

Abstract
AIMS
To describe the pharmacokinetics of chloroquine (CQ) and sulfadoxine (SDx), and to identify predictors of treatment response in children with malaria given the CQ + SDx and pyrimethamine (PYR) combination.
METHODS
Eighty-six Ugandan children with uncomplicated falciparum malaria, 6 months to 5 years old, were randomly treated with prepacked fixed-dose CQ + SDx/PYR. The youngest children (<24 months) received half strength and the older (>24 months) full strength treatment. The reported day 14 failure rates were 48% and 18%, respectively. Capillary blood (100 μl) applied on to filter paper was collected on eight occasions during 28 days of follow up. Concentrations of CQ and SDx were determined. A population approach was used for the pharmacokinetic analysis.
RESULTS
A two-compartment model adequately described the data for both CQ and SDx. For CQ, the typical apparent clearance (CL/F) and volume of distribution (VC/F) values were estimated to be 2.84 l h−1 and 230 l. The typical CL/F for SDx was 0.023 l h−1, while the factor relating its VC/F to normalized body weight was 1.6 l kg−1. Post hoc parameter estimates for both drugs showed lower maximum concentrations (Cmax) and concentration-time curve areas (AUC(0,336 h)) in younger children. The AUC(0,336 h) for SDx and CQ were independently significant factors for prediction of cure. Simulations suggest that giving the higher dose to the youngest children would result in higher CQ and SDx concentrations and improved outcome.
CONCLUSIONS
The study results suggest that full-strength combination to all children would improve the cure rate.
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Both chloroquine (CQ) and sulfadoxine/ pyrimethamine (SDx/PYR) remain important drugs in the control of malaria.
The available data on CQ, SDx and PYR are summary pharmacokinetic parameters based on classical/traditional methods, mostly in adults.
No study has described the population pharmacokinetics of a fixed-dose CQ + SDx/PYR combination in children with falciparum malaria.
WHAT THIS STUDY ADDS
This study presents population pharmacokinetic data on CQ and SDx in children with uncomplicated falciparum malaria.
The study demonstrates that in age-based fixed-dose regimens with CQ and SDx, drug exposures and outcomes may be correctly predicted, although correlation with body weight is poor.
The study proposes dose modification to improve response with the CQ + SDx/PYR combination.
Keywords: chloroquine, falciparum malaria, increased dose proposal, population pharmacokinetics, sulfadoxine
Introduction
Malaria continues to be a major health problem in most countries of the tropical world especially in sub-Saharan Africa. The WHO recommends artemisinin combination therapies (ACT) as the first line drugs for the treatment of uncomplicated Plasmodium falciparum malaria after wide spread resistance to chloroquine (CQ) and sulfadoxine/pyrimethamine (SDx/PYR) emerged in the last decade or so [1]. In certain situations both drugs have remained useful; for example, for malaria in pregnancy, SDX/PYR is recommended for the intermittent preventive treatment (IPT). Chloroquine may also still remain an important drug in falciparum malaria treatment [2], as reported from a recent study in Malawi, where a 99% efficacy of CQ against P. falciparum was shown, 12 years after it was withdrawn [3].
Treatment response in children with falciparum malaria depends on a number of factors, namely parasite resistance, host natural immunity, the drug quality and the pharmacokinetics of the administered drug. Of these factors, information on pharmacokinetics of antimalarials in children is rather limited. Most pharmacokinetic data on SDx/PYR and CQ have been from adult populations. Only a few studies have been performed in children with clinical malaria. In a recent study, the pharmacokinetics of SDx/PYR monotherapy in falciparum malaria was reported with a recommendation for increased dose in children 2–5 years old [4].
In Uganda a fixed-dose CQ plus SDx/PYR combination is currently employed in the home based management of fever (HBMF) program [5]. The formulation used is a generic product locally known as Homapak. The formulation is administered as ‘half-strength’ prepacked fixed-dose for younger children under 2 years, and as ‘full-strength’ for older children 2–5 years. We have previously reported that the children treated with the half-strength had higher day 14 total failure rates, 48.4% compared with 18.2% in the older children treated with the full-strength [6].
By employing the population approach to the pharmacokinetics of CQ and SDx/PYR in children with uncomplicated falciparum malaria, the rationale for dose design can be established. The population pharmacokinetics of multiple-dose SDx/PYR in small studies in children with congenital toxoplasmosis has recently been reported [7, 8]. However, there are no population pharmacokinetic studies in patients with falciparum malaria treated with the combination of CQ + SDx/PYR.
The present study was designed to describe the population pharmacokinetic parameters of CQ and SDx, and to identify predictors of treatment response in children with uncomplicated falciparum malaria. These data were then used to propose a modified dosage schedule for the fixed-dose CQ + SDx/PYR combination.
Methods
Study design and participants
This study was part of an antimalarial clinical trial during the months of July through November 2004, at Walukuba Health Centre, a peri-urban intense malaria transmission area, in Jinja district, Uganda. Efficacy results with fixed-dose CQ + SDx/PYR compared with amodiaquine + SDx/PYR in children between 6 months and 5 years with uncomplicated falciparum malaria have previously been presented [6]. For the present evaluation, only the children treated with fixed dose of CQ + SDx/PYR (n = 86) in the previous study were considered. The group comprised of 6–24 month (n = 31), and 25–60 month (n = 55) old children, given half-strength and full-strength of the fixed-dose formulation, respectively. The half-strength group were treated with SDx/PYR 250 mg/12.5 mg as a single dose combined with CQ 75 mg (base) daily for 3 consecutive days, while the full-strength group received SDx/PYR 500 mg/25 mg single dose combined with CQ 150 mg (base) for 3 consecutive days as previously described [6]. Treatment response in the 86 children was classed as failure (29.1%), which included early treatment failure, late clinical failure and late parasitological failure or as cure (70.9%) when adequate clinical and parasitological response (ACPR) was observed by day 14 [9]. Only children from whom pre- and post-treatment blood samples, and whose anthropometric measurements could be obtained at recruitment were included in the analysis (n = 83), irrespective of whether they completed the 28-day follow up period or not. Three children (3/86) were excluded from this analysis due to missing blood samples or anthropometric measurements.
Ethical considerations
The Faculty of Medicine Ethical Review Board, Makerere University, Uganda, and the Regional Ethical Review Board at Karolinska Institutet in Stockholm had previously approved the study. The Uganda National Council for Science and Technology (UNCST) gave permission to conduct the study in Uganda. Parents/guardians gave informed written consent.
Pharmacokinetic sampling
Using precision capillary tubes, blood samples from finger-pricks (100 μl) were collected predose on day 0, and then on days 1, 2, 3, 7, 14 and 28. On days 1 and 2, samples were collected just before the repeat CQ dose. Blood samples on the other days were collected at about the same hour of day for the individual participant. Additional postdose samples were collected on day 0, at 0.5, 1, 2, 3, 4, 5, 6, or 8 h, with each patient supplying at least one data point. The number of blood samples obtained from each child for the determination of SDx and CQ varied from 1 to 8. The samples were dried on Whatman no. 1 filter paper and stored at an average room temperature of 22°C in sealed envelopes for 3–6 months before analysis.
Bioanalytical methods
Samples were analyzed according to validated methods for CQ [10] and SDx [11]. For CQ, filter paper samples were cut into pieces and placed in 15 ml polyethylene tubes to which 0.5 ml of diethylamine (DEA) 0.5% in water, 25 μl of 2.9 μmol l−1 4-(4-dimethylamino-1-methylbutylamino)-7-chloroquinoline as the internal standard (I.S), 2 ml of potassium hydroxide (1 mol l−1), and 7 ml of di-isopropyl ether were added. Extraction was on a reciprocal shaker for 20 min and then centrifugation for 10 min at 3500 g. The organic phase was transferred to another 8 ml polypropylene tube and back-extracted with 150 μl of a phosphate buffer, pH 2.5. The organic layer was removed by aspiration, and 130 μl of the aqueous phase was then injected into the chromatographic system. The mobile phase consisted of a mixture of methanol : phosphate buffer : perchloric acid (at 250 : 747.5 : 2.5, v : v). Elution was at flow rate of 1.5 ml min−1.
For SDx, dried filter paper spots were cut into small pieces and placed in polypropylene tubes to which 1.5 ml of 0.1 mol l−1 sodium hydroxide, 100 μl of 750 μmol l−1 sulfamethoxazole (IS) were added and shaken for 15 min. Then 0.25 ml of 1 mol l−1 zinc sulphate was added to the mixture, shaken for 15 min, and centrifuged at 1000 g. An aliquot (20 µl) of the clear supernatant was injected into the chromatograph. The mobile phase consisted of acetonitrile : phosphate buffer, pH 3.0 (20 : 80, v : v). Due to lack of adequate samples the PYR concentrations were not determined.
Instrumentation consisted of a Gilson model 231 sample injector with 200 μl loop (Villiers le Bel, France), a Gilson model 118 UV-Vis absorbance detector set at 333 nm for chloroquine and 254 nm for sulfadoxine, and a Kontron model 422 high-pressure pump (Milan, Italy). A C18 reversed-phase column, Zorbax® SB, 75 × 4.6 mm I.D, 3.5 μm (Chrom Tech, Hägersten, Sweden) was used. The limit of quantification for CQ was 15.4 μg l−1 and for SDx 4.5 mg l−1, with the interassay coefficient of variation at 2.9% and 4.4%, respectively.
Pharmacokinetic analysis
A total of 498 blood samples were collected from the 83 participants, from which 380 CQ and 443 SDx concentration measurements were successfully analyzed. One hundred and eighteen CQ and 45 SDx concentration measurements were either below the limit of quantification, had no detectable concentrations or the samples did not have the required volume of blood at the time of sampling, and were therefore not included in the pharmacokinetic analysis. Five children had pretreatment SDx concentrations ranging from 3.9 to 34.1 μg ml−1, while 13 had pretreatment CQ concentrations ranging from 7.1 to 72.3 ng ml−1. These samples were also excluded from the PK analysis.
Figure 1 shows the observed concentration vs. time data for both drugs. Pooled data sets of chloroquine or sulfadoxine concentrations were used for modelling, using a mixed-effect modelling approach (NONMEM, version V, Globomax, Hanover, MD). In both models, the first-order conditional estimation with interaction option (FOCE INTERACTION) in NONMEM was used. Discriminations between nested models were based on the objective function value (OFV) provided by NONMEM at a significance level of P < 0.001, equal to a decrease of 10.8 in the OFV, graphical analysis of residuals, and predictions in model diagnostics using Xpose, version 3.1 [12]. Regression of individual PK parameters against the covariates was applied to assess the influence of sex, age, body weight, and height.
Figure 1.
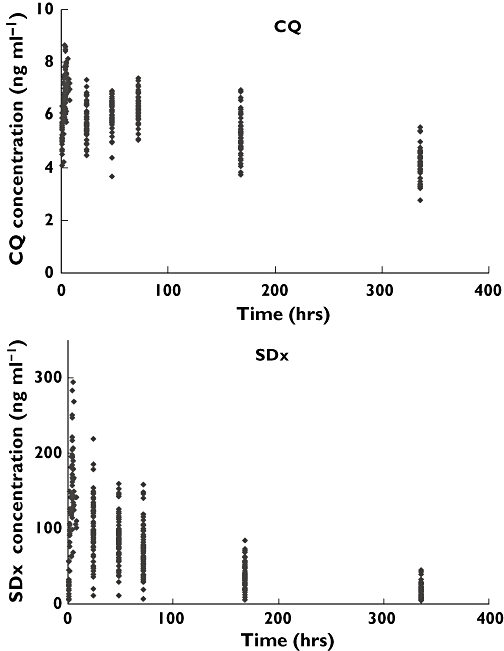
Pooled chloroquine (A) and sulfadoxine (B) concentrations vs. time
Chloroquine pharmacokinetic model
Various structural models were tested on normal or log-normalized data. These included a one-, two-, or a three-compartmental model with or without an absorption lag-time. Interindividual variability terms for different model parameters were tested in addition to additive or proportional residual error terms. A two-compartment model, including a first-order absorption rate constant, applied to the log-normalized blood chloroquine data described the time-course of chloroquine best. None of the tested covariates showed any effect on the model parameters. An exponential variance model was used to describe the interindividual variability in the clearance and central volume of distribution. The final model included proportional residual error model.
Sulfadoxine pharmacokinetic model
Several pharmacokinetic models were applied to normal or log-transformed sulfadoxine concentration data, including one-, two-, or three-compartment models, with or without an absorption lag-time. Interindividual variability terms for different model parameters were tested in addition to additive or proportional residual error terms. The final model consisted of a two-compartment pharmacokinetic model with a first-order absorption rate constant and an absorption lag-time. Body weight was found to correlate with the central volume of distribution (VC) and was incorporated in the final model:
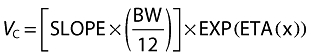 |
where VC is the apparent central volume of distribution of sulfadoxine, SLOPE is the factor relating VC to body weight (BW) in kg, and 12 is the median body weight of the studied population. ETA is the variability term associated with the model parameter VC. The final model included an additional interindividual variability term for sulfadoxine clearance. Proportional as well as additive residual error components were used to describe the error terms.
Statistical methods
Predictors of treatment response were identified using logistic regression including treatment response at 2 weeks (yes/no) as dependent variable. Independent variables tested were AUC(0,24 h) and AUC(0,336 h) for SDx, AUC(0,24 h), AUC(0,72 h) and AUC(0,336 h) for CQ, sex, age, weight and height, using Statistica version 7.0 (Statsoft Inc., Tulsa OK). Independent factors were included in a forward stepwise manner until no further significant factors were found. In the bi-variate analysis for individual effects, adjusted r2 values were provided when numbers of observations differed or were few. Categorical variables were compared using the chi square, and continuous variables by the independent samples t-test. A P value <0.05 was considered significant.
Results
The characteristics of the study participants are summarized in Table 1.
Table 1.
Baseline characteristics of study population
| Study population (n = 83) | |||
|---|---|---|---|
| Characteristics | Mean ± SD | Median | Range |
| Age (months) | 30 ± 14 | 29 | 6–60 |
| Weight (kg) | 11.9 ± 2.7 | 12 | 7–17 |
| Height (cm) | 87 ± 12 | 86 | 61–109 |
| Haemoglobin (mg dl−1) | 9.6 ± 1.7 | 9.8 | 6–13 |
| Number of blood samples/child | 5 ± 1 | 5 | 1–8 |
| Sex (% male) | 59 | ||
Pharmacokinetic results
The proposed pharmacokinetic models described both the chloroquine and the sulfadoxine blood concentration data well, as seen in the basic diagnostic plots of CQ and SDx models (Figure 2).
Figure 2.
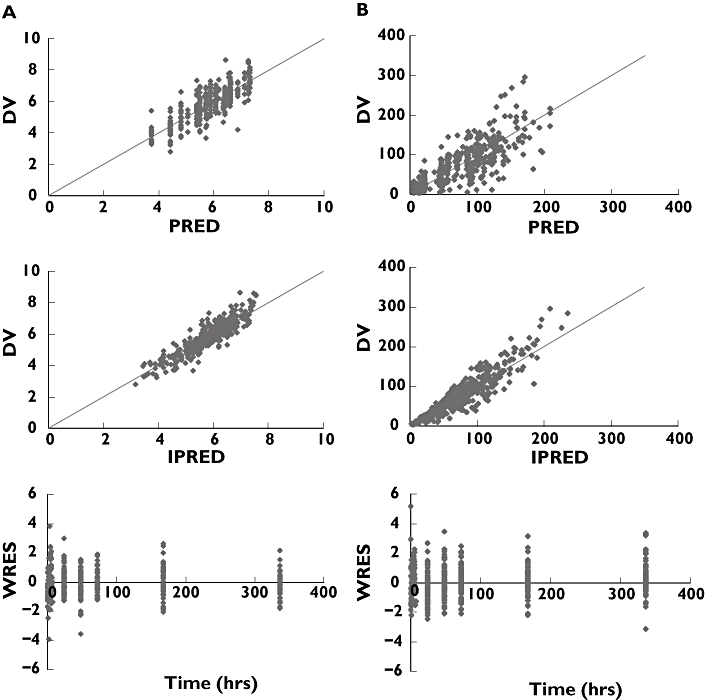
Basic goodness of fit plots (a) CQ and (b) SDx. DV: dependent variable (observed concentrations: log-normalized for the CQ data, linear for the SDx data), PRED, population prediction; IPRE, individual prediction; WRES, weighted residuals
Population pharmacokinetic parameter estimates for the whole group are presented in Table 2, while the post hoc secondary parameters estimates for each dose group are presented in Table 3 for SDx and Table 4 for CQ.
Table 2.
Population pharmacokinetic parameter estimates for SDx and CQ in 83 children aged 6–60 months
| Model parameters* | SDx estimate (RSE%) | IIV (RSE%) | CQ estimate (RSE%) | IIV (RSE%) |
|---|---|---|---|---|
| VC (l kg−1) | – | 31.8 (20) | 0.57 (67) | |
| Slope | 1.53 (18) | 0.47 (33) | – | – |
| CL (l h−1) | 0.023 (5.3) | 0.33 (35) | 2.84 (5.4) | 0.3 (35) |
| CLD2 (l h−1) | 0.13 (15) | – | 3.29 (11) | – |
| Vp (l kg−1) | 1.6 (16) | – | 230 (7.8) | – |
| ka (h−1) | 0.30 (24) | – | 0.14 (13) | – |
| Lag time (h) | 0.29 (14) | – | Not estimated | – |
| Additive error | 2.6 (100) | – | 0.5 (6.3) | – |
| Proportional error | 0.27 (12) | – | Not estimated | – |
CL, apparent clearance; CLD2, apparent intercompartmental clearance values; IIV, interindividual variability; ka, absorption rate constant; Lag-time, absorption lag-time; RSE, relative standard error in percent; Slope, factor relating the volume of distribution to normalized body weight (BW); VC, apparent central volume of distribution; Vp, apparent volume of peripheral compartment.
Table 3.
Descriptive statistics of individual post hoc secondary parameter estimates for SDx by dose group
| Dose 250 mg, age 6–24 months | Dose 500 mg, age 24–60 months | |||||
|---|---|---|---|---|---|---|
| Parameters‡ | Mean | SD | Min–max | Mean | SD | Min–max |
| Cmax† (μg ml−1) | 130 | 24 | 99–227 | 171 | 32 | 85–249 |
| tmax (h) | 5.7 | 0.60 | 3.6–6.9 | 7.0 | 0.68 | 4.8–8.0 |
| α-HL (h) | 3.1 | 0.48 | 1.5–3.9 | 4.2 | 0.49 | 3.0–5.0 |
| β-HL (h) | 113 | 37 | 51–217 | 98 | 32 | 18–177 |
| AUC(0,336 h)† (μg ml−1 h) | 12 500 | 3250 | 6 280–21 900 | 16 900 | 4690 | 2 840–27 500 |
| AUC(0,24 h)† (μg ml−1 h) | 2 310 | 305 | 1 830–3 330 | 3 240 | 560 | 1 680–4 510 |
Unpaired t-test for difference between low and high dose groups were all significant at P < 0.00005.
α-HL, initial disposition half-life; β-HL, terminal elimination half-life; Cmax, maximum blood concentration attained; tmax, the time to maximum blood concentration.
Table 4.
Descriptive statistics of individual post hoc secondary parameter estimates for CQ by dose group
| Dose 75 mg × 3, age 6–24 months | Dose 150 mg × 3, age 24–60 months | |||||
|---|---|---|---|---|---|---|
| Parameters‡ | Mean | SD | Min–max | Mean | SD | Min–max |
| Cmax† (μg ml−1)§ | 0.790 | 0.123 | 0.536–1.14 | 1.43 | 0.238 | 0.607–1.92 |
| tmax (h)§ | 6.5 | 1.2 | 4.5–9.4 | 6.9 | 1.5 | 4.9–13.2 |
| α-HL (h) | 3.43 | 1.22 | 1.68–6.82 | 3.97 | 1.95 | 1.92–14.1 |
| β-HL (h) | 115 | 19.7 | 87.5–166 | 108 | 10.4 | 81.5–131 |
| AUC(0,336 h)† (μg ml−1 h) | 77.5 | 17.5 | 50.3–121 | 140 | 19.1 | 87–184 |
| AUC(0,72 h)† (μg ml−1 h) | 44.0 | 5.65 | 33.9–56.4 | 81.9 | 7.7 | 58.7–99.3 |
| AUC(0,24 h)† (μg ml−1 h) | 11.9 | 1.37 | 9.38–15.1 | 22.2 | 2.45 | 11.8–26.8 |
Unpaired t-test for difference between low and high dose groups were all significant at P < 0.00005.
α-HL, early elimination half-life; β-HL, terminal elimination half-life; Cmax, maximum blood concentration attained; tmax, the time to maximum blood concentration.
Parameters estimated after first dose.
Treatment response
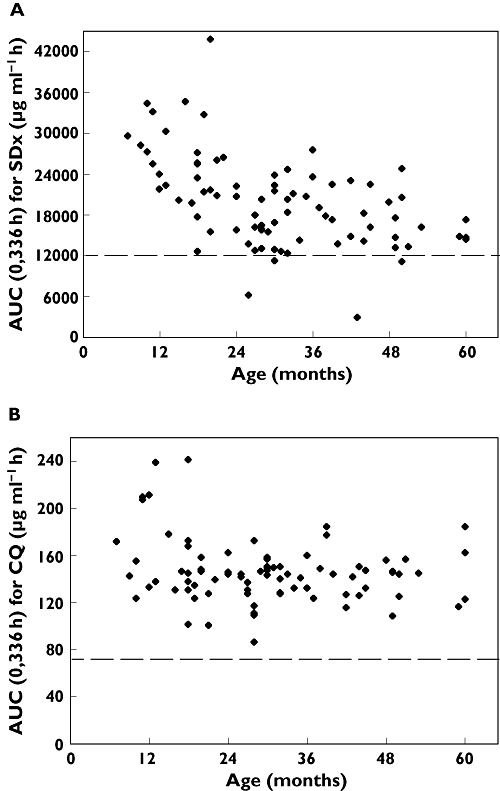
In the whole population there were 59 (71%) responders and 24 (29%) non-responders as observed 14 days after initiation of treatment. The logistic regression of treatment response vs. potential predictors identified AUC(0,336 h) for SDx and AUC(0,336 h) for CQ as independent and significant factors (P < 0.05 for both factors Wald). No other factor significantly predicted the response. Treatment failures were more common in children aged 24 months and younger who received the lower Homapak dose (15/31 = 48% non-responders), than in older children who received the higher dose (9/52 = 17% non-responders) with P < 0.01 using the Yates corrected chi-square test. The difference in treatment response was associated with lower exposures to both SDx and CQ in the younger age group as shown in Tables 3 and 4 and Figure 3.
Figure 3.
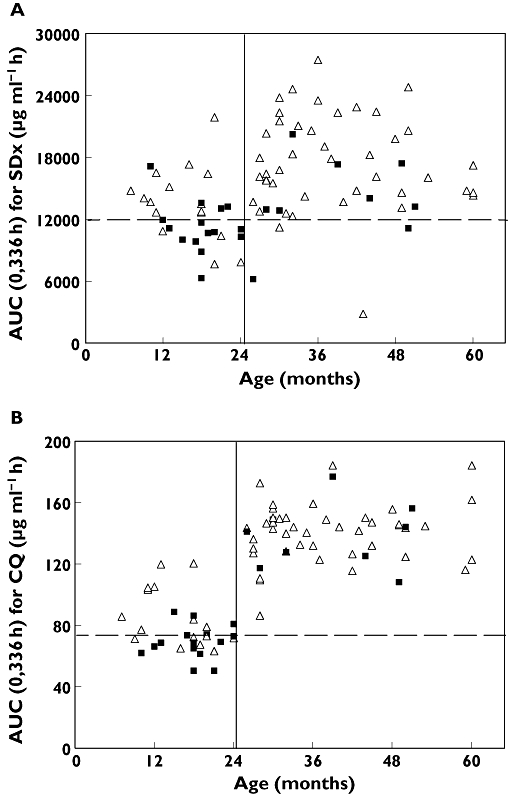
AUC(0,336 h) vs. age. (A) SDx and (B) CQ from the current regimen of the fixed-dose CQ + SP in the younger (6–24 months) and older (<24–60months) children. Treatment responders represented by open triangles (▵) and nonresponders by solid squares (▪). Higher Homapak dose administered at >24 months. Broken line depicts optimal cut-off value for AUC(0,336 h) to predict treatment response
Using logistic regression, the AUC(0,336 h) for SDx that best discriminated between responders and nonresponders was determined to be 12 000 μg ml−1 h with which 13/24 nonresponders (54%) and 53/59 responders (90%) were correctly predicted. Similarly for CQ the corresponding optimal AUC(0,336 h) cut-off value was 76 μg ml−1 h which correctly predicted 12/24 nonresponders (50%) and 52/59 responders (88%) (Figure 3).
Safety by age group
No adverse reactions were observed or reported in this small group of young children, indicating that the estimated exposure concentrations for Cmax and AUC are safe for both CQ and SDx.
Discussion
Pharmacokinetics of CQ and SDx in children with malaria
In the present study we have applied a population approach [13] to evaluate the pharmacokinetics of CQ and SDx in the fixed-dose CQ + SDx/PYR combination, using sparse data from children with uncomplicated falciparum malaria as the target patient population [14–16]. However, collection of field samples for pharmacokinetic studies in children has particular challenges since frequent sampling over a follow up period is required. Children tend to get less willing to have repeat finger pricks. When such situations arise, it may be difficult to obtain the correct blood volumes especially using capillary tubes. In this study such samples were discarded thereby reducing the number that were finally considered for analysis. Furthermore problems of blood sampling also led to failure to collect adequate samples for the analysis of PYR concentrations. Our discussion has therefore been limited to CQ and SDx only, and in this case SDx concentrations have been used as surrogates for PYR concentrations in the combination.
From the samples processed, our findings show that the summary pharmacokinetic parameters of CQ and SDx are within the range we had previously described in adult volunteers after Homapak administration [17]. Furthermore, we have shown that two-compartmental models best describe the pharmacokinetics of CQ and SDx in children treated for uncomplicated falciparum malaria. Chloroquine indeed does exhibit multicompartmental pharmacokinetics as was shown by other investigators [18–21], and in our own study [17]. Although in our healthy volunteers [17] and in a recent study by Barnes et al.[4] SDx disposition was reported as one-compartmental, the two compartmental pharmacokinetics of SDx in the present study are in agreement with the results reported by Weidekamm et al.[22]. Differences in the sampling periods or pharmacokinetic analysis approach (individual or population modelling) could explain the divergent compartmental dispositions in these studies.
Relationship between exposure (AUC(0,336 h)) and response
This study demonstrates that the drug exposures (AUC(0,336 h)) for CQ and SDx individually were significant predictors for cure. Combined exposures of the two drugs were also significant predictors for cure, thus showing that the exposure by 336 h may be important for therapy with CQ and SDx. Earlier exposures (AUC(0,24 h) and AUC(0,72 h)), did not significantly predict cure within the given dosages of CQ and SDx. A previous study found no significant difference in AUC(0,8 h) of SDx between those who were cured and those whose treatment failed [23]. We could not make a reasonable estimation of the AUC(0,∞) since our sampling period covered around two half-lives only, thus limiting the use of this parameter in the evaluation of response in this study. Even so, these conflicting results indicate that AUC may not be the best parameter for evaluating treatment outcomes with antimalarials, as it is dependent on the sampling duration and the given dose. Furthermore the relevant exposure (AUC) and duration to ascertain cure with either CQ or SDx is not well established. This may be very difficult to determine, since there are different drugs involved. Such a study would also require a large number of treatment failures, which would cause ethical concerns.
Proposed modification of dose regimen in children 6 months to 5 years
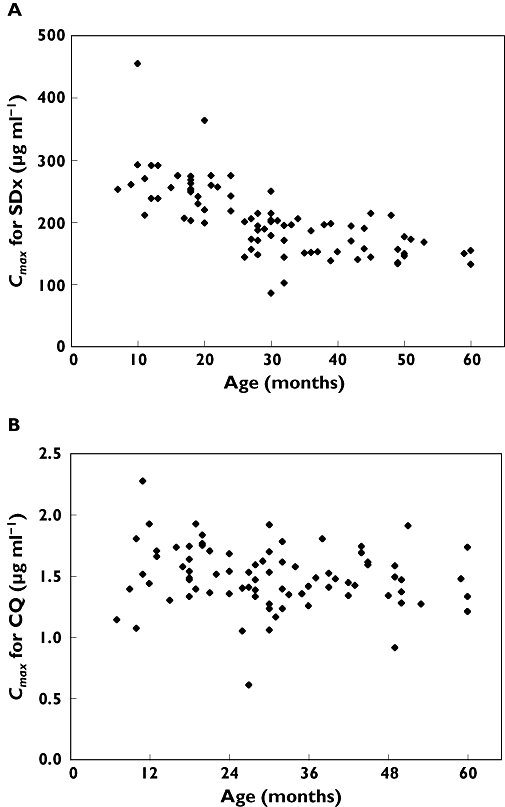
In this study, compartmental pharmacokinetic models were developed for CQ and SDx in children ranging from 6 to 60 months in age. The population pharmacokinetic model for CQ did not imply any correlation between body weight and age with the model parameters, while the SDx modelling results indicated an effect of body weight on the apparent central volume of distribution for the compound. The lack of covariate influence on CQ pharmacokinetics is an interesting finding and implies the current empirical age-based dosing to be irrational in this age range. Furthermore, these results suggest a weight-based dosing of SDx to be more appropriate than the current dosing based on age in children older than 6 months. Supporting this argument is the finding in this study that children receiving the lower Homapak dose had lower exposures to both SDx and CQ and a higher incidence of treatment failures, as compared with children receiving the higher Homapak dose. The current practice to administer 50% lower doses to children ≤24 months therefore seems suboptimal. Figure 4 shows distributions of AUC(0,336 h) for SDx (A) and CQ (B) acquired by simulations, where all children received the higher Homapak dose.
Figure 4.
Predicted AUC(0,336 h) vs. age (A) for SDx and (B) for CQ if the higher Homapak dose had been administered to all children 6 months to 5 years. Broken line depicts optimal cut-off value for AUC(0,336 h) to predict treatment response
If the higher dose is given to all, only four children would achieve SDx AUC(0,336 h) values below the suggested cut-off value for treatment response, and no child would have AUC(0,336 h) values below the corresponding cut-off value for CQ.
From a safety perspective it is important to avoid exceedingly high concentrations during the initial phase of dose intake. Figure 5 show the predicted Cmax values for SDx and CQ, respectively, after the first dose intake for a dose regimen with a higher Homapak dose (150 mg CQ (base) and 500 mg/25 mg of SDx/PYR, respectively) given to all the children between 6 months to 5 years. The modified dose regimen would generate Cmax concentrations for SDx which are not substantially higher in the age range 6–24 months than at >24 months, while for CQ no difference between the age ranges in the distribution of Cmax is apparent. By comparison, Barnes et al.[4] have suggested a much higher dose modification for SDx/PYR in children 2–5 years with uncomplicated falciparum malaria, doubling what we now propose. However, due to methodological differences between these two studies, it would be impossible to make comparisons of the predicted concentrations if all children in the present study were given 1000 mg/50 mg of SDx/PYR in the respective age groups.
Figure 5.
Predicted Cmaxvs. age (a) for SDx and (b) for CQ if the higher Homapak dose had been administered to all children 6 months to 5 years
We found a strong relationship between high drug exposure and cure, which in the model correlated well with body weight. In countries where these drugs are still relatively efficacious or where programs still use these combinations, the present study has shown that response to CQ + SDx/PYR can be improved by giving higher doses irrespective of body weight for children from 6 months to 5 years of age. A recommendation for children between 6 months to 24 months to be administered the higher dose regimen given to the older children can still be done within safe Cmax limits to avoid any possible adverse effects.
In conclusion, a population model applied to the pharmacokinetics of CQ and SDx in uncomplicated falciparum malaria showed that children who were given the lower dose achieved lower exposure and had poor response. A proposed dose modification to give the higher (full-strength) dose from 6 months to 5 years would greatly improve the exposure and the proportion of children who would be cured. However, since the exact relevant exposure is not well defined, a well-controlled study in a larger population of children would be needed to determine the exact exposure important for the prediction of cure.
We are grateful to Margareta Mahindi, Karolinska Institutet, Sweden, for her contribution in drug concentration analysis. This study was funded by SIDA/SAREC, Grant No. SWE 2004–098, Makerere University-Karolinska Institutet research collaboration.
REFERENCES
- 1.World Health Organization. Guidelines for the treatment of malaria. Geneva: WHO; 2006. WHO/HTM/MAL/2006.1108. [Google Scholar]
- 2.Ginsburg H. Should chloroquine be laid to rest? Acta Trop. 2005;96:16–23. doi: 10.1016/j.actatropica.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 3.Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, Taylor TE, Plowe CV. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355:1959–66. doi: 10.1056/NEJMoa062032. [DOI] [PubMed] [Google Scholar]
- 4.Barnes KI, Little F, Smith PJ, Evans A, Watkins WM, White NJ. Sulfadoxine-pyrimethamine pharmacokinetics in malaria: pediatric dosing implications. Clin Pharmacol Ther. 2006;80:582–96. doi: 10.1016/j.clpt.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Ministry of Health (MoH) Uganda. Implementation guidelines for the Home Based Management of Fever Strategy. 1. Kampala: Ministry of Health; 2002. [Google Scholar]
- 6.Obua C, Ntale M, Lundblad MS, Mahindi M, Gustafsson LL, Ogwal-Okeng JW, Anokbonggo WW, Hellgren U. Pharmacokinetic interactions between chloroquine, sulfadoxine and pyrimethamine and their bioequivalence in a generic fixed-dose combination in healthy volunteers in Uganda. Afr Health Sci. 2006;6:86–92. doi: 10.5555/afhs.2006.6.2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corvaisier S, Charpiat B, Mounier C, Wallon M, Leboucher G, Al Kurdi M, Chaulet JF, Peyron F. Population pharmacokinetics of pyrimethamine and sulfadoxine in children treated for congenital toxoplasmosis. Antimicrob Agents Chemother. 2004;48:3794–80. doi: 10.1128/AAC.48.10.3794-3800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trenque T, Simon N, Villena I, Chemla C, Quereux C, Leroux B, Jaussaud R, Remy G, Dupouy D, Millart H, Pinon JM, Urien S. Population pharmacokinetics of pyrimethamine and sulfadoxine in children with congenital toxoplasmosis. Br J Clin Pharmacol. 2004;57:735–41. doi: 10.1111/j.1365-2125.2004.02077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization. Assessment and monitoring of antimalarial drug efficacy for the treatment of uncomplicated falciparum malaria. Geneva: WHO; 2003. WHO/HTM/RBM/2003.50. [Google Scholar]
- 10.Minzi OM, Rais M, Svensson JO, Gustafsson LL, Ericsson O. High-performance liquid chromatographic method for determination of amodiaquine, chloroquine and their monodesethyl metabolites in biological samples. J Chromatogr B Anal Technol Biomed Life Sci. 2003;783:473–80. doi: 10.1016/s1570-0232(02)00727-4. [DOI] [PubMed] [Google Scholar]
- 11.Bergqvist Y, Hjelm E, Rombo L. Sulfadoxine assay using capillary blood samples dried on filter paper – suitable for monitoring of blood concentrations in the field. Ther Drug Monit. 1987;9:203–7. doi: 10.1097/00007691-198706000-00013. [DOI] [PubMed] [Google Scholar]
- 12.Jonsson EN, Karlsson MO. Xpose- an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 13.Beal SL, Sheiner LB. Estimating population kinetics. Crit Rev Biomed Eng. 1982;8:195–222. [PubMed] [Google Scholar]
- 14.Whiting B, Kelman AW, Grevel J. Population pharmacokinetics theory and clinical applications. Clin Pharmacokinet. 1986;11:387–401. doi: 10.2165/00003088-198611050-00004. [DOI] [PubMed] [Google Scholar]
- 15.Aarons L. Population pharmacokinetics: theory and practice. Br J Clin Pharmacol. 1991;32:669–70. [PMC free article] [PubMed] [Google Scholar]
- 16.Kauffman RE, Kearns GL. Pharmacokinetic studies in paediatric patients: clinical and ethical considerations. Clin Pharmacokinet. 1992;23:10–29. doi: 10.2165/00003088-199223010-00002. [DOI] [PubMed] [Google Scholar]
- 17.Obua C, Gustafsson LL, Aguttu C, Anokbonggo WW, Ogwal-Okeng JW, Chiria J, Hellgren U. Improved efficacy with amodiaquine instead of chloroquine in sulfadoxine/pyrimethamine combination treatment of falciparum malaria in Uganda: experience with fixed-dose formulation. Acta Trop. 2006;100:142–50. doi: 10.1016/j.actatropica.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Walker O, Dawodu AH, Adeyokunnu AA, Salako LA, Alvan G. Plasma chloroquine and desethylchloroquine concentrations in children during and after chloroquine treatment for malaria. Br J Clin Pharmacol. 1983;16:701–5. doi: 10.1111/j.1365-2125.1983.tb02244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frisk-Holmberg M, Bergkvist Y, Domeij-Nyberg B, Hellstrom L, Jansson F. Chloroquine serum concentration and side effects: evidence for dose-dependent kinetics. Clin Pharmacol Ther. 1979;25:345–50. doi: 10.1002/cpt1979253345. [DOI] [PubMed] [Google Scholar]
- 20.Gustafsson LL, Walker O, Alvan G, Beermann B, Estevez F, Gleisner L, Lindstrom B, Sjoqvist F. Disposition of chloroquine in man after single intravenous and oral doses. Br J Clin Pharmacol. 1983;15:471–9. doi: 10.1111/j.1365-2125.1983.tb01532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adelusi SA, Dawodu AH, Salako LA. Kinetics of the uptake and elimination of chloroquine in children with malaria. Br J Clin Pharmacol. 1982;14:483–7. doi: 10.1111/j.1365-2125.1982.tb02016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weidekamm E, Plozza-Nottebrock H, Forgo I, Dubach UC. Plasma concentrations of pyrimethamine and sulfadoxine and evaluation of pharmacokinetic data by computerised curve fitting. Bull World Health Organ. 1982;60:115–22. [PMC free article] [PubMed] [Google Scholar]
- 23.Dzinjamala FK, Macheso A, Kublin JG, Taylor TE, Barnes KI, Molyneux ME, Plowe CV, Smith PJ. Association between pharmacokinetics and in vivo therapeutic efficacy of sulfadoxine-pyrimethamine in Malawian children. Antimicrob Agents Chemother. 2005;49:3601–6. doi: 10.1128/AAC.49.9.3601-3606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]