating thresholds and receptor activity, will likely govern the cytotoxic potential of lipophilic TRPV1 agonists in vivo. Together, these data elicit a number of intriguing hypotheses regarding the potential role of TRPV1 in mediating airway toxicities by chemically and physically distinct substances and suggest a potential role for this receptor in mediating environmentally influenced airway diseases such as asthma, COPD, or ARDS.
FIGURE 7.
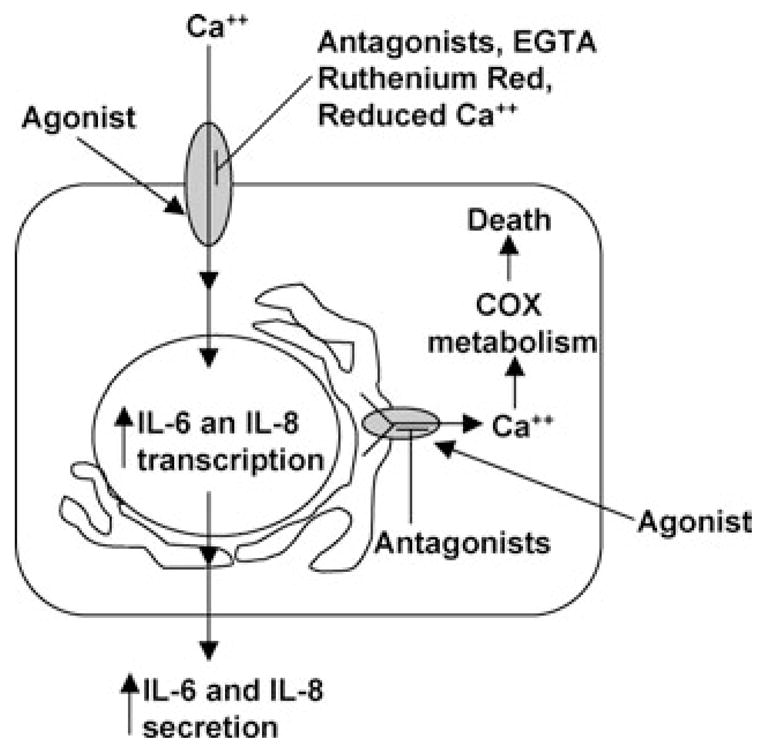
Schematic representation of the proposed mechanism(s) for extracellular calcium-dependent cytokine production and extracellular calcium-independent cell death in TRPV1-overexpressing cells. TRPV1 is represented as shaded ovals on the plasma membrane and endoplasmic reticulum of cells. TRPV1-selective antagonists and overt modifications to extracellular calcium content selectively inhibit cytokine responses, but not cell death. Only lipophilic TRPV1 antagonists prevent cell death, presumably by inhibiting intracellular, thapsigargin-sensitive TRPV1-mediated activities.
Acknowledgments
The authors would like to thank Drs. John Veranth, Alan Light, and Peter Blumberg for assistance with experimental procedures and insightful comments on this manuscript. We would also like to express our gratitude to Dr. Dan Simmons and Bryant Oliverson for providing data on cyclo-oxygenase inhibition.
Contract Grant Sponsor: National Heart, Lung, and Blood Institute.
Contract Grant Number: HL069813.
Footnotes
Christopher A. Reilly and Mark E. Johansen contributed equally to the work presented in this manuscript.
References
- 1.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 2.Kedei N, Szabo T, Lile JD, Treanor JJ, Olah Z, Iadarola MJ, Blumberg PM. Analysis of the native quaternary structure of vanilloid receptor 1. J Biol Chem. 2001;276:28613–28619. doi: 10.1074/jbc.M103272200. [DOI] [PubMed] [Google Scholar]
- 3.Benham CD, Gunthorpe MJ, Davis JB. TRPV channels as temperature sensors. Cell Calcium. 2004;33:479–487. doi: 10.1016/s0143-4160(03)00063-0. [DOI] [PubMed] [Google Scholar]
- 4.Hayes P, Meadows HJ, Gunthorpe MJ, Harries MH, Duckworth DM, Cairns W, Harrison DC, Clarke CE, Ellington K, Prinjha RK, Barton AJ, Medhurst AD, Smith GD, Topp S, Murdock P, Sanger GJ, Terrett J, Jenkins O, Benham CD, Randall AD, Gloger IS, Davis JB. Cloning and functional expression of a human orthologue of rat vanilloid receptor-1. Pain. 2000;88:205–215. doi: 10.1016/S0304-3959(00)00353-5. [DOI] [PubMed] [Google Scholar]
- 5.Southall MD, Li T, Gharibova LS, Pei Y, Nicol GD, Travers JB. Activation of epidermal vanilloid receptor-1 induces release of proinflammatory mediators in human keratinocytes. J Pharmacol Exp Ther. 2003;304:217–222. doi: 10.1124/jpet.102.040675. [DOI] [PubMed] [Google Scholar]
- 6.Veronesi B, Carter JD, Devlin RB, Simon SA, Oortgiesen M. Neuropeptides and capsaicin stimulate the release of inflammatory cytokines in a human bronchial epithelial cell line. Neuropeptides. 1999;33:447–456. doi: 10.1054/npep.1999.0761. [DOI] [PubMed] [Google Scholar]
- 7.Reilly CA, Taylor JL, Lanza DL, Carr BA, Crouch DJ, Yost GS. Capsaicinoids cause inflammation and epithelial cell death through activation of vanilloid receptors. Toxicol Sci. 2003;73:170–181. doi: 10.1093/toxsci/kfg044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agopyan N, Head J, Yu S, Simon SA. TRPV1 receptors mediate particulate matter-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L563–L572. doi: 10.1152/ajplung.00299.2003. [DOI] [PubMed] [Google Scholar]
- 9.Crandall M, Kwash J, Yu W, White G. Activation of protein kinase C sensitizes human VR1 to capsaicin and to moderate decreases in pH at physiological temperatures in Xenopus oocytes. Pain. 2002;98:109–117. doi: 10.1016/s0304-3959(02)00034-9. [DOI] [PubMed] [Google Scholar]
- 10.Olah Z, Karai L, Iadarola MJ. Protein kinase C (alpha) is required for vanilloid receptor 1 activation. Evidence for multiple signaling pathways. J Biol Chem. 2002;277:35752–35759. doi: 10.1074/jbc.M201551200. [DOI] [PubMed] [Google Scholar]
- 11.Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, t Gereau RW. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci USA. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Premkumar LS, Qi ZH, Van Buren J, Raisinghani M. Enhancement of potency and efficacy of NADA by PKC-mediated phosphorylation of vanilloid receptor. J Neurophysiol. 2004;91:1442–1449. doi: 10.1152/jn.00745.2003. [DOI] [PubMed] [Google Scholar]
- 13.Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem. 2004;279:25665–25672. doi: 10.1074/jbc.M311515200. [DOI] [PubMed] [Google Scholar]
- 14.Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol. 2004;123:53–62. doi: 10.1085/jgp.200308906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284–1288. doi: 10.1126/science.1083646. [DOI] [PubMed] [Google Scholar]
- 16.Veronesi B, Oortgiesen M, Carter JD, Devlin RB. Particulate matter initiates inflammatory cytokine release by activation of capsaicin and acid receptors in a human bronchial epithelial cell line. Toxicol Appl Pharmacol. 1999;154:106–115. doi: 10.1006/taap.1998.8567. [DOI] [PubMed] [Google Scholar]
- 17.Oortgiesen M, Veronesi B, Eichenbaum G, Kiser PF, Simon SA. Residual oil fly ash and charged polymers activate epithelial cells and nociceptive sensory neurons. Am J Physiol Lung Cell Mol Physiol. 2000;278:L683–L695. doi: 10.1152/ajplung.2000.278.4.L683. [DOI] [PubMed] [Google Scholar]
- 18.Veronesi B, Wei G, Zeng JQ, Oortgiesen M. Electrostatic charge activates inflammatory vanilloid (VR1) receptors. Neurotoxicology. 2003;24:463–473. doi: 10.1016/S0161-813X(03)00022-6. [DOI] [PubMed] [Google Scholar]
- 19.Agopyan N, Bhatti T, Yu S, Simon SA. Vanilloid receptor activation by 2- and 10-microm particles induces responses leading to apoptosis in human airway epithelial cells. Toxicol Appl Pharmacol. 2003;192:21–35. doi: 10.1016/s0041-008x(03)00259-x. [DOI] [PubMed] [Google Scholar]
- 20.Cochereau C, Sanchez D, Creppy EE. Tyrosine prevents capsaicin-induced protein synthesis inhibition in cultured cells. Toxicology. 1997;117:133–139. doi: 10.1016/s0300-483x(96)03561-5. [DOI] [PubMed] [Google Scholar]
- 21.Richeux F, Cascante M, Ennamany R, Saboureau D, Creppy EE. Cytotoxicity and genotoxicity of capsaicin in human neuroblastoma cells SHSY-5Y. Arch Toxicol. 1997;73:403–409. doi: 10.1007/s002040050680. [DOI] [PubMed] [Google Scholar]
- 22.Creppy EE, Richeux F, Carratu MR, Cuomo V, Cochereau C, Ennamany R, Saboureau D. Cytotoxicity of capsaicin in monkey kidney cells: Lack of antagonistic effects of capsazepine and Ruthenium red. Arch Toxicol. 2000;74:40–47. doi: 10.1007/s002040050650. [DOI] [PubMed] [Google Scholar]
- 23.Richeux F, Cascante M, Ennamany R, Sanchez D, Sanni A, Saboureau D, Creppy EE. Implications of oxidative stress and inflammatory process in the cytotoxicity of capsaicin in human endothelial cells: Lack of DNA strand breakage. Toxicology. 2000;147:41–49. doi: 10.1016/s0300-483x(00)00184-0. [DOI] [PubMed] [Google Scholar]
- 24.Seabrook GR, Sutton KG, Jarolimek W, Hollingworth GJ, Teague S, Webb J, Clark N, Boyce S, Kerby J, Ali Z, Chou M, Middleton R, Kaczorowski G, Jones AB. Functional properties of the high-affinity TRPV1 (VR1) vanilloid receptor antagonist (4-hydroxy-5-iodo-3-methoxyphenylacetate ester) iodo-resiniferatoxin. J Pharmacol Exp Ther. 2002;303:1052–1060. doi: 10.1124/jpet.102.040394. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Szabo T, Welter JD, Toth A, Tran R, Lee J, Kang SU, Suh YG, Blumberg PM. High affinity antagonists of the vanilloid receptor. Mol Pharmacol. 2002;62:947–956. doi: 10.1124/mol.62.4.947. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Kang M, Shin M, Kim JM, Kang SU, Lim JO, Choi HK, Suh YG, Park HG, Oh U, Kim HD, Park YH, Ha HJ, Kim YH, Toth A, Wang Y, Tran R, Pearce LV, Lundberg DJ, Blumberg PM. N-(3-Acyloxy-2-benzylpropyl)-N’-[4-(methylsulfonylamino)benzyl]thiourea analogues: Novel potent and high affinity antagonists and partial antagonists of the vanilloid receptor. J Med Chem. 2003;46:3116–3126. doi: 10.1021/jm030089u. [DOI] [PubMed] [Google Scholar]
- 27.Lee J, Kang SU, Choi HK, Lim JO, Kil MJ, Jin MK, Kim KP, Sung JH, Chung SJ, Ha HJ, Kim YH, Pearce LV, Tran R, Lundberg DJ, Wang Y, Toth A, Blumberg PM. Analysis of structure-activity relationships for the ‘B-region’ of N-(3-acyloxy-2-benzylpropyl)-N(’)-[4-(methylsulfonylamino)benzyl]thiourea analogues as vanilloid receptor antagonists: Discovery of an N-hydroxythiourea analogue with potent analgesic activity. Bioorg Med Chem Lett. 2004;14:2291–2297. doi: 10.1016/j.bmcl.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Biro T, Maurer M, Modarres S, Lewin NE, Brodie C, Acs G, Acs P, Paus R, Blumberg PM. Characterization of functional vanilloid receptors expressed by mast cells. Blood. 1998;91:1332–1340. [PubMed] [Google Scholar]
- 29.Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- 30.Wood JN, Winter J, James IF, Rang HP, Yeats J, Bevan S. Capsaicin-induced ion fluxes in dorsal root ganglion cells in culture. J Neurosci. 1988;8:3208–3220. doi: 10.1523/JNEUROSCI.08-09-03208.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordt SE, Tominaga M, Julius D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci USA. 2000;97:8134–8139. doi: 10.1073/pnas.100129497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olah Z, Szabo T, Karai L, Hough C, Fields RD, Caudle RM, Blumberg PM, Iadarola MJ. Ligand-induced dynamic membrane changes and cell deletion conferred by vanilloid receptor 1. J Biol Chem. 2001;276:11021–11030. doi: 10.1074/jbc.M008392200. [DOI] [PubMed] [Google Scholar]
- 33.Grant ER, Dubin AE, Zhang SP, Zivin RA, Zhong Z. Simultaneous intracellular calcium and sodium flux imaging in human vanilloid receptor 1 (VR1)-transfected human embryonic kidney cells: A method to resolve ionic dependence of VR1-mediated cell death. J Pharmacol Exp Ther. 2002;300:9–17. doi: 10.1124/jpet.300.1.9. [DOI] [PubMed] [Google Scholar]
- 34.Samet JM, Ghio AJ, Costa DL, Madden MC. Increased expression of cyclooxygenase 2 mediates oil fly ash-induced lung injury. Exp Lung Res. 2000;26:57–69. doi: 10.1080/019021400269961. [DOI] [PubMed] [Google Scholar]
- 35.Samet JM, Reed W, Ghio AJ, Devlin RB, Carter JD, Dailey LA, Bromberg PA, Madden MC. Induction of prostaglandin H synthase 2 in human airway epithelial cells exposed to residual oil fly ash. Toxicol Appl Pharmacol. 1996;141:159–168. doi: 10.1006/taap.1996.0272. [DOI] [PubMed] [Google Scholar]