

Summary
All-atom molecular dynamics simulations on increasingly powerful computers have been combined with experiments to characterize protein folding in more detail over wider time ranges. The folding of small ultrafast folding proteins is being simulated on μs timescales, leading to improved structural predictions and folding rates. To what extent is “closing the gap” between simulation and experiment for such systems providing insights into general mechanisms of protein folding?
Introduction
All-atom molecular dynamics (MD) simulations can generate atomic-resolution structural models for states that are only indirectly accessible by experiment [1,2]. Recently, increased computer power has been used to generate multiple microsecond (μs) simulations. Research groups using such simulations have reported refolding from unfolded states for a variety of small model proteins and peptides, using both explicit and implicit solvent models. We will discuss the implications of new long timescale simulations using these brute-force methods, as well as how simulation is being used to model poorly populated states. Parallel developments that use increasingly sophisticated models with simplifying features for capturing longer time scales in realistic detail [3–6] will not be discussed.
Partially folded states are heterogeneous, consisting of many rapidly exchanging conformations. Ensemble averaging from such states complicates the interpretation of experimental data that would normally be readily analyzed for well-folded systems. Structural signals detected by ensemble methods such as nuclear magnetic resonance (NMR) or optical spectroscopy may originate from many different structures (rather than a single average structure) or from a small fraction of folded structure in a large ensemble of unfolded structures [7–9]. All-atom MD complements experimental approaches by providing a molecular framework for interpretation of experiment and fleshing out the protein folding process.
All-atom MD simulations provide a detailed representation of protein dynamics on picosecond (ps) to microsecond (μs) timescales. Robust treatment of protein-protein and protein-solvent interactions can be achieved with explicit solvent MD simulations. The effects of solvent can also be modeled implicitly to increase computational efficiency [10], but it comes at the cost of accuracy. For example, protein folding involves significant displacement of solvent that is not taken into account by such models [11]. Nonetheless, structures generated with implicit solvent simulation can be useful provided they have been experimentally validated.
Comparison of simulation and experiment is complicated by differences in accessible timescale. MD simulations typically run for tens to hundreds of nanoseconds (ns), up to 1–2 μs. Ultrafast folding proteins such as the engrailed homeodomain and the villin headpiece have folding times on the order of μs near room temperature. Generally, refolding from extended states using explicit solvent has been out of reach at these timescales. Instead, methods that increase the probability of observing a transition between states are used [12–14]. Also, thermal or chemical denaturant unfolding simulations can be used to characterize events along the folding pathway. Alternatively, biasing potential terms can be applied to limit the time that simulations linger in local minima or to limit sampling of conformational space [2,15].
Where does protein folding start? Residual structure in denatured states
Chemically denatured states are generally highly expanded and obey a power law dependence between expansion, as measured by radius of gyration (Rg) and protein length [16]. But, denatured states generated by other methods typically display signals incompatible with sequence-independent residual structure [17]. Recently, Förster-resonance energy transfer (FRET) was used to measure protein expansion in chemical denaturants. Eaton et. al. used single-molecule FRET to determine the radius of gyration of Protein L and CspTm under a range of guanidinium chloride (GdmCl) concentrations [18]. The two proteins collapsed to different degrees at low denaturant concentrations, contrary to simple length dependence of the Rg. This contrasts with previous results showing no collapse for protein L at low denaturant concentrations using time-resolved SAXS [19]. The dyes used for the FRET studies may be perturbing the denatured state. To investigate this further, both proteins were simulated in water/urea mixtures and the Rg increased with increasing urea; however, comparison with experiment is complicated by the different solvent conditions.
Protein Unfolding: Observation of reversible results
According to the principle of microscopic reversibility, the unfolding pathway should be the reverse of the folding pathway, and we expect unfolding simulations to shed light on folding. All-atom MD simulations of chymotrypsin inhibitor 2 (CI2) have demonstrated microscopic reversibility of the folding pathway [20]. A CI2 simulation conducted at the protein’s Tm (348K) crosses the transition state (TS) to the denatured state, remains there for 36 ns, and then refolds (Figure 1). The structures of both the starting and refolded states are very similar based on physical properties, topology, and pairwise contact maps. Detailed analysis of the loss of interactions during unfolding demonstrates that the same contacts are gained in reverse during the folding process. Thus, this study demonstrates that folding and unfolding are microscopically reversible within a single trajectory. Additional demonstrations of microscopic reversibility are forthcoming. Such studies provide justification for studying protein folding by more computationally efficient unfolding simulations.
Figure 1.
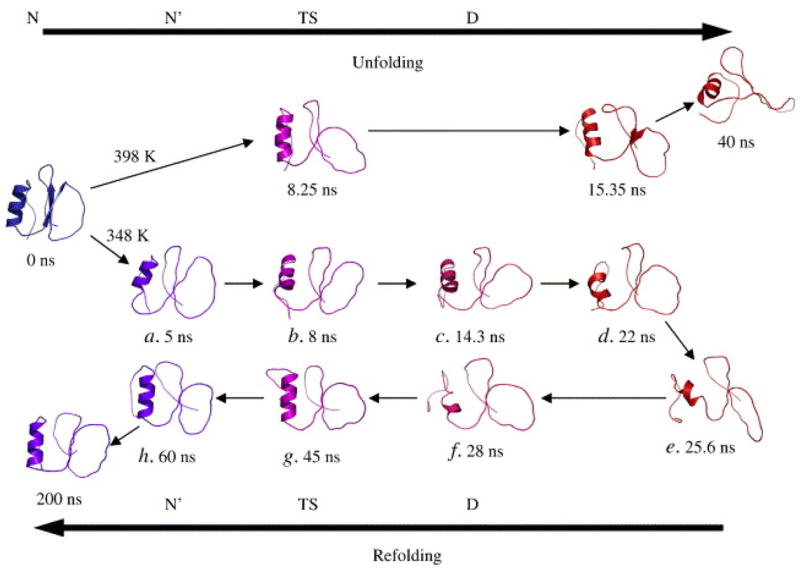
Chymotrypsin inhibitor 2 unfolds through its transition state to the denatured state, and then returns to a N′ state through the same transition state at 348K. This is the first observed case of microscopic reversibility in a simulation system.
A common criticism of simulation is that there is the difficulty in determining the properties of an ensemble of conformations from a limited number of simulations. Day et al. showed that 5–10, 20-ns thermal unfolding simulations capture the properties of a larger set of 100 simulations [21]. Further, the average unfolding/folding pathway delineated by these properties accurately describes the nucleation-condensation folding process of CI2, with simultaneous formation of secondary and tertiary contacts, in agreement with experiment.
Protein Refolding: Repeated simulation of microsecond timescales
Protein refolding from extended structures using MD is hindered by the approximations necessary to access long timescales. In many cases hydrophobic collapse can be observed, but the precise criteria used to assess refolding vary, measures such as backbone Cα RMSD or particular contacts in the hydrophobic core are used in an inconsistent fashion (see Table 1). Consider the landmark 1 μs simulation of Duan and Kollman in which hydrophobic collapse was achieved but the native state was not reached, as expected from the minimal 4 μs folding time [22, 23]. The seminal refolding prediction of Trp-cage by Simmerling et al was a notable exception to the difficulty of correctly replicating intramolecular contacts [24].
Table 1.
Protocols for refolding studies of proteins and peptides
| System | Simulation protcol | Refolding criteria | Starting state | Reference |
|---|---|---|---|---|
| HP-36 | Single 1.0 μs 300K | Rg, ss content, contacts, Global Cα RMSD, Local Cα RMSD | Thermally denatured | [22] |
| Trp-cage | Multiple 20 ns325K | Force field energy | Unknown | [24] |
| HP-35 | 20 1.0 μs 300K | Global Cα RMSD | Fully extended | [25] |
| HP-35NleNle | 410 ~ 1.0μs | Local Cα RMSd, contacts | Thermally denatured | [29] |
| Albumin- binding domain | 20 400ns 300K | Global Cα RMSD | Fully extended | [27] |
| α-spectrin SH3 | 26 200ns 300K | Global Cα RMSD, SASA, ss content, contacts | Transition state | [30] |
| CI2 | 1 200ns 348K | Global Cα RMSD, contacts, topology | Native state | [20] |
The 35-residue N-terminal region of the villin headpiece subdomain (HP-35) is a popular model system for simulation. A recent study by Lei et al. reported refolding on the μs time scale using implicit solvent [25]. Twenty individual μs simulations were started from fully extended states. Refolding was assessed by Cα RMSD to the X-ray structure. In 7 trajectories, structures with sub 1.0 Å RMSD are reached and maintained. A region identified by mutagenesis as vital to stability consistently forms early in these simulations [26]. After the initial collapse of the protein, trajectories fold on an average of 100 ns. The authors attribute the fast folding dynamics of the simulations to the lack of solvent viscosity. Lei et al. have also reported refolding for the wild-type and two mutants of an albumin-binding domain, a 47-residue three-helix bundle, using a similar protocol with 20, 400-ns simulations per structure [27]. The agreement of the best structures from the refolding simulations by Cα RMSD to the minimized average NMR structure is 2–2.5 Å with the wild-type closer than the mutants.
A variant of HP-35 containing non-natural amino acids exhibits sub-μs folding kinetics [28]. Ensign et al. have accumulated 410 μs of refolding simulations of this variant at 300K in water [29]. Refolding is observed from multiple thermally denatured starting conformations. In this case, refolding was defined both in terms of the individual Cα RMSDs of the three major α-helices of villin to their counterparts in an energy-minimized native structure and by the formation of three Phe contacts in the hydrophobic core of the protein. It would be interesting to see how well the refolded structures reproduce the experimental NOEs.
Refolding from a TS was reported for a single trajectory from a set of simulations of α-spectrin SH3 domain [30]. Nine putative TS structures were used as the starting structures. These structures were generated using MD with Φ-values as restraints [2]. Two candidates that displayed favorable refolding properties were simulated multiple times for 200 ns. Refolding was assessed by a combination of properties: solvent accessible surface area; native secondary structure; Cα RMSD from the X-ray model; and the Cα RMSD of subsets of structures. In one case, the Cα RMSD fell below 1.5 Å with native-like values for the remainder of the monitored properties. Refolding has also been observed from multiple engrailed homeodomain TS structures generated from thermal unfolding simulations [31].
Unfolding in small modular systems
The FBP28 WW domain is a 37-residue, three-stranded β-sheet protein that cooperatively folds on the μs timescale by an apparent two-state transition. FBP28 unfolds over a wide range of denaturant concentrations because its small size means that it has low sensitivity to denaturant concentration (low mD-N) and its low stability means only lower than average ΔΔG values for Φ-value analysis are accessible. TS structures for FBP28 WW were selected from unfolding MD simulations and validated by comparison to experimental Φ-values [32]. The FBP28 TS is similar to other WW domains; the first turn is native-like while the second turn is still being formed. However, the simulations indicate that the first turn does not contain the correct native backbone hydrogen bonds, and that the high Φ-values in the turn region are due to side chain packing. Further simulations of hYAPtm and Pin1 WW domains confirmed the conserved dynamic behavior of the first turn [33].
The formation of the first turn in Pin1 WW domain is rate-limiting. Jäger et al. argue that the unusual Pin1 six-residue first turn evolves because of functional considerations rather than for stability [34]. The first loop is more dynamic by NMR than in FBP28 [35]. Interestingly, redesign of the six-residue loop with the FPB28 five-residue loop yielded a variant that is both structurally similar and more stable, but binding to a functional model peptide is reduced. This is consistent with previous studies that have demonstrated that optimal folding behavior is not the sole constraint on protein function [34, 36].
Protein Folding: Early events can define folded structure
Both nucleation-condensation and framework folding mechanisms can be observed within the three-helix bundle homeodomain fold family [37]. The spectrum of behavior observed within this fold family has been explained as the result of the balance of secondary and tertiary forces acting upon a single underlying mechanism [38]. The c-Myb-transforming protein has a behavior that is a mixture of the two mechanisms. This was further demonstrated when a high-energy intermediate of c-Myb, predicted by simulation, was stabilized by mutation and characterized by experiment [39]. In contrast, the engrailed homeodomain (EnHD) is a 54-residue three-helix bundle that folds through an intermediate by a framework mechanism. This intermediate has been stabilized by mutation and characterized by NMR [40]. The structured helix-turn-helix (HTH) motif of the L16A EnHD intermediate was expressed independently and structurally determined by NMR [41]. The EnHD HTH motif has the same backbone structure as in the native structure. Moreover, the independent HTH domain possesses the same folding kinetics as the fast phase of the full-length protein. The expression and stability of the EnHD intermediate is further evidence that the stability of folding substructures is a key factor in the slide from nucleation-condensation to framework folding mechanisms. Interestingly, structures similar to the engrailed intermediate form a major cluster in Monte Carlo μ-potential simulations of folding [42]. Notably, this intermediate was first identified by MD and was later determined by NMR [40, 43].
Since the proposal of the nucleation-condensation model of folding, which unifies the more extreme framework and hydrophobic collapse models [44–46], there has been an active search for additional proteins that display characteristics of different mechanisms, as with the 3-helix bundle proteins mentioned above. The PDZ domain, PDZ2, is a small αβ protein that folds through a high-energy intermediate. Φ-values were obtained for both the early transition from the denatured to intermediate state and the late transition from the intermediate to the native state [47]. Restrained MD simulations were conducted using these Φ-values as restraints[48]. The early transition is characteristic of nucleation-condensation, whereas the late transition is consistent with the framework model.
Im7 and Im9 are four-helix bundle proteins that fold by different apparent mechanisms: Im7 folds through an intermediate whereas Im9 folds in a two-state manner. The intermediate state of Im7 was stabilized by mutation and characterized [49]. In addition, restrained MD simulations were conducted using hydrogen-exchange protection factors as restraints [50]. Structures generated by these methods were similar, suggesting that the Im7 intermediate contains three helices with the fourth helix unstructured. A double mutant of Im9 (dubbed SIm9) was designed that displays similar three-state folding. The fractional Φ-values observed for SIm9 are suggestive of a nucleation-condensation mechanism. Structures of the Im9 TS generated by MD with Φ-value restraints are more heterogeneous than those of Im7; SIm9 structures display a similar level of heterogeneity. While TS models generated with a very limited number of restraints can be useful, it must be remembered that the models are underdetermined and contain no pathway information.
The Paracelsus challenge, proposed by Rose and Creamer [51], resulted in a number of protein pairs with high sequence identity and different topologies. Recently, rather than directly engineering a single protein into another fold, Orban et al. engineered two proteins from different folds towards higher sequence identity [52]. This resulted in a protein G (β-sheet packed against an α-helix) variant (G311) and a protein A (three-helix bundle) variant (A219) with 59% sequence identity but different topologies. The folding pathways of both of these structurally divergent proteins were studied by MD unfolding simulations [53]. Viewing unfolding simulations of G311 in reverse, the canonical β3/β4 protein G hairpin forms early, and residual helical structure is located in regions predicted from chemical shift deviations. Interestingly, much of the sequence dissimilarity between the two proteins is located in the region of the β3/β4 hairpin (35% sequence identity). The loss of the β3/β4 turn in A219 guides it to late helix formation rather than early β-hairpin formation. Oddly in A219, the denatured state is far more unstructured, lacking much of the residual helix observed in the G311 denatured state. In short, early nucleating events in both proteins (strong α-helical propensities in G311, early β-turn formation in A219) are nonnative but direct the folding pathway to its conclusion.
Titin is suggested to fold by two different TSs: nucleation-condensation and framework [54]. These TSs exist on multiple independent pathways, indicated by the unusual nonlinear increase in the unfolding rate under increasing denaturant concentrations [54]. The TS ensemble was modeled for each pathway using Φ-value analysis and restrained MD [55]. The ensemble at low denaturant concentrations has native-like topology by Cα RMSD, while possessing few native contacts characteristic of nucleation-condensation. The TS ensemble generated from Φ-values of the dominant folding pathway at high denaturant concentrations contain many regions of native-like secondary structure in addition to regions containing little secondary structure.
Conclusions
All-atom MD simulations of protein folding is providing important information on the early events in protein folding for small proteins; however, the major information about folding pathways, their transition states and intermediates is still coming from simulation of protein unfolding or simulations of folding using simplified models.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li A, Daggett V. Characterization of the transition state of protein unfolding by use of molecular dynamics: chymotrypsin inhibitor 2. Proc Natl Acad Sci U S A. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindorff-Larsen K, Vendruscolo M, Paci E, Dobson CM. Transition states for protein folding have native topologies despite high structural variability. Nat Struct Mol Biol. 2004;11:443–449. doi: 10.1038/nsmb765. [DOI] [PubMed] [Google Scholar]
- 3.Weinkam P, Zong C, Wolynes PG. A funneled energy landscape for cytochrome c directly predicts the sequential folding route inferred from hydrogen exchange experiments. Proceedings of the National Academy of Sciences. 2005;102:12401–12406. doi: 10.1073/pnas.0505274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okazaki K-i, Koga N, Takada S, Onuchic JN, Wolynes PG. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proceedings of the National Academy of Sciences. 2006;103:11844–11849. doi: 10.1073/pnas.0604375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubner IA, Deeds EJ, Shakhnovich EI. High-resolution protein folding with a transferable potential. Proceedings of the National Academy of Sciences. 2005;102:18914–18919. doi: 10.1073/pnas.0502181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung MS, Thirumalai D. Effects of Crowding and Confinement on the Structures of the Transition State Ensemble in Proteins. J Phys Chem B. 2007;111:8250–8257. doi: 10.1021/jp068201y. [DOI] [PubMed] [Google Scholar]
- 7.Dyson HJ, Wright PE. Unfolded proteins and protein folding studied by NMR. Chem Rev. 2004;104:3607–3622. doi: 10.1021/cr030403s. [DOI] [PubMed] [Google Scholar]
- 8.Eaton WA, Munoz V, Hagen SJ, Jas GS, Lapidus LJ, Henry ER, Hofrichter J. Fast kinetics and mechanisms in protein folding. Annual Review of Biophysics and Biomolecular Structure. 2000;29:327–359. doi: 10.1146/annurev.biophys.29.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fredriksson K, Louhivuori M, Permi P, Annila A. On the interpretation of residual dipolar couplings as reporters of molecular dynamics. J Am Chem Soc. 2004;126:12646–12650. doi: 10.1021/ja048287d. [DOI] [PubMed] [Google Scholar]
- 10.Bashford D, Case DA. Generalized Born models of macromolecular solvation effects. Annual Review of Physical Chemistry. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 11.Plaxco KW, Baker D. Limited internal friction in the rate-limiting step of a two-state protein folding reaction. Proceedings of the National Academy of Sciences. 1998;95:13591–13596. doi: 10.1073/pnas.95.23.13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pande VS, Baker I, Chapman J, Elmer SP, Khaliq S, Larson SM, Rhee YM, Shirts MR, Snow CD, Sorin EJ, et al. Atomistic protein folding simulations on the submillisecond time scale using worldwide distributed computing. Biopolymers. 2003;68:91–109. doi: 10.1002/bip.10219. [DOI] [PubMed] [Google Scholar]
- 13.Zhang W, Wu C, Duan Y. Convergence of replica exchange molecular dynamics. Journal of Chemical Physics. 2005;123 doi: 10.1063/1.2056540. [DOI] [PubMed] [Google Scholar]
- 14.Settanni G, Gsponer J, Caflisch A. Formation of the Folding Nucleus of an SH3 Domain Investigated by Loosely Coupled Molecular Dynamics Simulations. Biophys J. 2004;86:1691–1701. doi: 10.1016/S0006-3495(04)74238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paci E, Lindorff-Larsen K, Dobson CM, Karplus M, Vendruscolo M. Transition State Contact Orders Correlate with Protein Folding Rates. Journal of Molecular Biology. 2005;352:495–500. doi: 10.1016/j.jmb.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 16.Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci U S A. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarney ER, Kohn JE, Plaxco KW. Is there or isn’t there? The case for (and against) residual structure in chemically denatured proteins. Crit Rev Biochem Mol Biol. 2005;40:181–189. doi: 10.1080/10409230591008143. [DOI] [PubMed] [Google Scholar]
- 18.Merchant KA, Best RB, Louis JM, Gopich IV, Eaton WA. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc Natl Acad Sci U S A. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plaxco KW, Millett IS, Segel DJ, Doniach S, Baker D. Chain collapse can occur concomitantly with the rate-limiting step in protein folding. Nat Struct Mol Biol. 1999;6:554–556. doi: 10.1038/9329. [DOI] [PubMed] [Google Scholar]
- 20.Day R, Daggett V. Direct observation of microscopic reversibility in single-molecule protein folding. J Mol Biol. 2007;366:677–686. doi: 10.1016/j.jmb.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day R, Daggett V. Ensemble versus single-molecule protein unfolding. Proc Natl Acad Sci U S A. 2005;102:13445–13450. doi: 10.1073/pnas.0501773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 23.Kubelka J, Eaton WA, Hofrichter J. Experimental Tests of Villin Subdomain Folding Simulations. Journal of Molecular Biology. 2003;329:625–630. doi: 10.1016/s0022-2836(03)00519-9. [DOI] [PubMed] [Google Scholar]
- 24.Simmerling C, Strockbine B, Roitberg AE. All-atom structure prediction and folding simulations of a stable protein. Journal of the American Chemical Society. 2002;124:11258–11259. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 25.Lei H, Duan Y. Two-stage Folding of HP-35 from Ab Initio Simulations. Journal of Molecular Biology. 2007;370:196–206. doi: 10.1016/j.jmb.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frank BS, Vardar D, Buckley DA, McKnight CJ. The role of aromatic residues in the hydrophobic core of the villin headpiece subdomain. Protein Sci. 2002;11:680–687. doi: 10.1110/ps.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei H, Duan Y. Ab Initio Folding of Albumin Binding Domain from All-Atom Molecular Dynamics Simulation. J Phys Chem B. 2007;111:5458–5463. doi: 10.1021/jp0704867. [DOI] [PubMed] [Google Scholar]
- 28.Kubelka J, Chiu TK, Davies DR, Eaton WA, Hofrichter J. Sub-microsecond Protein Folding. Journal of Molecular Biology. 2006;359:546–553. doi: 10.1016/j.jmb.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 29.Ensign DL, Kasson PM, Pande VS. Heterogeneity even at the speed limit of folding: large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. Journal of Molecular Biology. doi: 10.1016/j.jmb.2007.09.069. In Press, Accepted Manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xavier Periole MV, Mark AE. Molecular dynamics simulations from putative transition states of alpha-spectrin SH3 domain. Proteins: Structure, Function, and Bioinformatics. 2007;9999 doi: 10.1002/prot.21491. NA. [DOI] [PubMed] [Google Scholar]
- 31.Beck DAC, Daggett V. A One-Dimensional Reaction Coordinate for Identification of Transition States from Explicit Solvent Pfold-Like Calculations. Biophysical Journal. 2007;93:8832–3391. doi: 10.1529/biophysj.106.100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrovich M, Jonsson AL, Ferguson N, Daggett V, Fersht AR. Phi-analysis at the experimental limits: mechanism of beta-hairpin formation. J Mol Biol. 2006;360:865–881. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 33.Sharpe T, Jonsson AL, Rutherford TJ, Daggett V, Fersht AR. The role of the turn in beta-hairpin formation during WW domain folding. Protein Sci. 2007;16:2233–2239. doi: 10.1110/ps.073004907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jager M, Zhang Y, Bieschke J, Nguyen H, Dendle M, Bowman ME, Noel JP, Gruebele M, Kelly JW. Structure-function-folding relationship in a WW domain. Proceedings of the National Academy of Sciences. 2006;103:10648–10653. doi: 10.1073/pnas.0600511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macias MJ, Gervais V, Civera C, Oschkinat H. Structural analysis of WW domains and design of a WW prototype. Nat Struct Mol Biol. 2000;7:375–379. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 36.Jager M, Dendle M, Fuller AA, Kelly JW. A cross-strand Trp Trp pair stabilizes the hPin1 WW domain at the expense of function. Protein Sci. 2007;16:2306–2313. doi: 10.1110/ps.072904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gianni S, Guydosh NR, Khan F, Caldas TD, Mayor U, White GWN, DeMarco ML, Daggett V, Fersht AR. Unifying features in protein-folding mechanisms. Proceedings of the National Academy of Sciences. 2003;100:13286–13291. doi: 10.1073/pnas.1835776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daggett V, Fersht AR. Is there a unifying mechanism for protein folding? Trends Biochem Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 39.White GW, Gianni S, Grossmann JG, Jemth P, Fersht AR, Daggett V. Simulation and experiment conspire to reveal cryptic intermediates and a slide from the nucleation-condensation to framework mechanism of folding. J Mol Biol. 2005;350:757–775. doi: 10.1016/j.jmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Religa TL, Markson JS, Mayor U, Freund SM, Fersht AR. Solution structure of a protein denatured state and folding intermediate. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 41.Religa TL, Johnson CM, Vu DM, Brewer SH, Dyer RB, Fersht AR. The helix-turn-helix motif as an ultrafast independently folding domain: the pathway of folding of Engrailed homeodomain. Proc Natl Acad Sci U S A. 2007;104:9272–9277. doi: 10.1073/pnas.0703434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hubner IA, Deeds EJ, Shakhnovich EI. Understanding ensemble protein folding at atomic detail. Proceedings of the National Academy of Sciences. 2006;103:17747–17752. doi: 10.1073/pnas.0605580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayor U, Johnson CM, Daggett V, Fersht AR. Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13518–13522. doi: 10.1073/pnas.250473497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itzhaki LS, Otzen DE, Fersht AR. The Structure of the Transition State for Folding of Chymotrypsin Inhibitor 2 Analysed by Protein Engineering Methods: Evidence for a Nucleation-condensation Mechanism for Protein Folding. Journal of Molecular Biology. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 45.Otzen DE, Itzhaki LS, ElMasry NF, Jackson SE, Fersht AR. Structure of the Transition State for the Folding/Unfolding of the Barley Chymotrypsin Inhibitor 2 and Its Implications for Mechanisms of Protein Folding. Proceedings of the National Academy of Sciences. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daggett V, Fersht AR. Is there a unifying mechanism for protein folding? Trends in Biochemical Sciences. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 47.Gianni S, Calosci N, Aelen JMA, Vuister GW, Brunori M, Travaglini-Allocatelli C. Kinetic folding mechanism of PDZ2 from PTP-BL. Protein Engineering, Design and Selection. 2005;18:389–395. doi: 10.1093/protein/gzi047. [DOI] [PubMed] [Google Scholar]
- 48.Gianni S, Geierhaas CD, Calosci N, Jemth P, Vuister GW, Travaglini-Allocatelli C, Vendruscolo M, Brunori M. A PDZ domain recapitulates a unifying mechanism for protein folding. Proceedings of the National Academy of Sciences. 2007;104:128–133. doi: 10.1073/pnas.0602770104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spence GR, Capaldi AP, Radford SE. Trapping the On-pathway Folding Intermediate of Im7 at Equilibrium. Journal of Molecular Biology. 2004;341:215–226. doi: 10.1016/j.jmb.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 50.Gsponer J, Hopearuoho H, Whittaker SBM, Spence GR, Moore GR, Paci E, Radford SE, Vendruscolo M. Determination of an ensemble of structures representing the intermediate state of the bacterial immunity protein Im7. Proceedings of the National Academy of Sciences. 2006;103:99–104. doi: 10.1073/pnas.0508667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose GD, Creamer TP. Protein-folding - Predicting Predicting. Proteins-Structure Function and Genetics. 1994;19:1–3. doi: 10.1002/prot.340190102. [DOI] [PubMed] [Google Scholar]
- 52.Alexander PA, Rozak DA, Orban J, Bryan PN. Directed Evolution of Highly Homologous Proteins with Different Folds by Phage Display: Implications for the Protein Folding Code. Biochemistry. 2005;44:14045–14054. doi: 10.1021/bi051231r. [DOI] [PubMed] [Google Scholar]
- 53.Scott KA, Daggett V. Folding Mechanisms of Proteins with High Sequence Identity but Different Folds. Biochemistry. 2007;46:1545–1556. doi: 10.1021/bi061904l. [DOI] [PubMed] [Google Scholar]
- 54.Wright CF, Lindorff-Larsen K, Randles LG, Clarke J. Parallel protein-unfolding pathways revealed and mapped. Nat Struct Mol Biol. 2003;10:658–662. doi: 10.1038/nsb947. [DOI] [PubMed] [Google Scholar]
- 55.Geierhaas CD, Best RB, Paci E, Vendruscolo M, Clarke J. Structural Comparison of the Two Alternative Transition States for Folding of TI I27. Biophys J. 2006;91:263–275. doi: 10.1529/biophysj.105.077057. [DOI] [PMC free article] [PubMed] [Google Scholar]