

Summary
In response to invading pathogens, Toll-like receptors (TLR) play a critical role in the initiation of the innate immune response, which can be either beneficial or detrimental to the host. In the present study, we demonstrated that central nervous system (CNS) glial cells are activated by Lymphocytic Choriomeningitis Virus (LCMV) in a TLR2-MyD88/Mal-dependent manner. Specifically, in response to LCMV, both astrocytes and microglial cells isolated from wild type (WT) mice produced chemokines, such as MCP-1, RANTES and TNF-α. Similar responses occurred in TLR3 KO and TLR4 KO glial cells. In striking contrast, both astrocytes and microglial cells isolated from mice deficient in TLR2, MyD88, and Mal did not produce any of these chemokines. In addition, LCMV infection of glial cells induced up-regulation of TLR2, MHC-class-I and II, CD40, CD86 in a MyD88-dependent manner. These results define a functional role for TLR signaling in viral infection-induced activation of CNS glial cells as well as for the immunopathology in the CNS.
Keywords: Lymphocytic Choriomeningitis Virus (LCMV), Toll-like receptors, CNS glial cells
Introduction
Toll-like receptors (TLRs) are important components of innate immunity (Finberg and Kurt-Jones, 2004; Medzhitov, 2001; Takeda and Akira, 2005). More than 10 TLRs have been identified so far (Kawai and Akira, 2007). Through the Toll/IL-1 receptor (TIR) domain, TLRs interact with their ligands, which are widely expressed in a variety of pathogens. All TLRs, with the exception of TLR3, use Myeloid Differentiation factor 88 (MyD88) as the adaptor molecule to activate the downstream signaling pathways. TLR3 ligand activates a TRIF-dependent signaling pathway. In addition, another TIR-domain containing adaptor, Mal (TIR domain-containing adaptor protein), is essential for both TLR2 and TLR4 activated MyD88-dependent signaling pathways. Following engagement with TLR ligands, TLR-dependent signals lead to the activation of the transcriptional factors nuclear factor κB (NF-κB), activator protein-1 (AP-1), or Interferon Regulatory Factors (IRFs), thus coordinately regulating host defense against microbial infections. There is increasing evidence that TLR/MyD88 mediated signals play a central role in the activation of the innate and adaptive immune response to microbial pathogens (Palliser et al., 2004; Pasare and Medzhitov, 2004; Schnare et al., 2001; Zhou et al., 2005). However, the exact mechanisms by which TLRs regulate viral pathogenesis are poorly defined (Lang et al., 2006). Astrocytes and microglial cells are the predominant non-neuronal parenchymal cells in the brain and are essential for maintaining CNS homeostasis (Hertz et al., 1990). They are crucial for the induction of innate immune responses, including secreting chemokines within the brain to protect neuronal cells from invading pathogens (Dong and Benveniste, 2001; Frei et al., 1994; Olson and Miller, 2004). On the other hand, glial cell-mediated inflammation in the brain can also contribute to damage of the CNS (Lehnardt et al., 2006). Chemokines are involved in the development of viral infection-mediated inflammatory diseases of the CNS and peripheral tissues (Allan et al., 1987; de Jong et al., 2006; Kamperschroer and Quinn, 2002). The production of chemokines in the brain parenchyma acts as a signal to attract pathogen-specific CD4+ and CD8+ T cells to the brain and therefore contributes to the control of infection but also to immune-mediated diseases (Kamperschroer and Quinn, 2002). Recent studies have demonstrated that both CNS astrocytes and microglial cells express TLRs (Bowman et al., 2003; Farina et al., 2005; McKimmie and Fazakerley, 2005). The role of TLRs in both innate and adaptive immunity suggest that TLRs could be involved in the initiation of CNS inflammation in response to viral pathogens.
LCMV is a noncytolytic virus and most of the diseases associated with LCMV infection in mice are mediated by innate and acquired immune responses rather than direct cytopathic effects of the virus (Allan et al., 1987; Oldstone, 2002; Zinkernagel et al., 1985). LCMV is used as a model to evaluate viral infection-induced protective immune responses and immune-mediated pathogenesis. It has been found that intracranial LCMV infection leads to up-regulation of several chemokine genes in the brain, including MCP-1, TNF-α, RANTES (Asensio and Campbell, 1997; Campbell et al., 1994). Recent studies have demonstrated that the production of these chemokines occurs in a TLR-dependent manner (Takeda and Akira, 2005), suggesting that TLRs may also be involved in LCMV-induced chemokine responses within the brain. Although it has been known for years that intracranial LCMV infection in immunocompetent mice induces a robust chemokine gene expression and a MHC-class-I-restricted CD8 T cell response, which is related to LCMV-induced death, the mechanisms associated with the immunopathological damage in the brain and death are not fully understood (Allan et al., 1987; Andersen et al., 1991; Wacher et al., 2007). The aim of this study was to explore the role of the innate immune system in the activation of CNS glial cells in response to LCMV. Our studies demonstrate that the TLR2-MyD88/Mal pathway is required for the activation and induction of chemokines in mouse CNS glial cells.
Experimental Procedures
Virus
The Armstrong strain of LCMV (LCMV-Arm) was originally obtained from Dr. Liisa K. Selin (University of Massachusetts Medical School, Worcester, MA). LCMV-Arm was propagated on BHK-21 cells (ATCC) at low MOI (0.01). Viral titer was determined with an immunological focus assay (Battegay et al., 1991). Rat anti-LCMV NP antibody was kindly provided by Dr. Demetrius Moskophidis (Medical College of Georgia, Augusta, GA).
Mice
TLR2, TLR3, TLR4, MyD88, and Mal knockout (KO) mice were a gift of Dr. S Akira (Osaka University, Osaka, Japan). MyD88 KO and TLR2 KO mice were backcrossed with C57BL/6 mice at least 10 generations. Mice genotypes were determined by PCR of tail DNA. Mice were bred and maintained under specific-pathogen-free conditions. IL-1R1 KO (C57BL/6 mice background. Stock number: 003245) and C57BL/6 mice (wild-type control mice) were purchased from the Jackson Laboratories (Bar Harbor, Maine).
Reagents
Pam2CSK4 (TLR2/6 ligand) and Pam3CSK4 (TLR2/1 ligand) were obtained from Sigma (St. Louis, Mo.). The lipopolysaccharide (LPS) was repurified by phenol extraction prior to use to remove lipopeptides, as described previously (Compton et al., 2003). Recombinant mouse TNF-α and IL-1β were purchased from R&D Systems (Minneapolis, Minn). Polyinosinic-polycytidylic acid (poly(I:C), TLR3 ligand) was from Amersham Biosciences.
Primary brain mixed glial cultures and enrichment of the primary microglial cells
Primary brain mixed glial cells were prepared according to the methods described by Frei et al (Frei et al., 1989). Briefly, 1-3 day old neonatal mice were euthanized and decapitated. Brains were homogenized with a steel mesh and washed twice with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FCS. Cells were cultured for 9 days in DMEM supplemented with 10% FCS. After 2 more passages (8 days), cells were used as “CNS primary mixed glial cells” (> 85% are astrocytes) (Frei et al., 1989). Astrocytes were identified with antibody against glial fibrillary acidic protein (GFAP) (Dako) (Frei et al., 1989; McKimmie and Fazakerley, 2005). To enrich the primary microglial cells, cells were positively isolated using anti-CD11b antibody conjugated microbeads following the instructions supplied by manufacturer (Miltenyi Biotec, Auburn, CA). Purity of CD11b+ T cells was > 85% as assessed by flow cytometry.
Immortalization of microglial cells
Immortalized microglial cell lines were established by infecting primary mixed glial cells with J2, a recombinant retrovirus encoding for the viral oncogenes v-myc and v-raf, based on the description by Blasi et al(Blasi et al., 1990). Briefly, primary mixed glial cell were cultured in 2.5 ml 0.45 micron filtered ΨCRE-J2 cell supernatant, as the source of J2 retrovirus, supplemented with 5μg of Polybrene (Sigma)/ml and 2.5 ml of DMEM/10% FCS/ 10ug/ml Ciprofloxacin in a T25 flask. After 24 h at 37°C, supernatants were removed and cells were maintained in DMEM/10% FCS/ 10ug/ml Ciprofloxacin. 14-21 days after infection with J2, colonies of proliferating cells were observed and cultures were vigorously shaken and pipetted for detachment of microglial cells. Afterwards, immortalized cells were passaged 4-5 times and subcloned by limiting dilution using conditioned medium. Characterization of the immortalized clonal microglial cells with immunofluorescence and flow cytometry revealed them to be 100% CD11b positive and GFAP negative.
Detection of chemokines (ELISA and intracellular chemokine staining)
For stimulation, cells were seeded in 24-well plates at a density of 1×105/well and allowed to grow overnight to about 70% confluence. Cells were incubated with no stimulus or with live or UV-inactivated LCMV-Arm. Positive controls included LPS (100 ng/ml, TLR4 ligand), Pam3 (10 μg/ml, TLR1 and TLR2 ligand), poly IC (25μg/ml, TLR3 ligand), mouse recombinant TNF-α (100 ng/ml, TLR independent control). The cultures were incubated for additional 19 h at 37°C. The levels of MCP-1 and RANTES in the culture supernatants were measured using the OptEIA dual-antibody detection ELISA assay (BD Pharmingen, San Diego) following the manufacturer’s recommendations. Results are shown as pg/ml. For intracellular MCP-1 and TNF-α staining, cells were first incubated with different stimuli for 1 h, then Brefeldin A (BFA) (final concentration: 1 μg/ml) was added and cells were incubated for overnight. The intracellular expression of MCP-1 and TNF-α was determined following our routine intracellular cytokine staining protocol (Zhou et al., 2005). Anti-mouse MCP-1 antibody (Clone: 2H5) and TNF-α antibody were purchased from BD PharMingen. To analyze the expression of TLR2, MHC class-II (I-Ab), class-I (H-2Db), co-stimulatory molecules (CD40, CD86) in CNS glial cells, cells were washed once with PBS (pH 7.2) and stained with PE-conjugated anti-mouse TLR2 (clone TL2.1) or other antibodies as indicated (BD Pharmingen). After washing three times with PBS, sample data were acquired on a BD-LSR-II flow cytometer (Becton Dickinson). Data were analyzed by using FlowJo software (Treestar)
For the detection of GFAP, the intracellular staining (ICS) protocol was used. Briefly, cells were first permeabilized with cytofix buffer (BD) for 15 min at 4 °C, washed once, then the unconjugated anti-mouse GFAP (DAKO) was added at a dilution of 1:200. After incubation for 1 h, the secondary antibody, PE-conjugated goat anti-rabbit IgG or isotype antibody was added and cells were incubated for another 30 min. Samples were washed and analyzed on a BD-LSR-II flow cytometer (Becton Dickinson). Data were analyzed by using FlowJo software (Treestar).
Confocal Microscopy
For confocal microscopy, primary mixed glial cells from wildtype and MyD88 KO mice were grown on Poly-L-lysine (Sigma) coated glass cover slips, infected with LCMV as described above and fixed in 4% Paraformaldehyde. Cells were blocked and permeabilized with 0.1% saponin/1% bovine serum albumin/10% normal goat serum/PBS (pH 7.4) and incubated with rat anti-CD11b (Serotec) and rabbit anti-GFAP (DAKO) followed by Alexa Fluor® 488 goat anti-rabbit IgG and Alexa Fluor® 647 goat anti-rat IgG (Invitrogen) as well as PE-conjugated anti-TNF antibody (BD Pharmingen, San Diego). Nuclei were stained with Hoechst 33258 (1:1000, Invitrogen). After mounting, samples were imaged with a Leica TCS SP2 AOBS laser scanning confocal microscope.
Statistical analysis
Data were evaluated using the two-tailed Student’s t test. Results were expressed as means +/- standard errors. Values of P <0.05 were regarded as significant.
Results
LCMV induced chemokine responses in mouse CNS astrocytes and microglial cells are TLR2/MyD88/Mal-dependent
We have previously shown that the TLR2-MyD88 pathway is essential for LCMV-induced chemokine and cytokine responses ex vivo in mouse peritoneal macrophages and in vivo in intravenously (i.v.) LCMV-infected mice (Zhou et al., 2005). To determine whether the TLR2-MyD88 signaling pathway is also important for cytokine production in brain glial cells, we established primary brain mixed glial cell cultures from WT mice as well as mice deficient in TLR2 or the TLR2 adaptor molecules MyD88 and MAL. As TLR specific controls, we used primary brain glial cells from mice deficient in TLR3 (TRIF-dependent), TLR4 (MyD88/TRIF-dependent), and IL-1R1 (MyD88-dependent and TLR-independent).
The subsets of cells present in the primary mixed glial cell cultures were identified by flow cytometry. Expression of GFAP and CD11b were used to identify astrocytes and microglial cells, respectively. GFAP+ astrocytes accounted for greater than 80% of the total glial cells (Fig 1a), while the percentage of CD11b+ microglial cells varied between 5-20%. Thus, we separated the cells present in the mixed primary glial cell culture into two populations: CD11b-GFAP+ astrocytes and CD11b+ GFAP- microglial cells.
Fig 1. LCMV-induced chemokine production in mouse CNS astrocytes is TLR2-MyD88-dependent.


A: Primary CNS mixed glial cells were surface-stained with CD11b and intracellularly stained with anti-GFAP antibody. B: Primary CNS glial cells (passage 2) were challenged with live LCMV-Arm or UV-inactivated LCMV-Arm for 18-24 hours. As controls, cells were treated with medium, mouse recombinant TNF-α (TLR-independent), Pam2 (TLR2 ligand) and poly:IC (TLR3 ligand). The levels of MCP-1 and RANTES in the supernatants were measured by ELISA. One representative of at least five experiments is shown.
Next, we determined whether TLRs were involved in LCMV-induced chemokine and cytokine responses in CNS glial cells using both ELISA and single cell-based intracellular cytokine staining (ICS) approaches. LCMV challenge of WT primary brain mixed glial cells predominantly induced MCP-1 and RANTES (Fig 1b), whereas very little TNF-a and IL-6 was produced (data not shown). In contrast, LCMV challenge of primary glial cells deficient in TLR2, MyD88, or Mal did not induce any of these chemokines (Fig 1b and data not shown). In addition, in response to LCMV, TLR3 KO and TLR4 KO primary mixed glial cells produced similar patterns of chemokines as WT glial cells (data not shown). These results revealed that TLR2-MyD88/Mal-dependent signaling is essential for LCMV-induced chemokine response in CNS glial cells.
To further characterize possible differences in astrocytic and microglial activation through the LCMV-induced TLR2-MyD88/Mal signaling pathway, the production of two representative chemokines, MCP-1 and TNF-α, was analyzed by both intracellular staining (ICS) and confocal microscopy. Interestingly, ICS demonstrated that WT astrocytes (CD11b- subset) responded to multiple TLR ligands, including LCMV, by producing MCP-1 (Fig 2a). In contrast, microglial cells (CD11b+ subset) in the same culture produced predominately TNF-α and much lower levels of MCP-1(Fig 2b and 2c), suggesting that these two major glial cell populations differ in their cytokine production induced by LCMV. Experiments assessing the expression of cytokines by individual cells using confocal microscope confirmed these results revealing expression of TNF-α in glial cells after infection with LCMV (Fig 2d). Of note, brain glial cells deficient in TLR2 and the TLR2 adaptor molecules, MyD88 and Mal, did not respond to LCMV but did respond to the MyD88-independent TLR3 ligand, poly I:C (Fig 2a), which indicated that these TLR2-MyD88/Mal deficient astrocytes were not intrinsically defective in their ability to produce cytokines.
Fig 2. In response to PAMPs, including LCMV, primary astrocytes primarily produce MCP-1, whereas primary microglial cells produce TNF-α.




LCMV effects are mediated via TLR2/MyD88/Mal; CNS primary glial cells (passage 2 to 3) were challenged with LCMV and other TLR ligands for 18-24 hours in the presence of Brefeldin A at a concentration of 1 μg/ml. Cells were first stained with CD11b, which was used to distinguish microglial cells (CD11b positive subset) from astrocytes (CD11b negative subset). The intracellular expression of MCP-1 and TNF-α was determined by double staining with antibodies against mouse MCP-1 or TNF-α (Fig 2B). The expression of CD11b was used to distinguish microglial cells (CD11b+) from astrocytes (CD11b-). The percentage of each subset of cells expressing either MCP-1 (A) or TNF-α (B) was gated, analyzed and shown as bar graphs. Representatives of the FACS analysis were shown in the bottom panels of Fig 2a, 2b. LCMV-infected primary microglial cells were stained with antibodies against LCMV-NP, CD11b, and TNF-α. Cells were first gated on LCMV+CD11b+ and LCMV+CD11b- subsets of cells. The production of TNF-α by these two populations was separately gated (C). TLR3 and/or TLR4 KO primary mixed glial cells were included as TLR specificity controls. Values on the upper right quadrant represent the percentage of glial cells producing cytokines. One representative of at least five experiments is shown. (D) Confocal Microscopy: Cells were treated as described in the text, fixed with Paraformaldehyde, permeabilized and stained with anti-CD11b antibody (a marker for microglial cells) and anti-GFAP antibody (as a marker for astrocytes). Additionally, cells were incubated with anti-TNF-α antibodies for intracellular TNF production and Hoechst 33258 to stain nuclei.
Moreover, by measuring the expression of LCMV-NP, we demonstrated that both TNF-α and MCP-1 positive populations were limited to the infected cells, implying that LCMV infection directly correlated with the induction of both MCP-1 and TNF-α in glial cells (Fig 3a and 3b). While TLR2, MyD88 and Mal deficient brain glial cells did not respond to LCMV, brain glial cells deficient in TLR3 or TLR4 responded to LCMV challenge comparable to WT glial cells (Fig 2a, 2b, 2c). Thus, these studies further confirmed that the TLR2-MyD88/Mal-dependent signaling pathway is essential for LCMV-induced chemokine expression in astrocytes and microglial cells despite the fact that these populations differ in the cytokines they produced.
Fig 3. LCMV infected mixed glial cells synthesize MCP-1 and TNF-α in a TLR2/MyD88/Mal dependent fashion.

LCMV-infected primary mixed glial cells were challenged with LCMV as described in Fig 2 and stained with antibodies against LCMV-NP together with either MCP-1 (A) or TNF-α (B). Values on the upper right quadrant represent the percentage of glial cells producing cytokines.
Since the number of CD11b+ microglial cells in primary mixed brain glial cell cultures is comparatively low (5-20% of total mixed glial cells), we used two different approaches to validate the role of TLR2 and MyD88 in microglial cells in response to LCMV. As a first approach, WT and MyD88 KO primary microglial cells in the primary mixed glial cells were isolated using CD11b microbeads. Here, we demonstrated that enriched WT primary microglial cells produced a large amount of TNF-α and lower levels of MCP-1 in response to LCMV challenge (Fig 4a), whereas enriched MyD88 KO primary microglial cells did not produce any of these cytokines (Fig 4a). The second approach involved the use of immortalized microglial cells. Consistent with the data shown in Fig 3a, LCMV challenge of immortalized microglia induced a large amount of TNF-α and a relatively smaller amount of MCP-1. Importantly, LCMV-induced expression of both TNF-α and MCP-1 in microglial cells was dependent on the presence of TLR2 and MyD88 on brain microglial cells (Fig 4b). Taken together, our data clearly demonstrated that LCMV-induced chemokine responses in mouse glial cells (both astrocytes and microglia) are TLR2-MyD88/Mal-dependent, suggesting that LCMV infection directly activates brain glial cells via the TLR2-dependent signaling pathway. Furthermore, the results from primary cells as well as immortalized cells indicated that production of TNF-α predominates in microglial cells, whereas astrocytes primarily produce MCP-1 in response to LCMV infection.
Fig 4. Both enriched primary microglial cells and immortalized microglial cells synthesize more TNF-α than MCP-1 in response to LCMV infection, TLR2 and MyD88 deficient cells fail to respond.
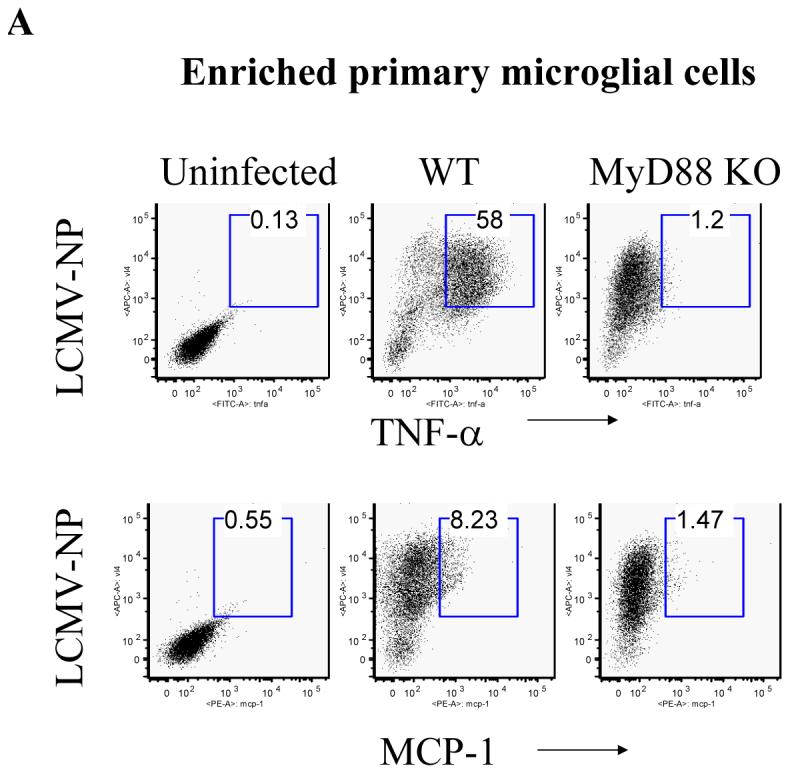

(A) WT and MyD88 KO primary microglial cells were enriched with CD11b microbeads and challenged with LCMV. Cells were separately stained with antibodies against LCMV together with either TNF-α or MCP-1. Values on the upper right quadrant represent the percentage of microglial cells producing TNF-α; (B) Immortalized microglial cells were challenged with LCMV and other TLR ligands as described above. Cells were first stained with CD11b and then subjected to the intracellular staining of MCP-1 and TNF-α. Values on the upper left quadrant represent the percentage of microglial cells producing TNF-α; Values on the upper right quadrant represent the percentage of microglial cells producing both TNF-α and MCP-1; Values on the low right quadrant represent the percentage of microglial cells producing MCP-1. One representative of at least five experiments is shown.
LCMV induced up-regulation of TLR2 in CNS glial cells is MyD88-dependent
CNS astrocytes and microglial cells constitutively express low levels of TLR2 (Ebert et al., 2005; McKimmie and Fazakerley, 2005). Our previous (Zhou et al., 2005) and current studies have demonstrated that TLR2 and MyD88 are required for LCMV-induced chemokine and cytokine production. Because an increase in expression levels of TLR2 could alter and facilitate the response to LCMV in glial cells, we investigated whether the infection with LCMV itself could modulate the expression of TLR2 in brain glial cells. In astrocytes, LCMV challenge dramatically up-regulated the expression of TLR2 in WT as well as in IL-1R1 KO cells that were used as control cells, but not in MyD88 KO astrocytes (Fig 5a). Importantly, both TNF-α (TLR-independent) and poly I:C (TLR3-dependent but MyD88-independent) challenge significantly up-regulated TLR2 expression in MyD88 KO astrocytes. Similarly, a MyD88-dependent up-regulation of TLR2 could be seen in microglial cells after infection with LCMV (Fig 5b). Therefore, we demonstrated that both astrocytes and microglia show an increase in TLR2 expression in response to LCMV infection and that this up-regulation in both cell types is dependent on the presence of MyD88. These data are consistent with the concept that LCMV and other PAMPs utilize the same “activation-induced feedback loop” to regulate the expression of TLR2 (Esen and Kielian, 2006; Jack et al., 2005).
Fig 5. LCMV induces up-regulation of TLR2 in CNS astrocytes and microglial cells in a MyD88-dependent fashion.


A: Primary CNS glial cells were challenged with LCMV and various controls for 18-24 hours. IL-1R1 KO primary glial cells were included as a control. The expression of TLR2 was measured by flow cytometry. Cells were gated on CD11b negative cells. B: Immortalized WT and MyD88 KO microglial cells were seeded into 24-well plates and challenged with LCMV as well as various controls for 18-24 hours. The expression of TLR2 was determined by flow cytometry. One representative of at least three experiments is shown.
LCMV-induced activation of microglial cells is TLR2-MyD88 dependent
As CNS astrocytes and microglial cells constitutively express MHC class I and II (ref?) molecules and various TLRs, including TLR2, they may function as both active inflammatory cells and as APC-like cells to modulate the adaptive immune responses in the brain. To determine if LCMV challenge activates immune function and antigen-presenting properties in CNS glial cells via the TLR2 signaling pathway, the expression of immune-related cell surface markers characteristic of activation was examined. LCMV challenge resulted in the up-regulation of MHC class I and class II, CD40 and CD86 in WT microglial cells, while LCMV infection did not induce any of these changes in TLR2 KO (Fig 6a, 6b) nor in MyD88 KO microglial cells (data not shown). As a TLR2 and MyD88-independent control, poly:IC induced up-regulation of these molecules in WT as well as in TLR2 KO and MyD88 KO microglial cells. In summary, these data demonstrated that TLR2-MyD88 signaling is essential for LCMV-induced expression of immunoregulatory molecules on microglial cells, that are important for antigen presentation and thus may be a pre-requisite to the induction of virus specific adaptive immunity.
Fig 6. LCMV-induced up-regulation of MHC and co-stimulatory molecules in microglial cells is in a TLR2-dependent fashion.


Immortalized WT and TLR2 KO microglial cells were seeded into 24-well plates and stimulated with LCMV and other TLR ligands for 18-24 hours as indicated. The expression of MHC-class I (A, bottom panel), class II (A, top panel), CD40 (B, top panel), CD86 (B, bottom panel) was analyzed by FACS staining. One representative of at least three experiments is shown.
Discussion
The main findings of the current study are as follows: 1) TLR2-MyD88/Mal signaling pathways play a central role in LCMV-induced chemokine (cytokine) responses in CNS glial cells. 2) TLR2-MyD88-dependent signaling is essential for the LCMV-induced activation of both astrocytes and microglial cells, 3) Microglial cells and astrocytes differ in the type of chemokines (cytokines) they produce in response to viral infection (microglial cells produce primarily TNF-α and astrocytes produce predominantly MCP-1).
LCMV is a non-cytolytic virus and LCMV infection in vivo does not directly result in tissue damage. LCMV-induced pathologic changes and clinical symptoms are exclusively associated with the initial inflammatory response and later, with adaptive immune responses (Oldstone, 2002; Zinkernagel et al., 1985). Although it has been known for decades that i.c. LCMV infection in mice causes fatal meningitis within 6-8 days post-infection, with the infiltrating LCMV-specific CD8 T cells playing a decisive role, the physiologic mechanisms that lead to meningitis and death have not been fully investigated. The proposed mechanisms include the following aspects (Allan et al., 1987; Andersen et al., 1991): 1. LCMV-specific mechanical distension / edema / compression that is inflammation-mediated and CD8+ T cell-dependent. 2. Release of chemical mediators. 3. LCMV-specific CD8+ T cell-mediated killing or cytotoxicity. Our current studies may provide new insights that will allow a better understanding of LCMV-induced immunopathology in the brain.
It has been demonstrated that LCMV infection in mouse brain induces robust chemokine expression (Asensio and Campbell, 1997; Wacher et al., 2007), which not only causes the recruitment and infiltration of leukocytes into the brain but also contributes to LCMV-induced immunopathogenesis in the CNS. Astrocytes and microglial cells are the two important CNS non-neuronal parenchymal cells, and they function as the major innate immune cells in the brain (Hertz et al., 1990; Olson and Miller, 2004). Furthermore, in a LCMV infection model in neonatal rats it has been shown that the virus predominantly infects astrocytes and, at lower levels, microglial cells, and that the infection then spreads to other regions of the brain and eventually reaches neurons (Bonthius et al., 2002), which underlines the importance of glial cells for LCMV infections in vivo.
Our studies clearly demonstrated that TLR2-MyD88/Mal dependent innate immune signaling is essential for LCMV challenge-induced chemokine and cytokine responses in these CNS glial cells. In response to invading microbial pathogens, including LCMV, brain resident glial cells contribute to the production of MCP-1, RANTES, TNF-α, IL-6 (Sandberg et al., 1994). Production of these chemokines and cytokines in turn attract more viral-specific T cells, specifically CD8+ T cells into the brain, and exacerbate the inflammation that may eventually lead to death (Andersen et al., 1991; Ward et al., 1998).
The important roles played by astrocytes and microglial cells in various CNS diseases, such as multiple sclerosis (MS), Alzheimer disease, HIV-associated CNS diseases, have already attracted enormous attention (Ambrosini et al., 2005; Bajetto et al., 2002; Clarke et al., 2006). As the major glial cells in the brain, astrocytes play an essential role in the formation of the BBB and protection of the CNS from various invading microbial pathogens. Upon exposure to microbial components or other inflammatory molecules, both astrocytes and microglial cells participate to the inflammatory process in the brain by inducing a variety of chemokines and cytokines and by recruiting inflammatory cells from the periphery. Following the discovery of TLRs expression in CNS glial cells, the mechanism by which brain glial cells respond to invading microbial pathogens and inflammatory molecules, has been better defined (Lehnardt et al., 2006; McKimmie and Fazakerley, 2005; Olson and Miller, 2004). In the present study, we found that the expression of TLR2 and MyD88/Mal in CNS glial cells are essential for LCMV-induced activation and chemokine responses, which is consistent with our previous studies in mouse PECs (peritoneal exudates cells)(Zhou et al., 2005). We further demonstrated that LCMV challenge is able to up-regulate the expression of TLR2, MHC molecules, co-stimulatory molecules expression in both astrocytes and microglial cells and this upregulation is also dependent on the MyD88 signaling.
The TLR2-MyD88/Mal dependent chemokine and cytokine production and activation in CNS glial cells may either act locally to induce a disorder of neurons or attract more inflammatory cells into the brain to exacerbate the CNS pathologic process. Moreover, the up-regulated TLR2, MHC, and costimulatory molecules in CNS glial cells could activate the infiltrated inflammatory cells, such as CD8+ and CD4+ T cells, to amplify the inflammation in the brain.
In summary, this study demonstrated that the innate TLR2-MyD88/Mal signaling pathway is essential for LCMV-induced activation and production of chemokines in mouse CNS glial cells. It is possible that the known polymorphisms of TLRs (including TLR2) could account for the differences in the susceptibility of people to viral encephalitis or meningitis.
ACKNOWLEDGMENTS
We thank Junko Kato for excellent secretarial assistance. This work was supported by NIAID Regional Center of Excellence Grant AI 057159, NIH grants R01 AI 49309 and P01 AI 0577484 (RWF), NIH grant AI 51415 (EAKJ) and the German Academic Exchange Program (AH).
Abbreviations used
- MyD88
Myeloid Differentiation factor 88
- TLR
Toll-like receptor
- LCMV
Lymphocytic Choriomeningitis Virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan JE, Dixon JE, Doherty PC. Nature of the inflammatory process in the central nervous system of mice infected with lymphocytic choriomeningitis virus. Curr Top Microbiol Immunol. 1987;134:131–143. doi: 10.1007/978-3-642-71726-0_6. [DOI] [PubMed] [Google Scholar]
- Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F, Coccia EM. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol. 2005;64:706–715. doi: 10.1097/01.jnen.0000173893.01929.fc. [DOI] [PubMed] [Google Scholar]
- Andersen IH, Marker O, Thomsen AR. Breakdown of blood-brain barrier function in the murine lymphocytic choriomeningitis virus infection mediated by virus-specific CD8+ T cells. J Neuroimmunol. 1991;31:155–163. doi: 10.1016/0165-5728(91)90021-x. [DOI] [PubMed] [Google Scholar]
- Asensio VC, Campbell IL. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol. 1997;71:7832–7840. doi: 10.1128/jvi.71.10.7832-7840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem. 2002;82:1311–1329. doi: 10.1046/j.1471-4159.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- Battegay M, Cooper S, Althage A, Banziger J, Hengartner H, Zinkernagel RM. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J Virol Methods. 1991;33:191–198. doi: 10.1016/0166-0934(91)90018-u. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Mahoney J, Buchmeier MJ, Karacay B, Taggard D. Critical role for glial cells in the propagation and spread of lymphocytic choriomeningitis virus in the developing rat brain. J Virol. 2002;76:6618–6635. doi: 10.1128/JVI.76.13.6618-6635.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- Campbell IL, Hobbs MV, Kemper P, Oldstone MB. Cerebral expression of multiple cytokine genes in mice with lymphocytic choriomeningitis. J Immunol. 1994;152:716–723. [PubMed] [Google Scholar]
- Clarke JN, Lake JA, Burrell CJ, Wesselingh SL, Gorry PR, Li P. Novel pathway of human immunodeficiency virus type 1 uptake and release in astrocytes. Virology. 2006;348:141–155. doi: 10.1016/j.virol.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77:4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Ebert S, Gerber J, Bader S, Muhlhauser F, Brechtel K, Mitchell TJ, Nau R. Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J Neuroimmunol. 2005;159:87–96. doi: 10.1016/j.jneuroim.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol. 2006;176:6802–6811. doi: 10.4049/jimmunol.176.11.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol. 2005;159:12–19. doi: 10.1016/j.jneuroim.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Finberg RW, Kurt-Jones EA. Viruses and Toll-like receptors. Microbes Infect. 2004;6:1356–1360. doi: 10.1016/j.micinf.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Frei K, Lins H, Schwerdel C, Fontana A. Antigen presentation in the central nervous system. The inhibitory effect of IL-10 on MHC class II expression and production of cytokines depends on the inducing signals and the type of cell analyzed. J Immunol. 1994;152:2720–2728. [PubMed] [Google Scholar]
- Frei K, Malipiero UV, Leist TP, Zinkernagel RM, Schwab ME, Fontana A. On the cellular source and function of interleukin 6 produced in the central nervous system in viral diseases. Eur J Immunol. 1989;19:689–694. doi: 10.1002/eji.1830190418. [DOI] [PubMed] [Google Scholar]
- Hertz L, McFarlin DE, Waksman BH. Astrocytes: auxiliary cells for immune responses in the central nervous system? Immunol Today. 1990;11:265–268. doi: 10.1016/0167-5699(90)90106-j. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Kamperschroer C, Quinn DG. The role of proinflammatory cytokines in wasting disease during lymphocytic choriomeningitis virus infection. J Immunol. 2002;169:340–349. doi: 10.4049/jimmunol.169.1.340. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem (Tokyo) 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- Lang KS, Georgiev P, Recher M, Navarini AA, Bergthaler A, Heikenwalder M, Harris NL, Junt T, Odermatt B, Clavien PA, Pircher H, Akira S, Hengartner H, Zinkernagel RM. Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J Clin Invest. 2006;116:2456–2463. doi: 10.1172/JCI28349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- McKimmie CS, Fazakerley JK. In response to pathogens, glial cells dynamically and differentially regulate Toll-like receptor gene expression. J Neuroimmunol. 2005;169:116–125. doi: 10.1016/j.jneuroim.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Oldstone MB. Biology and pathogenesis of lymphocytic choriomeningitis virus infection. Curr Top Microbiol Immunol. 2002;263:83–117. doi: 10.1007/978-3-642-56055-2_6. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Palliser D, Ploegh H, Boes M. Myeloid differentiation factor 88 is required for cross-priming in vivo. J Immunol. 2004;172:3415–3421. doi: 10.4049/jimmunol.172.6.3415. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Sandberg K, Kemper P, Stalder A, Zhang J, Hobbs MV, Whitton JL, Campbell IL. Altered tissue distribution of viral replication and T cell spreading is pivotal in the protection against fatal lymphocytic choriomeningitis in mice after neutralization of IFN-alpha/beta. J Immunol. 1994;153:220–231. [PubMed] [Google Scholar]
- Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Wacher C, Muller M, Hofer MJ, Getts DR, Zabaras R, Ousman SS, Terenzi F, Sen GC, King NJ, Campbell IL. Coordinated regulation and widespread cellular expression of interferon-stimulated genes (ISG) ISG-49, ISG-54, and ISG-56 in the central nervous system after infection with distinct viruses. J Virol. 2007;81:860–871. doi: 10.1128/JVI.01167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SG, Bacon K, Westwick J. Chemokines and T lymphocytes: more than an attraction. Immunity. 1998;9:1–11. doi: 10.1016/s1074-7613(00)80583-x. [DOI] [PubMed] [Google Scholar]
- Zhou S, Kurt-Jones EA, Mandell L, Cerny A, Chan M, Golenbock DT, Finberg RW. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol. 2005;35:822–830. doi: 10.1002/eji.200425730. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, Pfau CJ, Hengartner H, Althage A. Susceptibility to murine lymphocytic choriomeningitis maps to class I MHC genes--a model for MHC/disease associations. Nature. 1985;316:814–817. doi: 10.1038/316814a0. [DOI] [PubMed] [Google Scholar]