

Abstract
Systemic lupus erythematosus (SLE, lupus) results from immune-mediated damage to multiple organs. Its pathogenesis should be viewed as a series of steps, beginning with impaired immune regulation that permits self-reactive T–B-cell activation, which results in the production of autoantibodies. Activated T and B cells then infiltrate tissues, which along with autoanti-body and immune complex deposition, triggering local events that ultimately cause organ damage. Although improved understanding of early autoimmune events might open up avenues for disease prevention, future investigations must focus on the mechanisms of end-organ damage in model systems and how to translate this knowledge into human disease. Understanding the mechanisms of each pathogenetic step would provide a rational basis for the development of disease stage-specific diagnostic markers and treatments.
The challenge
Systemic lupus erythematosus (SLE) is the most heterogeneous autoimmune disease that affects multiple organs [1]. The disease progresses through four broad stages, that is, the presence of autoantibodies against a variety of ubiquitous self-antigens, deposition of autoantibodies and immune complexes in tissues, development of tissue inflammation and finally, tissue damage and fibrosis. Although there has been a marked improvement in five-year survival from <50% in the 1950s to >90% in the 1990s, many patients still suffer from disease flares (i.e. uncontrolled disease with exacerbations) and complications, such as infections, premature atherosclerosis and cognitive dysfunction [2]. Strikingly, the incidence of SLE has nearly tripled in Minnesota over the past four decades, which is not necessarily a result of improved recognition of mild disease because the percentages of most major manifestations remain similar in 1950–1979 versus 1980–1992 cohorts [3]. Hence, there is an urgent need to assess our current research paradigms. Despite advances in our understanding of the role of autoimmunity and genetic factors in the pathogenesis of lupus, mechanisms of disease development and progression remain largely unknown. The companion review in this issue of Trends in Immunology by Croker and Kimberly highlights certain challenges facing the clinician and clinical scientist dealing with SLE. This opinion will begin with an overview of animal models of lupus and discuss what has been learnt from model systems about this disease. The opinion hopes to convey how animal model investigations can provide fundamental insights relevant to human disease and how this knowledge can be translated to assist in the development of new diagnostic markers and targeted treatments.
Heterogeneity of SLE: different animal models represent its various stages and subsets
Although clinical criteria have helped clinicians in making the diagnosis of SLE, the marked heterogeneity in disease expression has posed a difficulty in clearly defining the disease and formulating mechanistic investigations. Consequently, many investigators have turned toward animal models, which develop a homogeneous disease recapitulating the serological and histopathological features of SLE [4]. Examples of such models include the (NZBxNZW)F1 (BWF1), MRL-MpJ and NZM.2410 mouse strains.
Up to one-third of the general population might have some antibodies reactive against self-antigens [5]. Yet, <1% of the general population develops full-blown systemic autoimmune disease, such as SLE [6] (Figure 1). Thus, most individuals who have autoantibodies are protected from their pathological consequences. Some individuals do have tissue deposition of autoantibodies but have no inflammation; others experience immune deposition as well as inflammation but have no chronic tissue damage, whereas still others develop immune deposition and inflammation as well as progressive tissue destruction. Serendipitous discoveries or deliberate attempts have led to the identification of animal models that appear to recapitulate these various steps or stages of disease (Figure 2). For example, BALB/c mice immunized with a DNA surrogate peptide develop autoantibodies and extensive immune deposition but have no renal inflammation [7], whereas BALB/c mice injected with the hydrocarbon oil pristane develop immune deposition and a limited kidney inflammation but no kidney failure [8,9]. However, genetically lupus-prone mouse strains develop lethal renal disease spontaneously, with strain-dependent variation in disease patterns and severity [4,10]. For example, kidney disease in BWF1 mice progresses from mild focal glomerulonephritis in early stages to generalized inflammation and ultimately to chronic fibrosis, characterized by glomerulosclerosis and tubulo-interstitial fibrosis. MRL-MpJ-Faslpr/lpr (MRL-lpr) mice develop massive glomerular and interstitial inflammation but have generally limited glomerulosclerosis and fibrotic changes, whereas NZM.2410 mice develop profound glomerulosclerosis without much glomerular inflammation [10].
Figure 1.

Autoimmunity is common in the general population, whereas autoimmune diseases are rare. The presence of self-reactive T and B cells and autoantibodies is common in the general population. Only a small percentage of these individuals develop inflammatory disease and this is generally self-limiting. Some of these individuals, however, develop a more persistent but mild and usually localized, autoimmune disease, which is referred to as undifferentiated autoimmune or connective tissue disease. Finally, a small percentage of patients develop a full-blown autoimmune syndrome, such as SLE. The determinants of disease progression from the initiation of autoimmunity to full-blown disease expression can include the loss of suppressor mechanisms and the gain of pathogenic factors.
Figure 2.
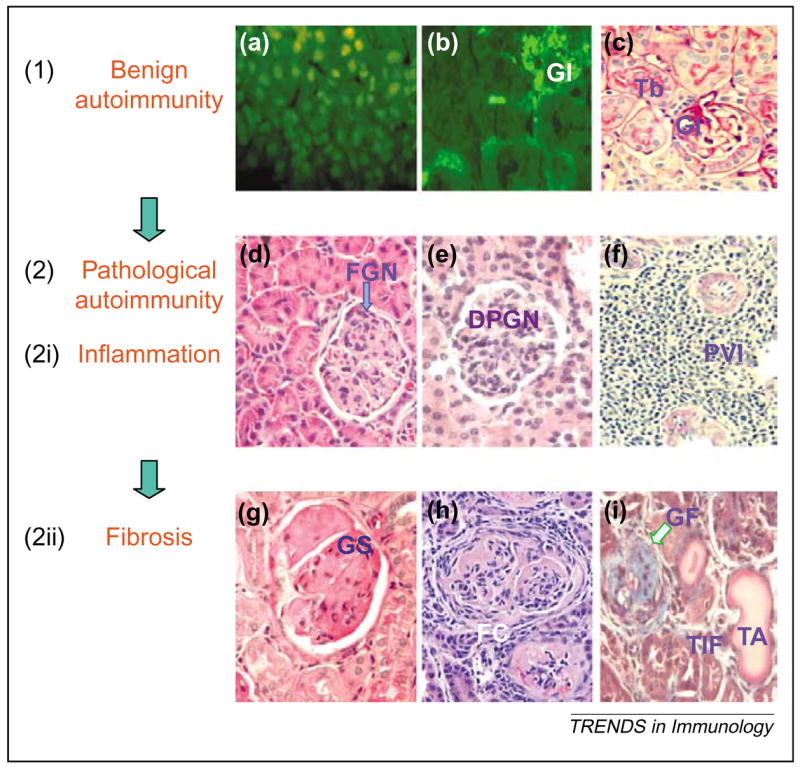
Major stages of disease progression in SLE (e.g. lupus nephritis). The disease course can be envisioned in two broad stages, namely (1) benign and (2) pathological autoimmunity. Each of these broad stages can be further sub-classified into two stages; the benign autoimmunity stage into the stage of (a) autoantibody development and of (b) autoantibody and immune complex deposition in tissues, and the pathological autoimmunity stage into stages of (2i) (d–f) inflammation and (2ii) (g–i) fibrosis. (a) Anti-nuclear antibody in the serum of an SLE patient, detected by an indirect immunofluorescence assay using Hep-2 cells; stained nuclei appear green/yellow. (b) Glomerular (GI) and tubular (Tb) deposition of IgG, detected by a direct immunofluorescence assay; stained tissues appear pale green. (c–i) Renal histology in (c,f–h) periodic acid-Schiff (PAS), (d,e) hematoxylin and eosin (H&E) (i) and Masson’s trichrome stained sections. (c) Normal glomerulus and Tb. (d–f) Stages of renal inflammation in sections showing localized glomerular inflammation [focal glomerulonephritis (FGN)] in (d), diffuse proliferative glomerulonephritis (DPGN) with inflammatory cell infiltration and proliferation of glomerular cells in (e) and intense perivascular infiltration (PVI) in (f). (g–i) Stages of renal fibrosis in sections showing glomerulosclerosis (GS) (i.e. extracellular matrix deposition in glomeruli) in (g), fibrous crescent (FC) (i.e. periglomerular fibrosis) in (h) and advanced glomerular fibrosis with scarring (GF), dilated atrophic tubules (TA) and tubulo-interstitial fibrosis (TIF) (fibrosis in the interstitium of kidneys with loss of tubules) in (i). The disease course might serially progress from the steps (a) to (i) in some animal models, such as BWF1 mice. In other models, however, the disease might be limited to early steps, for example, BALB/c mice immunized with a DNA surrogate peptide develop steps (a) and (b) but have no inflammation (c) [7] and BALB/c mice injected with pristane develop steps (a) and (b) and have limited glomerular inflammation (d) [8,22]. In some other models, the disease advances further but exhibits predominance of one of the other steps, for example, MRL-lpr mice develop massive glomerular (e) and interstitial (f) inflammation but have generally limited glomerulosclerosis and fibrotic changes, whereas NZM.2410 mice develop profound glomerulosclerosis (g) without much glomerular inflammation [10]. These different animal models might represent different subsets or stages of human SLE.
Consistent disease patterns in individual mouse strains and the relative ease in testing renal disease have driven extensive investigations into lupus nephritis. Model investigations into lupus involvement of other organs, however, have been limited, perhaps owing to the variability in disease expression or the lack of appropriate tools to investigate these manifestations. For example, neuropsychiatric lupus is frustrating to manage because there are no reliable disease markers and treatment remains largely empiric [11]. Although some forms of neuropsychiatric lupus develop in BWF1, MRL-lpr and NZM.2410 mice [12], animal investigations have been limited.
An increased risk of cardiovascular disease is another major problem in SLE [13]. Studies in NZW/BXSB F1 males, which develop myocardial infarction as a result of immune complex deposition [14], have shed some light on this manifestation. However, it is unclear whether a similar mechanism characterizes human disease. MRL mice develop dermatitis, arthritis and vasculitis, although the incidence of these manifestations is variable [15,16]. Thus, although the animal investigations have helped in understanding some steps in the pathogenesis of SLE, mouse models have not fully recapitulated the waxing and waning nature and the full spectrum of human SLE, suggesting a need for a continuing search for additional model systems. For example, similar to human SLE, SLE in dogs is a chronic disease with alternating periods of remission and relapses and manifests with fever, polyarthritis, glomerulonephritis, mucocutaneous lesions, lymphadenopathy and splenomegaly [17].
Tracing the steps of the pathogenesis of SLE
The clinical syndrome, known as human lupus, might actually encompass several diseases that have similar clinical features, yet with different mechanisms of pathogenesis. Here, we envision the natural course and pathogenesis of SLE as a series of steps that are narrated in the following working roadmap (Figure 3). It is hoped that a better understanding of this roadmap will facilitate the development of treatment strategies that can block the progression of disease at each step in most patients.
Figure 3.

Roadmap to disease development and progression in SLE. According to our working model, the lupus disease progresses through a series of steps: (1) immune dysregulation and (2) immunity against self generally occur long before the onset of the first symptoms that are manifestations of (3) autoimmune ‘invasion’ and (4) consequent inflammation. The autoimmune disease up to this stage is generally amenable to correction by the defenses of the body or by anti-inflammatory or immunosuppressive treatments. (5) Some patients, however, exhibit impaired tissue response to inflammation or have local or systemic factors that perpetuate tissue fibrosis and organ damage. The patients at stage (5) and beyond generally do not respond to currently available treatments.
Step 1: insufficient immune regulation in SLE –harnessing the capacity of inhibitory or suppressor T cells to suppress lupus
Otherwise healthy, non-autoimmune mice can be induced to develop antibodies to DNA and mild nephritis by in vivo stimulation of the Th cells that are capable of promoting autoantibody production [9]. These animals, however, completely recover from such an episode of autoimmunity, despite persistent exposure to autoreactive Th cells. Recovery from disease in these mice correlates temporally with the appearance of certain CD8+ T, CD4+CD25+ T and natural killer T (NKT) cells that are capable of suppressing autoantibody production (Figure 4). This suggests that self-reactive B and Th cells exist in the normal immune repertoire but are kept in control to avoid pathological autoimmunity. These control mechanisms are defective in lupus mice, which have impaired activation of such inhibitory, suppressor and regulatory T (Treg) cells [9]. Impairments in CD8+ T-cell suppressor functions have also been described in human SLE [18]. These observations might have therapeutic implications because we can correct this impairment in lupus mice by modifying the delivery of peptide antigens, for example, by DNA vaccination with minigenes that encode certain T-cell epitopes that are derived from anti-DNA variable regions [19,20]. Vaccination with nucleosome-derived peptides can also induce suppressor CD8+ and CD4+CD25+ T cells, which suppress disease in lupus mice [21]. Because similar T-cell epitopes exist in humans, strategies described here might be useful in therapy of the human disease.
Figure 4.

Inhibitory and suppressor mechanisms that protect from the development of pathological autoimmunity. (a) Lupus-prone BWF1 and (b) normal CWF1 mice were immunized with anti-DNA antibody V-region peptides and monitored for the development of anti-DNA antibodies (and disease). Most T-cell lines generated from immunized BWF1 mice are CD4+ Th cells that promote anti-DNA antibody formation in vitro [9]. By contrast, although CWF1 mice develop CD4+ Th cells during initial immunizations, most T-cell lines, including CD8+ cells, CD4+CD25+Treg cells and NKT cells, generated from CWF1 mice recovering from disease suppress anti-DNA antibody production by lupus (BWF1) B cells [9,19,20]. Abbreviation: CTLs, cytotoxic T lymphocytes.
NKT cells that co-express the NK receptor and invariant T-cell receptor (TCR) are reduced in numbers and exhibit impaired functions in lupus mice before the onset of clinical disease [15,22], suggesting a possible protective role for these cells. In fact, treatment with α-galactosylceramide (α-GalCer), which activates NKT cells, suppresses lupus dermatitis [15] and nephritis [23,24], whereas loss of NKT cells in CD1d-null mice exacerbates lupus dermatitis or nephritis [16,22,24]. Evidence also suggests a protective role for these cells in humans. First, patients with SLE have reduced numbers of NKT cells [25,26]. Second, CD161, a marker of NK and NKT cells, is the most significantly decreased lineage marker in the blood from SLE patients [27]. Third, SLE disease activity appears to correlate inversely with circulating NKT-cell numbers [26]. Thus, pharmacological modulation of NKT cells might be a target of therapeutic intervention.
Thus, animal studies show clearly that the hyperactive interaction between Th cells and autoimmune B cells can be interrupted by the induction of suppressor T cells. Studies have also begun to identify impairments in the numbers, functions and genes of suppressor cells in humans with SLE [18,25–27]. Identification of factors [e.g. reduced transforming growth factor-β (TGF-β)] that contribute to impaired suppressor T-cell function would be an important area of investigation [28], so that these cells can be modulated in vivo by correcting these factors.
Step 2: activation of autoreactive T and B cells
Strikingly, impairments in almost every step of the immune response occur in SLE. As narrated in the following substeps, vigorous attempts are underway to characterize these impairments in animal models and to translate this knowledge into human disease.
Step 2a: autoantigens in SLE and the prospects of antigen-specific therapies
Immunologists have been fascinated with the idea of identifying disease-specific autoantigens and using them in antigen-specific therapies [29,30]. This task becomes particularly difficult in SLE, where, in contrast to organ-specific autoimmune diseases, there is no organ-specific autoantigen target. Because many pathogenic autoantibodies bind DNA, there have been attempts to tolerize or turn off DNA-specific B cells. For example, more than 30 years ago, Borel and colleagues showed that it is possible to prevent lupus in an animal model by inducing tolerance to denatured DNA [31]. About 15 years later, this finding was translated into human disease by showing that a DNA–human IgG conjugate inhibits the formation of anti-dsDNA antibodies in vitro by lymphoid cells from SLE patients [32]. Such studies eventually led to a clinical trial to evaluate a dsDNA-directed B-cell tolerogen, a synthetic molecule with the ability to bind dsDNA antibodies, thus leading to anergy or apoptosis of B cells, which has shown delayed renal flares and reduction of anti-dsDNA antibodies in a subgroup of patients [33].
Since the 1980s, when anti-DNA production was shown to require T-cell help [30,34], there have been arduous and ingenious efforts to identify pathogenic T-cell epitopes in the nucleosome and anti-DNA Ig variable regions, which activate autoreactive T cells that exacerbate autoantibody production and murine lupus [30,35,36]. Importantly, treatment with these peptides that tolerize pathogenic Th cells, suppresses murine lupus [21,30,36]. Ongoing studies are trying to map such epitopes in humans [37,38] and to initiate clinical trials using analogous peptides [30,39,40]. In fact, a Phase II clinical trial using anti-DNA antibody-derived peptides is undergoing design by Teva Pharmaceuticals. It is important to emphasize that a rigorous characterization of the exquisite T-cell epitopes that activate suppressor T cells, versus those that stimulate pathogenic Th cells, in humans is crucial to ensure that we do not run into premature disappointment, as seen in some other antigen therapy trials. Complicating the selection of peptides for treatment, our murine studies suggest that suppressor and Th-cell epitopes might co-localize or overlap [20], as if nature has done a fine balancing act by putting together the ‘protective’ and ‘pathogenic’ epitopes. Further complicating the issue of antigen therapies is the presence of polymorphisms in the HLA regions, thus requiring formulation and testing of expensive individualized therapy. Anticipating this problem, investigators are attempting to develop consensus and/or promiscuous autoantigenic epitopes that can bind many HLA molecules and modulate a broad repertoire of self-reactive T cells [20,41–43]. Parallel efforts in this area in humans and mice could be helpful in developing antigen-specific therapies.
Step 2b: antigen-processing and -presentation abnormalities
Other studies have begun to identify impairments in antigen-presenting cell (APC) functions in lupus [44–46]. For example, dendritic cells (DCs) and macrophages display increased maturation and contribute to T-cell hyperactivity in a mouse model of lupus [46]. Further, Langerhans cells in MRL-lpr mice exhibit impaired migration to draining lymph nodes [45]. Importantly, correction of this defect results in improved lupus dermatitis [15,45]. Translation of these findings into human disease might open up novel therapeutic options.
Step 2c: T-cell abnormalities in SLE
T cells from patients and mice with SLE exhibit intrinsic T-cell abnormalities, such as diminished T-cell activation thresholds and dysregulated apoptosis. For example, TCR–CD3-mediated stimulation of SLE T cells shows aberrant calcium flux and reduced interleukin-2 (IL-2) production [47], which might be due to an anti-TCR–CD3 complex autoantibody that can activate the Ca2+–calmodulin kinase IV (CaMKIV) signaling cascade in T cells, thus resulting in downregulation of IL-2 transcription [48]. Epigenetic regulation of gene expression, such as histone acetylation and methylation, might also contribute to impaired SLE T-cell function [49]. Treatment with histone deacetylase inhibitors, such as trichostatin A, which correct these impairments and suppress lupus in mice [50], holds promise for humans.
An imbalance in the proapoptotic–antiapoptotic mechanisms contributes to the persistence of autoreactive clones. T cells from SLE patients resist anergy and activation-induced apoptosis by upregulating cyclooxygenase-2 (COX-2), along with the antiapoptotic molecule cellular FLICE (caspase-8)-inhibitory protein (c-FLIP) [51]. Because certain COX-2 inhibitors block pathogenic autoantibody production by causing the death of autoimmune Th cells [51], understanding these mechanisms can open new avenues for treatment.
Step 2d: uncontrolled T-cell help to B cells in SLE
T cells from patients and mice with SLE provide help to autoreactive B cells. Animal studies have clearly shown a role of excessive co-stimulation between T cell–B cell–APC through the CD40–CD40L or CD28–B7 pathways in lupus (reviewed in Ref. [52]). In fact, these studies have prompted a human SLE trial of soluble cytotoxic T lymphocyte-associated antigen 4 (CTLA4)–Ig fusion protein that blocks T cell-dependent B-cell functions [53]. Thus, blockade of T cell-mediated co-stimulation in human SLE might provide additional management options.
Step 2e: B-cell abnormalities in SLE
Genetic deletion of B cells leads to amelioration of murine lupus, suggesting a crucial role for B cells in lupus (reviewed in Ref. [54]). Given this evidence, B-cell depletion has emerged as a therapeutic option. The anti-CD20 antibody rituximab, which eliminates many B-cell subsets, reduces SLE disease activity [55], probably by suppressing the antigen-presenting effect of B cells on T-cell activation. Given the pivotal role of B cells in normal immune defenses, future studies should identify pathogenic B-cell subsets so that they can be targeted selectively for therapy. For example, genetic vaccination with MHC I-binding epitopes in the VH of anti-DNA antibodies preferentially ablates anti-DNA antibody-producing B cells and suppresses nephritis in lupus mice without affecting normal IgG levels [19]. Ongoing efforts to develop model systems to test such approaches for human disease could be useful.
Step 3: autoantibodies can cause SLE lesions
Although patients with SLE have autoantibodies against several different specificities, they are usually restricted to a recurring set of autoantigens. Such restricted polyclonality might be related to a unique case of molecular mimicry, whereby variable regions of autoantibodies contain shared T-cell epitopes [56]. Thus, T cells stimulated by a peptide derived from one autoantibody can stimulate several different B cells that express the shared epitope [20,56]. The importance of these observations is that recognition of the ‘first’ or dominant epitopes might enable their selective targeting to turn off the ‘spreading’ of pathological autoimmunity. However, because such spreading of autoimmune responses occurs mostly before clinical disease onset, such an approach would only be feasible in humans, if we have access to patient samples before disease onset. Indeed, a recent case-controlled study of U.S. Armed Forces personnel showed that at least one lupus-related autoantibody was present in 88% of patients several years before they were diagnosed with SLE [57]. Similar to murine SLE, the autoantibody specificities diversified over time in these individuals, before the onset of symptoms. A caveat (as illustrated in Figure 1) is that most individuals with autoantibodies do not develop autoimmune disease. Nonetheless, prospective studies of autoantibody-positive patient cohorts are warranted to identify biomarkers, such as a panel of autoantibodies or some other immune or genetic markers, which might enable the prediction of disease development. Should biomarkers with strong predictive value for disease development be identified, studies in this cohort and parallel animal investigations would be warranted to explore potential disease preventive approaches, such as peptide and peptide gene vaccination, which have shown remarkable efficacy in preventing disease in animals [20,21,29,30,36,42].
The presence of autoantibodies before disease onset and their correlation with disease activity suggest their pathogenic role. In fact, in vivo transfer of certain autoantibodies initiates lupus in otherwise normal mice [30,34,54]. Perhaps, the most powerful evidence for the pathogenicity of autoantibodies in humans is the transient development of lupus in newborns born to mothers with anti-Ro/SSA (Sjogren’s syndrome A) antibodies [58]. Hence, future investigations should be directed at identifying and eliminating pathogenic autoantibodies.
Step 4: tissue inflammation and disease activity
Although ample evidence supports a pathogenic role for autoantibodies, it is unclear how they cause the myriad lesions of lupus. Multiple mechanisms have been proposed. For example, certain murine anti-DNA antibodies interact with antigens in glomerular basement membrane or hippocampal neurons, thus initiating death or dysfunction of local cells, such as podocytes or neurons, respectively [59,60]. The autoantibody and immune complex deposition triggers the activation of the complement cascade, chemotaxis and infiltration with immune cells, resulting in local inflammation. A clear dissection of these events in animal models in vivo and in humans, using tissue biopsy samples from patients at different stages of disease, could be helpful in designing specific biomarkers and treatments.
Despite overwhelming data supporting the pathogenic role of anti-DNA antibodies, there are several instances in which the occurrence of tissue lesions can be ‘uncoupled’ from the presence of autoantibodies. For example, MRL-lpr mice rendered deficient in secreted IgG still develop some glomerulonephritis [61]. Similarly, congenic mice have been derived that contain certain lupus susceptible loci that predispose to autoantibody production but no tissue lesions, whereas strains harboring other loci develop tissue lesions but no autoantibodies [62]. Sometimes mice have extensive immune deposition but no pathological lesions [7]. This suggests that autoimmunity contributes to, but might not be sufficient to develop, tissue lesions, and that local factors, such as cytokines and chemokines, might have a crucial role in the development of tissue lesions. For example, antibody or pharmacological blockade or germ line deletion of certain cytokines, such as IL-4 and TGF-β, suppresses the development of tissue lesions, without any obvious effects on autoantibody levels [10,63,64]. These studies, however, do not exclude the possibility that the more relevant pathogenic auto-antibodies might differ in antigen specificity, binding affinity and Ig isotype and/or subclass, making their detection more difficult. It is also possible that autoantibodies might contribute to tissue lesions in some individuals but not in others. Additionally, autoimmunity might have a role in the initiation, but not in perpetuation, of tissue lesions. Conditional gene targeting approaches, where autoantibody production can be switched on and off at different time points, might clarify these issues.
Step 5: tissue fibrosis and organ damage
Results of repeated kidney biopsies in SLE patients and longitudinal analysis of renal pathology in BWF1 mice suggest that lupus nephritis progresses generally from focal glomerular infiltration to diffuse proliferative glomerulonephritis to glomerulosclerosis and eventually to tubulo-interstitial fibrosis [65]. Some models and patients, however, have only localized renal inflammation [8,9,22], whereas others have marked inflammation but not much renal fibrosis, and still others have severe glomerulosclerosis and fibrosis without much glomerular inflammation [10], as described earlier. Further, genetic studies in NZM mice suggest that congenic strains harboring some susceptibility loci develop acute glomerulonephritis, whereas strains bearing other loci develop chronic renal lesions [62]. Such heterogeneity in patterns of disease expression and progression, despite having similar underlying systemic autoimmunity, implies the contribution of local factors at the tissue level that influence the ultimate outcome of disease process. Identification of these factors will be crucial in designing disease stage- or subset-targeted treatments. In this regard, MRL-lpr mice that develop predominant inflammatory disease have high levels of interferon-γ (IFN-γ), whereas NZM.2410 mice that develop severe glomerulosclerosis have high in vivo levels of IL-4 [10]; and the BWF1 strain that experiences a ‘typical’ progression of nephritis from inflammation to fibrosis have increased TGF-β expression in kidneys. Importantly, treatment with angiontensin-converting enzyme inhibitors that reduces type 2 cytokines, such as IL-4 and TGF-β, suppresses chronic lupus nephritis without affecting glomerular activity score (inflammation) or anti-DNA antibody levels [64]. Direct antibody blockade of IL-4 or TGF-β, or genetic deletion of the type 2 cytokine transcription factor STAT6 (signal transducer and activator of transcription 6) also suppresses renal fibrosis, without reducing renal inflammation [10].
Further dissection of the role of these cytokines in the development of various stages of disease will require sophisticated conditional gene-targeting approaches. The translation of these animal studies into human disease will require a large collection of tissue biopsies, as well as the use of approaches, such as protein and gene microarrays, to identify differential responsiveness to, or production of, cytokines and chemokines. These studies might identify diagnostic and prognostic markers to distinguish inflammatory versus fibrotic stages of disease, as well as to identify therapeutic target molecules for late stages of lupus disease. These investigations are clearly necessary because there are currently no tests, short of an invasive kidney biopsy procedure, to predict the status of kidney disease. Further, there are no effective treatments for the tubulo-interstitial fibrotic stage of disease, which is a strong negative predictor of renal survival in patients with SLE [66].
Synthesis
Parallel developments in human disease observations and animal model investigations have helped in tracing some pathogenetic steps that lead to the manifestations of lupus. Some observations made in animal models are already being translated into human clinical trials. However, past experience has taught us that a rush to clinical trials must not occur without a full realization that the biological basis by which an intervention might suppress disease in an animal model might not be translatable directly to humans. Thus, although full elucidation of human lupus pathogenesis and the most effective therapies to suppress or prevent disease will clearly require intense investigations using model systems, a more intensive effort will be needed to translate this knowledge towards the development of disease stage-specific biomarkers and treatments for human disease.
Acknowledgments
I am supported by grants from the NIH/NIAMS and NIDDK (AR47322, DK69282 and AR50797). I thank Robert Kimberly sincerely for suggestions.
References
- 1.Malaviya AN, et al. Systemic lupus erythematosus in northern India: a review of 329 cases. J Assoc Physicians India. 1988;36:476–480. 484. [PubMed] [Google Scholar]
- 2.Borchers AT, et al. Surviving the butterfly and the wolf: mortality trends in systemic lupus erythematosus. Autoimmun Rev. 2004;3:423–453. doi: 10.1016/j.autrev.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Uramoto KM, et al. Trends in the incidence and mortality of systemic lupus erythematosus, 1950–1992. Arthritis Rheum. 1999;42:46–50. doi: 10.1002/1529-0131(199901)42:1<46::AID-ANR6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 4.Andrews BS, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teubner A, et al. Prevalence of circulating autoantibodies in healthy individuals. Med Klin. 2002;97:645–649. doi: 10.1007/s00063-002-1207-z. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson DL, et al. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 7.Deocharan B, et al. Differential effects of interleukin-4 in peptide induced autoimmunity. Clin Immunol. 2003;108:80–88. doi: 10.1016/s1521-6616(03)00096-2. [DOI] [PubMed] [Google Scholar]
- 8.Richards HB, et al. Interferon-γ is required for lupus nephritis in mice treated with the hydrocarbon oil pristane. Kidney Int. 2001;60:2173–2180. doi: 10.1046/j.1523-1755.2001.00045.x. [DOI] [PubMed] [Google Scholar]
- 9.Singh RR, et al. Induction of autoantibody production is limited in nonautoimmune mice. J Immunol. 2002;169:587–594. doi: 10.4049/jimmunol.169.1.587. [DOI] [PubMed] [Google Scholar]
- 10.Singh RR, et al. Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J Immunol. 2003;170:4818–4825. doi: 10.4049/jimmunol.170.9.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanly JG. Neuropsychiatric lupus. Rheum Dis Clin North Am. 2005;31:273–298. doi: 10.1016/j.rdc.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Brey RL, et al. Animal models for nervous system disease in systemic lupus erythematosus. Ann N Y Acad Sci. 1997;823:97–106. doi: 10.1111/j.1749-6632.1997.tb48382.x. [DOI] [PubMed] [Google Scholar]
- 13.Nikpour M, et al. Premature atherosclerosis in systemic lupus erythematosus. Rheum Dis Clin North Am. 2005;31:329–354. doi: 10.1016/j.rdc.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Kirzner RP, et al. Prevention of coronary vascular disease by transplantation of T-cell-depleted bone marrow and hematopoietic stem cell preparation in autoimmune-prone w/BF(1) mice. Biol Blood Marrow Transplant. 2000;6:513–522. doi: 10.1016/s1083-8791(00)70022-x. [DOI] [PubMed] [Google Scholar]
- 15.Yang JQ, et al. Repeated α-galactosylceramide administration results in expansion of NK T cells and alleviates inflammatory dermatitis in MRL-lpr/lpr mice. J Immunol. 2003;171:4439–4446. doi: 10.4049/jimmunol.171.8.4439. [DOI] [PubMed] [Google Scholar]
- 16.Yang JQ, et al. CD1d deficiency exacerbates inflammatory dermatitis in MRL-lpr/lpr mice. Eur J Immunol. 2004;34:1723–1732. doi: 10.1002/eji.200324099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DR. Canine systemic lupus erythematosus: new insights and their implications. J Comp Pathol. 1993;108:215–228. doi: 10.1016/s0021-9975(08)80286-1. [DOI] [PubMed] [Google Scholar]
- 18.Filaci G, et al. Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J Immunol. 2001;166:6452–6457. doi: 10.4049/jimmunol.166.10.6452. [DOI] [PubMed] [Google Scholar]
- 19.Fan GC, Singh RR. Vaccination with minigenes encoding V(H)-derived major histocompatibility complex class I-binding epitopes activates cytotoxic T cells that ablate autoantibody-producing B cells and inhibit lupus. J Exp Med. 2002;196:731–741. doi: 10.1084/jem.20020223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh RR. Prevention and control of reciprocal T–B cell diversification: implications for lupus-like autoimmunity. Mol Immunol. 2004;40:1137–1145. doi: 10.1016/j.molimm.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 21.Kang HK, et al. Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol. 2005;174:3247–3255. doi: 10.4049/jimmunol.174.6.3247. [DOI] [PubMed] [Google Scholar]
- 22.Yang JQ, et al. Immunoregulatory role of CD1d in the hydrocarbon oil-induced model of lupus nephritis. J Immunol. 2003;171:2142–2153. doi: 10.4049/jimmunol.171.4.2142. [DOI] [PubMed] [Google Scholar]
- 23.Singh AK, et al. The natural killer T cell ligand α-galactosylceramide prevents or promotes pristane-induced lupus in mice. Eur J Immunol. 2005;35:1143–1154. doi: 10.1002/eji.200425861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh RR, Yang J. CD1d-reactive NKT cells inhibit autoreactive B cells. Arthritis Rheum. 2004;50:S589. [Google Scholar]
- 25.Kojo S, et al. Dysfunction of T cell receptor AV24AJ18+, BV11+ double-negative regulatory natural killer T cells in autoimmune diseases. Arthritis Rheum. 2001;44:1127–1138. doi: 10.1002/1529-0131(200105)44:5<1127::AID-ANR194>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 26.Oishi Y, et al. Selective reduction and recovery of invariant Vα24JαQ T cell receptor T cells in correlation with disease activity in patients with systemic lupus erythematosus. J Rheumatol. 2001;28:275–283. [PubMed] [Google Scholar]
- 27.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horwitz DA, et al. The potential of human regulatory T cells generated ex vivo as a treatment for lupus and other chronic inflammatory diseases. Arthritis Res. 2002;4:241–246. doi: 10.1186/ar414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh RR. The potential use of peptides and vaccination to treat systemic lupus erythematosus. Curr Opin Rheumatol. 2000;12:399–406. doi: 10.1097/00002281-200009000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Datta SK. Major peptide autoepitopes for nucleosome-centered T and B cell interaction in human and murine lupus. Ann N Y Acad Sci. 2003;987:79–90. doi: 10.1111/j.1749-6632.2003.tb06035.x. [DOI] [PubMed] [Google Scholar]
- 31.Borel Y, et al. Prevention of murine lupus nephritis by carrier-dependent induction of immunologic tolerance to denatured DNA. Science. 1973;182:76–78. doi: 10.1126/science.182.4107.76. [DOI] [PubMed] [Google Scholar]
- 32.Borel Y, Borel H. Oligonucleotide linked to human gammaglobulin specifically diminishes anti-DNA antibody formation in cultured lymphoid cells from patients with systemic lupus erythematosus. J Clin Invest. 1988;82:1901–1907. doi: 10.1172/JCI113808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strand V, et al. Improvement in health-related quality of life in systemic lupus erythematosus patients enrolled in a randomized clinical trial comparing LJP 394 treatment with placebo. Lupus. 2003;12:677–686. doi: 10.1191/0961203303lu440oa. [DOI] [PubMed] [Google Scholar]
- 34.Hahn BH. Antibodies to DNA. N Engl J Med. 1998;338:1359–1368. doi: 10.1056/NEJM199805073381906. [DOI] [PubMed] [Google Scholar]
- 35.Singh RR, et al. T cell determinants from autoantibodies to DNA can upregulate autoimmunity in murine systemic lupus erythematosus. J Exp Med. 1995;181:2017–2027. doi: 10.1084/jem.181.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh RR, et al. Immune tolerance to autoantibody-derived peptides delays development of autoimmunity in murine lupus. J Clin Invest. 1995;96:2990–2996. doi: 10.1172/JCI118371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu L, et al. Major peptide autoepitopes for nucleosome-specific T cells of human lupus. J Clin Invest. 1999;104:345–355. doi: 10.1172/JCI6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalsi JK, et al. Peptides from antibodies to DNA elicit cytokine release from peripheral blood mononuclear cells of patients with systemic lupus erythematosus: relation of cytokine pattern to disease duration. Lupus. 2004;13:490–500. doi: 10.1191/0961203303lu1060oa. [DOI] [PubMed] [Google Scholar]
- 39.Sthoeger ZM, et al. Modulation of autoreactive responses of peripheral blood lymphocytes of patients with systemic lupus erythematosus by peptides based on human and murine anti-DNA autoantibodies. Clin Exp Immunol. 2003;131:385–392. doi: 10.1046/j.1365-2249.2003.02058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mauermann N, et al. Amelioration of lupus manifestations by a peptide based on the complementarity determining region 1 of an autoantibody in severe combined immunodeficient (SCID) mice engrafted with peripheral blood lymphocytes of systemic lupus erythematosus (SLE) patients. Clin Exp Immunol. 2004;137:513–520. doi: 10.1111/j.1365-2249.2004.02559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh RR, et al. Designing consensus ‘super-determinants’ that strongly influence autoantibody production in lupus. J Invest Med. 1998;46:230A. [Google Scholar]
- 42.Hahn BH, et al. Treatment with a consensus peptide based on amino acid sequences in autoantibodies prevents T cell activation by autoantigens and delays disease onset in murine lupus. Arthritis Rheum. 2001;44:432–441. doi: 10.1002/1529-0131(200102)44:2<432::AID-ANR62>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 43.Shi Y, et al. Promiscuous presentation and recognition of nucleosomal autoepitopes in lupus: role of autoimmune T cell receptor α chain. J Exp Med. 1998;187:367–378. doi: 10.1084/jem.187.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koller M, et al. Phenotypic and functional deficiencies of monocyte-derived dendritic cells in systemic lupus erythematosus (SLE) patients. Int Immunol. 2004;16:1595–1604. doi: 10.1093/intimm/dxh160. [DOI] [PubMed] [Google Scholar]
- 45.Eriksson AU, Singh RR. Impaired Langerhans dendritic cell functions in lupus-prone MRL-lpr mice can be restored by NKT cells. Arthritis Rheum. in press. [Google Scholar]
- 46.Zhu J, et al. T cell hyperactivity in lupus as a consequence of hyperstimulatory antigen-presenting cells. J Clin Invest. 2005;115:1869–1878. doi: 10.1172/JCI23049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsokos GC. Immune cell signaling and gene transcription in human lupus: the time has come. Int Rev Immunol. 2004;23:221–224. doi: 10.1080/08830180490452530. [DOI] [PubMed] [Google Scholar]
- 48.Juang YT, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest. 2005;115:996–1005. doi: 10.1172/JCI200522854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brooks WH. Autoimmune disorders result from loss of epigenetic control following chromosome damage. Med Hypotheses. 2005;64:590–598. doi: 10.1016/j.mehy.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 50.Mishra N, et al. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003;111:539–552. doi: 10.1172/JCI16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu L, et al. Human lupus T cells resist inactivation and escape death by upregulating COX-2. Nat Med. 2004;10:411–415. doi: 10.1038/nm1005. [DOI] [PubMed] [Google Scholar]
- 52.Hoffman RW. T cells in the pathogenesis of systemic lupus erythematosus. Clin Immunol. 2004;113:4–13. doi: 10.1016/j.clim.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 53.Finck BK, et al. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–1227. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 54.Shlomchik MJ, Madaio MP. The role of antibodies and B cells in the pathogenesis of lupus nephritis. Springer Semin Immunopathol. 2003;24:363–375. doi: 10.1007/s00281-003-0119-1. [DOI] [PubMed] [Google Scholar]
- 55.Looney RJ, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a Phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004;50:2580–2589. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 56.Singh RR, et al. Evidence for multiple mechanisms of polyclonal T cell activation in murine lupus. J Clin Invest. 1998;102:1841–1849. doi: 10.1172/JCI3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arbuckle MR, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 58.Lee LA. Transient autoimmunity related to maternal autoantibodies: neonatal lupus. Autoimmun Rev. 2005;4:207–213. doi: 10.1016/j.autrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 59.Kowal C, et al. Cognition and immunity; antibody impairs memory. Immunity. 2004;21:179–188. doi: 10.1016/j.immuni.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 60.Izui S, et al. In vitro demonstration of a particular affinity of glomerular basement membrane and collagen for DNA. A possible basis for a local formation of DNA–anti-DNA complexes in systemic lupus erythematosus. J Exp Med. 1976;144:428–443. doi: 10.1084/jem.144.2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan OT, et al. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waters ST, et al. Breaking tolerance to double stranded DNA, nucleosome, and other nuclear antigens is not required for the pathogenesis of lupus glomerulonephritis. J Exp Med. 2004;199:255–264. doi: 10.1084/jem.20031519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh RR, et al. Mechanisms of target organ damage in lupus: role of type 2 cytokines and TGF β. Clin Immunol Suppl. 2003;1:S52. [Google Scholar]
- 64.De Albuquerque DA, et al. An ACE inhibitor reduces Th2 cytokines and TGF-β1 and TGF-β2 isoforms in murine lupus nephritis. Kidney Int. 2004;65:846–859. doi: 10.1111/j.1523-1755.2004.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bajaj S, et al. Serial renal biopsy in systemic lupus erythematosus. J Rheumatol. 2000;27:2822–2826. [PubMed] [Google Scholar]
- 66.Daniel L, et al. Tubular lesions and tubular cell adhesion molecules for the prognosis of lupus nephritis. Kidney Int. 2001;60:2215–2221. doi: 10.1046/j.1523-1755.2001.00055.x. [DOI] [PubMed] [Google Scholar]