

Abstract
Ample evidence suggests a role of TGF-β in preventing autoimmunity. Multiorgan inflammatory disease, spontaneous activation of self-reactive T cells, and autoantibody production are hallmarks of autoimmune diseases, such as lupus. These features are reminiscent of the immunopathology manifest in TGF-β1-deficient mice. In this study, we show that lupus-prone (New Zealand Black and White)F1 mice have reduced expression of TGF-β1 in lymphoid tissues, and TGF-β1 or TGF-β1-producing T cells suppress autoantibody production. In contrast, the expression of TGF-β1 protein and mRNA and TGF-β signaling proteins (TGF-β receptor type II and phosphorylated SMAD3) increases in the target organs, i.e., kidneys, of these mice as they age and develop progressive organ damage. In fact, the levels of TGF-β1 in kidney tissue and urine correlate with the extent of chronic lesions that represent local tissue fibrosis. In vivo TGF-β blockade by treatment of these mice with an anti-TGF-β Ab selectively inhibits chronic fibrotic lesions without affecting autoantibody production and the inflammatory component of tissue injury. Thus, TGF-β plays a dual, seemingly paradoxical, role in the development of organ damage in multiorgan autoimmune diseases. According to our working model, reduced TGF-β in immune cells predisposes to immune dysregulation and autoantibody production, which causes tissue inflammation that triggers the production of anti-inflammatory cytokines such as TGF-β in target organs to counter inflammation. Enhanced TGF-β in target organs, in turn, can lead to dysregulated tissue repair, progressive fibrogenesis, and eventual end-organ damage.
Multiorgan inflammatory disease, spontaneous activation of self-reactive T cells, and autoantibody production are hallmarks of autoimmune diseases, such as systemic lupus erythematosus (SLE)5 (1–4). Interestingly, these features are reminiscent of the immunopathology manifest in TGF-β1-deficient mice (5–9). Deficiency of molecules involved in TGF-β signaling, such as TGF-β receptor (TβR) type II, also causes spontaneous lymphocyte activation, autoantibody production, and multiorgan inflammation (10–12). Thus, overwhelming evidence supports an immune regulatory role of TGF-β against autoimmunity. In fact, patients with autoimmune diseases, such as SLE, have reduced TGF-β production in their peripheral blood cell cultures (13). Hence, reduced TGF-β production by immune cells might predispose to autoreactive T cell activation and autoantibody production in autoimmune diseases.
In autoimmune diseases, infiltration with T cells or deposition of autoantibody-containing immune complexes in target organs, such as kidneys, causes early inflammatory lesions. The early immune-mediated injury is believed to trigger a series of events, including complement activation, chemokine production, further inflammatory cell infiltration, and inflammatory cytokine release, eventually resulting in deposition of extracellular matrix (14, 15). It is this fibrotic process that predicts the clinical outcome in autoimmune diseases, such as lupus (16–18). TGF-β appears to be a common end-stage pathway in the development of tissue fibrosis in a variety of conditions (14, 19–22). In fact, mice with transgenic expression of TGF-β1 exhibit fibrogenesis in various organs (23). Consequently, we hypothesize that TGF-β plays a dual role during the development and progression of immune-mediated inflammatory diseases. Although reduced TGF-β production by immune cells predisposes to immune dysregulation and development of autoimmunity in early life, the enhanced TGF-β production in tissues induces local fibrogenesis and ultimately causes end-stage organ disease. In this study, we tested this hypothesis by determining the expression of TGF-β and its signaling molecules in immune vs target tissues and by examining the role of TGF-β in the development of autoantibodies and damage of target organs, i.e., kidneys, using (New Zealand Black and White (NZB x NZW))F1 (BWF1) mice (24) that develop systemic autoimmune disease characterized by spontaneous T cell activation, autoantibody production, and fatal nephritis. Our data suggest a dual, seemingly paradoxical, role of TGF-β in the development of systemic autoimmune diseases.
Materials and Methods
Animals
Tgfb1−/− BALB/c mice were generated by introgressing Tgfb1-null genotype (5) onto the BALB/c background for eight generations. Breeding pairs of NZB, NZW, and BALB/c mice were purchased from The Jackson Laboratory and intercrossed to generate BWF1 and BALB/c x NZW F1 (CWF1) mice. Kidneys from TGF-β1-transgenic mice (23) were provided by Dr. J. Kopp. Animal experiments were performed in accordance with approved institutional guidelines.
Spleen cell culture for cytokine assays
Single-cell suspension of splenocytes was prepared, and red cells were lysed with 1 × NH4Cl buffer. A total of 5 × 106 cells per ml were cultured for 48 h in AIM-V medium for TGF-β measurements or in medium containing 10% FCS with 5 μg/ml Con A for other cytokines.
Preparation of kidney eluates for TGF-β analysis
Before harvesting tissues animals were perfused with PBS. Kidney tissue was homogenized in PBS, cells and solid fractions were removed by centrifugation at 1300 rpm for 10 min, and the eluate was stored at −20°C. Total protein content in the eluate was determined by the Pierce bicinchoninic acid method (Pierce).
Urine for TGF-β analysis
Fresh urine was collected from BALB/c and BWF1 mice of various ages and stored at −20°C. Before analysis for total TGF-β1 concentration, specimens were acidified with 1N HCl to achieve a pH of 2.6 for 1 h and then neutralized to pH 7.6 with 1N NaOH.
Measurement of cytokines
A sandwich ELISA was used to determine TGF-β1 levels (25), following the manufacturer’s recommendations (Promega). Briefly, ELISA plates were coated with anti-mouse TGF-β mAb overnight at 4°C, blocked, and washed once. The acid treated or untreated diluted samples (for total and active or endogenously free TGF-β1, respectively) and serially diluted TGF-β standard were added in duplicate and plates incubated at 37°C for 90 min. Plates were washed 5 times and then incubated for 2 h with polyclonal anti-TGF-β1 Ab. Plates were washed again five times, followed by incubation with HRP conjugate for 2 h, another washing, and the addition of HRP substrate TMB to the plates. The reaction was terminated after 15 min by the addition of 1N HCl. Plates were read using a Multiskan MS ELISA reader at 450 nm. Cytokines IL-2, IL-4, and IFN-γ were measured by ELISA using mAb pairs and recombinant cytokines (BD Pharmingen), as described (26).
Western blotting
Kidney tissue was lysed in lysis buffer containing protease inhibitors on ice. Protein concentration was estimated in kidney lysates using the Pierce protein assay kit (Pierce). Protein extracts from the kidneys of different mice were loaded onto a bis-tris gel (Invitrogen Life Technologies) after boiling and reduction with DTT and subjected to electrophoresis at constant 200 V for 30 min. Immediately after separation, proteins were immobilized onto a polyvinylidene difluoride membrane using an XCell a blot module system (Invitrogen Life Technologies) at constant 30 V for 1 h, blocked with milk powder, and probed with 1/1000 dilution of anti-TGF-β1 polyclonal Ab (Promega) for 1 h at room temperature or overnight at 4°C. Membranes were washed and then incubated with anti-rabbit IgG-HRP for 1 h at room temperature. Membranes were washed and developed using a chemiluminiscent detection system (Invitrogen Life Technologies).
Immunohistochemistry and immunofluorescence
Expression of TGF-β, TβRII, SMAD3, and phosphorylated (phospho)-SMAD3 was detected by immunohistochemistry using the peroxidase Vectastain ABC kit (Vector Laboratories), as described (27). SMAD3 was also detected using the Vectastain ABC-AP kit (Vector Laboratories). Briefly, animals were perfused, kidneys harvested, fixed in 4% paraformaldehyde, and paraffin embedded. Four-μm sections were fixed to negatively charged immunohistochemistry slides. Following deparaffinization and rehydration, slides were placed in 0.1M sodium citrate for 10 min at 95°C, blocked with goat sera, followed by an avidin and biotin block (Vector Labs), and then incubated with the rabbit polyclonal IgG anti-SMAD3 Ab (Santa Cruz Biotechnology) overnight. Biotinylated secondary Ab, avidin-conjugated alkaline phosphatase (AP), and AP substrate were added sequentially, and the slides were counterstained with Gill’s hematoxylin. Irrelevant, IgG was used as a negative control. Renal Ig deposition was detected using fluorescein-conjugated goat anti-mouse IgG (Sigma-Aldrich), as described (27).
In situ hybridization
In situ hybridization for TGF-β1 was performed on proteinase K-treated, 4-μm paraffin kidney sections, as described (27). The 35S label in the samples was detected by autoradiography, with the slides being exposed to NTB-2 emulsion (Kodak) for 7–14 days, per the manufacturer. Sections were counterstained with H&E, mounted in Permount, air-dried, and examined using a light microscope.
Semiquantitative RT-PCR
Total RNA was isolated from the kidneys using Trizol single step RNA isolation reagent (Invitrogen Life Technologies, BRL) and subjected to reverse transcription using access RT-PCR system (Promega), per manufacturer’s instructions. The PCR primers for TGF-β-1, -2, -3, IL-4, and pro-α1 collagen I and IV are described in Table I (28). The reaction mixture was heated for 45 min at 48°C and then 94°C for 2 min for reverse transcription, followed by the following protocol: 94°C for 30 s denaturation, 60°C for 1 min annealing, 68°C for 2 min extension for 40 cycles, and 1 cycle of 7 min for final extension in a PerkinElmer PCR machine. PCR products were visualized on 2% agarose gel (Promega), and band density was measured using National Institutes of Health image software version 6.0. The results are expressed as the relative density for each PCR product as compared with GAPDH (housekeeping) gene.
Table I.
RT-PCR oligonucleotides
| Sense (5′-3′) | Antisense (5′-3′) | Product Size (bp) | |
|---|---|---|---|
| TGFβ1 | ATACAGGGCTTTCGATTCAGC | GTCCAGGCTCCAAATATATAG | 360 |
| TGFβ2 | AGAATCGTCCGCTTTGATGTC | TCTTCGCTTTTATTCGGGATG | 382 |
| TGFβ3 | GTGCGCGAGTGGCTGTTGAGGAGA | GTGTCTGCGCTGCGGAGGTATGG | 446 |
| Pro-α1 collagen | TCGTGACCGTGACCTTGCG | AGGCACAGACGGCTGAGTAGG | 255 |
| Collagen IV | CCAAAGTCGCGGACCTGTA | GGCTCGATCCCTTCCTAACC | 220 |
| GAPDH | GGCCTTGACTGTGCCGTTGAATTT | ACAGCCGCATCTTCTTGTGCAGTG | 226 |
Anti-TGF-β Ab treatment
Animals were treated with an anti-TGF-β mAb (1.D.11.16, Celltrix), which neutralizes all three TGF-β isoforms (29). A dosage regimen of 500 μg per mouse twice a week, which has been shown to inhibit tissue fibrosis (22), was used. Control animals received the same dosage regimen of mouse IgG (Zymed Laboratories).
Detection of autoantibodies and IgG
Serum anti-dsDNA Ab and total IgG levels were detected by ELISA, as described (30). IgG antinuclear Ab (ANA) was assessed using the QUANTA Lite ANA ELISA (INOVA Diagnostics), adapted to test mouse samples. For detection of rheumatoid factor (RF), ELISA plates were coated with 4-hydroxy-3-iodo-5-nitrophenylacetic acid-BSA (10 μg/ml) for 1 h at 37°C. Plates were then washed with PBST, and anti-IgG2a anti-4-hydroxy-3-iodo-5-nitrophenylacetic acid (23.3) was added (diluted in 1% BSA at 1:1 ratio). Plates were washed again, and serum samples and standard were added for 1 h at 37°C. After washing, AP-conjugated anti-mouse Ig κ L chain (1/1000 dilution in 1% BSA) was added for 1 h at room temperature. Plates were developed with pNPP and read using an ELISA reader. For detection of anti-cardiolipin Ab, cardiolipin Ag (5 mg/ml, Sigma-Aldrich) was added to 10 ml of 200 proof ethyl alcohol (Daigger). One hundred μl of Ag was added to the ELISA plates. Vehicle (200 proof ethyl alcohol) served as control. Plates were then dried under the hood for 30 min and blocked for 1 h at room temperature with 1% BSA. Samples and standard were added onto plates for 2 h at room temperature. Plates were washed with PBST, and secondary Ab (goat-anti-mouse IgG) was added for 1 h at room temperature. Plates were read at 450 nm.
Assessment of renal disease
Proteinuria was measured on a 0–3+ scale using a colorimetric assay strip for albumin (Albustix; Bayer), where 0 = absent, 1+ ≤ 30 (mild), 2+ = 100 (moderate), and 3+ ≥ 300 mg/dl (severe). Severe proteinuria was defined as ≥300 mg/dl on two consecutive examinations (30). Paraffin kidney sections stained with H&E, periodic acid-Schiff (PAS), Masson’s trichrome, and Jones silver were scored for various histological features in a blind fashion, as described in Table II (27, 31, 32). The mean scores for individual features were summed to obtain the three main indices (glomerular activity index (GAI), tubulointerstitial index (TII), and chronicity index (CI)), which were then averaged and summed to determine a composite biopsy index. Arbitrary cut-off numbers were assigned to designate low vs high indices, which represent absent to mild vs moderate to severe disease, respectively.
Table II.
Renal biopsy scoring system
| Indices and Individual Components | Scale |
|---|---|
| GAI | |
| Glomerular hypercellularity | 0–3+ |
| Karyorrhexis/fibrinoid necrosis | (0–3+) × 2 |
| Cellular crescents | (0–3+) × 2 |
| Inflammatory cell infiltrate | 0–3+ |
| Hyaline deposits | 0–3+ |
| Maximum = 21 | |
| TII | |
| Tubular cell pyknosis/nuclear activation | 0–3+ |
| Tubular cell necrosis | 0–3+ |
| Tubular cell flattening | 0–3+ |
| Interstitial inflammation | 0–3+ |
| Macrophages/epithelial cells in tubular lumen | 0–3+ |
| Maximum = 15 | |
| CI | |
| Mesangial matrix deposition | 0–3+ |
| Glomerulosclerosis | 0–3+ |
| Glomerular scars | 0–3+ |
| Fibrous crescents | 0–3+ |
| Tubular atrophy | 0–3+ |
| Interstitial fibrosis | 0–3+ |
| Maximum = 18 | |
| Composite Biopsy Index 3 GAI/7 plus TII/5 plus CI/6 | Maximum = 9 |
Statistical analysis
Depending on the distribution of continuous data, Student’s t or Mann-Whitney U tests were used to compare various parameters between groups using GraphPad InStat software. Differences in the time to onset of protein-uria between groups were tested for significance using survival analysis and the log-rank test. Differences of categorical data between groups were assessed using the two-sided Fisher’s exact test and the log-likelihood ratio test.
Results
Dichotomy in TGF-β expression in the lymphoid vs renal tissues of BWF1 mice
Germline deficiency of TGF-β1 results in activation of self-reactive T cells and multiorgan inflammation (5, 7, 9). To understand the role of TGF-β in the development of spontaneous multiorgan inflammatory disease, we first assessed the expression of TGF-β1 in the lymphoid (spleen) and target organs (kidneys) of genetically autoimmune-prone BWF1 mice. TGF-β1 levels, as detected by Western blot, were lower in the spleen, but higher in the kidneys, of BWF1 mice than in age-matched BALB/c mice (Fig. 1, A and B). Cultured spleen cells also produced less TGF-β1 (active), as detected by ELISA, in diseased BWF1 mice than in age-matched male BWF1 mice, prenephritic female BWF1 mice, and age/sex/MHC-matched normal CWF1 mice (Fig. 1, C and D). In contrast, renal TGF-β1 protein, as detected by immunohistochemistry (Fig. 1E) and Western blot (Fig. 1, F and G), and Tgfb1 mRNA expression, as detected by in situ hybridization (Fig. 1H), were higher in BWF1 mice than in BALB/c mice. Renal TGF-β1 expression increased with age and disease progression in BWF1 mice (Fig. 1, E–H). Thus, lupus mice exhibit a dichotomous pattern of TGF-β1 expression in the lymphoid vs target (renal) tissues. Surface TGF-β1 as detected by flow cytometry was lower in splenic T cells from BWF1 mice than BALB/c mice (our unpublished data). T cell lines raised from the spleen of BWF1 mice also produced less TGF-β1 than T cell lines raised from normal CWF1 mice (data not shown; 25). These data clearly suggest a dichotomous expression of TGF-β1 in T cells vs renal tissues of lupus mice.
FIGURE 1.

Dichotomy in TGF-β1 expression in the lymphoid vs renal tissues of lupus mice. A and B, Spleen and kidneys from 33-wk-old healthy control BALB/c and lupus-prone BWF1 mice were analyzed for TGF-β1 and β-actin. A representative Western blot in two mice per group are shown in A. TGF-β1 band densities normalized to β-actin are expressed as the mean ± SE units in B. TGF-β1 levels were lower in the spleen, but higher in the kidneys, of BWF1 mice (■) than in BALB/c mice (□). *, p < 0.05–0.01; n = 5–10 per group, all females. C and D, Spleen cells from diseased BWF1 or control healthy mice were cultured without any stimulation. Supernatants were assayed for active and total (latent plus active) TGF-β1 by ELISA. In C, 38-wk-old female BWF1 mice were compared with age-matched male and with prenephritic 12-wk-old female BWF1 mice. In D, 21-wk-old female BWF1 mice were compared with age/sex-matched CWF1 mice (*, p < 0.05; n = 3–7 mice per group). E, Immunohistochemistry was performed to compare renal TGF-β1 expression (brown color) between 35-wk-old healthy BALB/c (left panel), 24–32-wk-old BWF1 mice with early disease (middle panel), and 46-wk-old BWF1 mice with advanced disease (right panel) (n = 5–10 per group, all females). F and G, Kidneys from BALB/c and BWF1 mice of different ages were analyzed for TGF-β1 by Western blot. TGF-β1 band densities normalized to β-actin are shown as the mean ± SE. *, p < 0.05–0.001 compared with BALB/c; n = 4–9 per group, all females. H, In situ hybridization for Tgfb1 mRNA expression in the kidneys. H&E-counterstained sections using dark-field (a–e) or bright-field (f) microscopy are shown. Bright areas in dark-field microscopy (a–e) and dark dots in bright field microscopy (f) indicate Tgfb1 expression. Representative sections from BWF1 mice with early (b) or advanced (a and c–f) nephritis are shown. No expression was seen with the control sense probe (a). Weak Tgfb1 mRNA expression was seen in the glomeruli of mice with early nephritis (b), whereas it was quite intense in advanced nephritis (c). As shown in high magnification views (d–f), expression was mostly in the glomeruli (e and f) and in inflammatory cell infiltrates (Inf in d). Minimal to no Tgfb1 mRNA expression was seen in BALB/c kidneys (not shown in the figure) (n = 3–7 mice per group). Results are representative of at least three independent experiments. (Magnification: E, ×20–30; H, a–c ×4, d–f ×20–30 G, glomeruli).
Expression of TGF-β signaling molecules in the kidneys of lupus mice
TGF-β has a complex receptor and intracellular signaling system. Binding of TGF-β ligands to TβRII results in a heteromeric complex formation with TβRI causing phosphorylation of TβRI. TβRI kinase activity, in turn, causes phosphorylation of the cytoplasmic proteins SMAD2 and SMAD3 (33). Phospho-SMAD2 and -SMAD3 then form a heterodimer, which, in association with the nuclear translocation cofactor SMAD4, enters the nucleus and interacts with DNA transcription factors to coordinate the expression of TGF-β-target genes (33). Consequently, evidence for nuclear localization of SMAD3 and increased expression of phospho-SMAD3 would suggest activation of the TGF-β system. To investigate whether increased TGF-β1 expression in the kidneys of BWF1 mice is associated with the activation of the TGF-β system, we assessed the expression of TβRII, SMAD3, and phospho-SMAD3 (Fig. 2). Expression of TβRII, SMAD3, and phospho-SMAD3 was higher in the glomeruli (Fig. 2, B, E, and H) and infiltrating cells (Fig. 2, C, F, and I) of BWF1 mice than in BALB/c mice (Fig. 2, A, D, and G). Increased renal TβRII expression by immunohistochemistry in lupus kidneys was further confirmed in Western blot assays (Fig. 2J). As renal disease advances further in BWF1 mice, glomerular cells are replaced by extracellular matrix-causing glomerulosclerosis, which is followed by changes in tubular cells causing tubulointerstitial fibrosis (data not shown). At this late stage of disease, the expression of SMAD3 becomes less intense in the glomeruli and increases in the tubular cells (Fig. 2K), which is similar to the renal expression of SMAD3 in Tgfb1 transgenic mice (data not shown) that develop severe glomerulosclerosis and tubulointerstitial diseases (23). Control Tgfb1−/− kidneys exhibited minimal to no staining for SMAD3 (data not shown). The intense nuclear localization of phospho-SMAD3 (Fig. 2L) and intense nuclear localization with weak cytoplasmic staining of SMAD3 (Fig. 2, M and N) clearly suggest the activation of the TGF-β system in lupus kidneys. Thus, all components of the TGF-β system, including the ligand, its receptor, and signaling molecules, as well as nuclear localization of signaling molecules increase in the kidneys of lupus mice.
FIGURE 2.
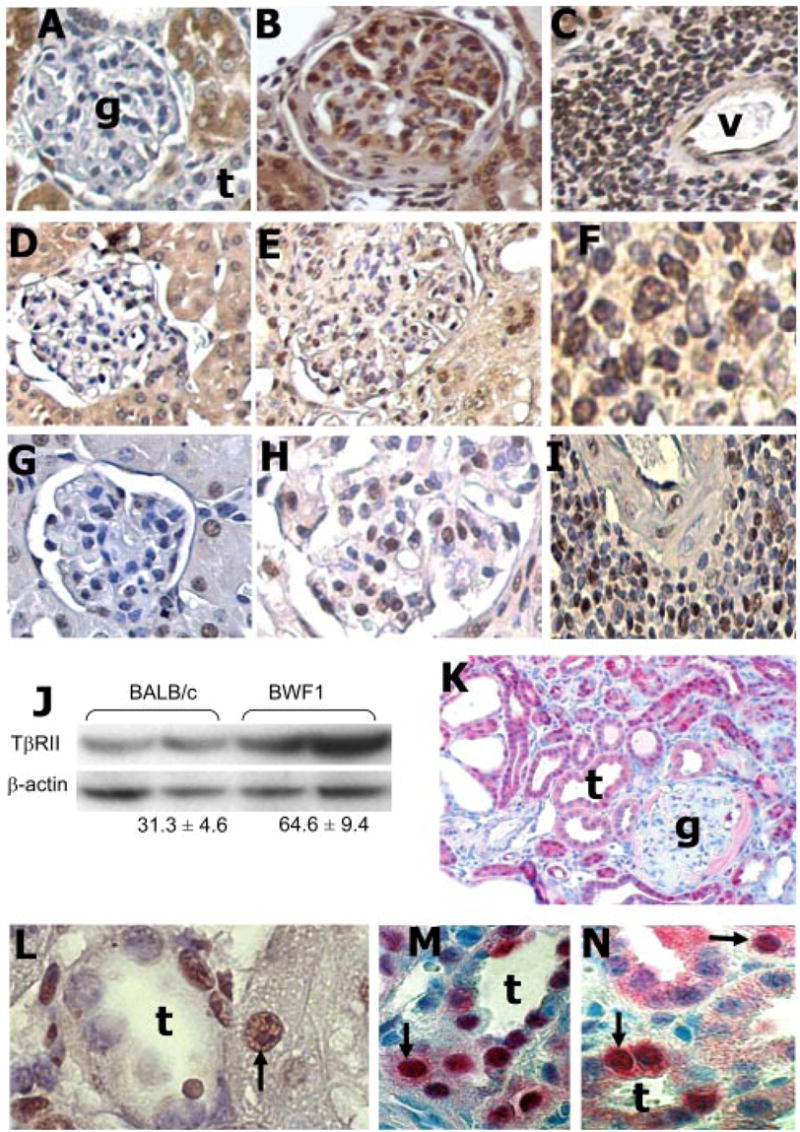
Expression of TGF-β signaling proteins in the kidneys. A–I, Immunohistochemical analysis for TβRII (A–C), SMAD3 (D–F), and phospho-SMAD3 (G–I) in renal sections from 35–46-wk-old BALB/c mice (left panels) and 25–35-wk-old BWF1 mice with diffuse proliferative nephritis (middle and right panels) (n = 6 mice per group). Brown staining denotes expression of these molecules in the glomeruli and tubules (left and middle panels) or in perivascular infiltrates (right panels). J, Western blot analysis for TβRII in protein extracts from the kidneys of 33–46-wk-old BALB/c or BWF1 mice. Mean ± SE band density for TβRII normalized to β-actin is noted under the Western blots (p < 0.01, n = 5 mice per group). (K–N) Renal sections from 36–46-wk-old BWF1 mice with chronic renal lesions, including glomerulosclerosis and tubulointerstitial nephritis, were stained to assess the expression of SMAD3 (red staining; K, M, and N) and phospho-SMAD3 (brown staining; L) by immunohistochemistry. Higher magnification views (F and L–N) clearly show intense nuclear staining for phospho-SMAD3 or intense nuclear with weak cytoplasmic staining for SMAD3 (black arrows) in tubular epithelial cells (L–N) and inflammatory cells (F). (Magnification: A–E and G–I, ×20–30; K, ×10; F, L–N, ×40–80; g, glomerulus; t, tubule; v, blood vessel).
Renal and urinary TGF-β levels correlate with chronic lesion scores
To determine whether increased renal TGF-β1 expression correlates with advancing renal disease, we measured TGF-β1 levels by ELISA in kidney eluates from BWF1 mice (Fig. 3A). We found that TGF-β1 was elevated in kidney tissues of animals having a raised composite kidney biopsy index. Whereas renal TGF-β1 levels did not correlate with the GAI that represents glomerular inflammation, the levels correlated strongly with TII and CI indices that reflect the presence of local fibrosis (Fig. 3A).
FIGURE 3.
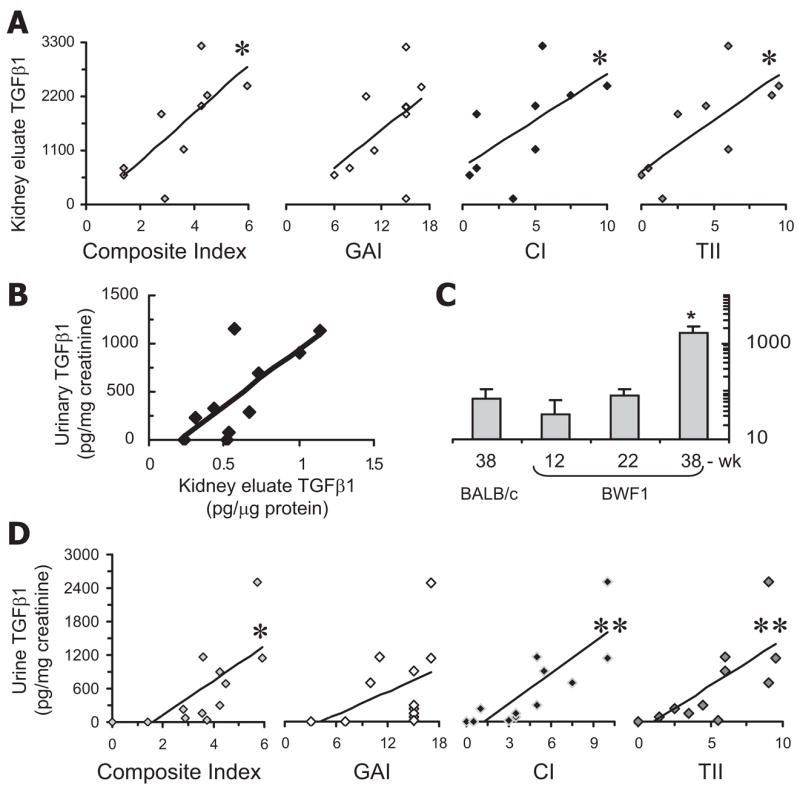
Renal and urine TGF-β1 levels correlate with chronic renal lesions in lupus mice. After collection of urine samples, BWF1 mice were sacrificed on the same day and their kidneys harvested. Half of each kidney was fixed, sectioned, and sections stained with H&E, PAS, Masson’s trichrome, and Jones silver. The stained sections were scored and the scores converted into individual renal indices, as described in Table II. Eluates from the remaining half of each kidney and urine samples were tested for total TGF-β1, protein, and creatinine levels. A, TGF-β1 levels (pg/mg protein) in kidney eluates correlate with the composite kidney biopsy, CI and TII (*R2 = 0.4–0.5, p < 0.05; n = 9 mice per group), but not with the GAI (R2 = 0.2). TGF-β1 levels from individual BWF1 mice are plotted against various kidney biopsy indices. Results represent two to four experiments. B, TGF-β1 levels in kidney eluates correlate with urinary TGF-β1 levels (R2 = 0.55). C, TGF-β1 (pg/mg creatinine) in the urine of control BALB/c and in BWF1 mice at different stages of disease [prenephritic (12-wk), early nephritic (22-wk), and advanced nephritic (38-wk)].*, p < 0.01 as compared with all other three groups; n = 7–8 mice per group; mean ± SE. D, Urinary TGF-β1 levels in BWF1 mice correlate with composite biopsy index (*R2 = 0.5, p < 0.05) and with the CI (**R2 = 0.66, p < 0.001) and TII (**R2 = 0.56, p < 0.01), but not with GAI (n = 12 mice per group).
Next, we reasoned that urinary TGF-β1 levels might reflect a rise in the activity of the TGF-β system in kidney cells, thus serving as a diagnostic marker for the advanced phase of lupus nephritis. Indeed, we found a significant correlation between renal and urinary TGF-β1 levels (Fig. 3B), and TGF-β1 levels were markedly increased in the urine of mice with advanced disease (Fig. 3C). No TGF-β1 protein was detected when microtiter ELISA plates were coated with an isotype-matched control Ig, demonstrating specificity of the assay for urinary TGF-β1 (data not shown). Next, we sought a correlation between urinary TGF-β1 levels and individual renal lesions. Although there was no correlation between urinary TGF-β1 levels and GAI, we found a strong correlation between urinary TGF-β1 levels, CI, and TII (Fig. 3D). Furthermore, whereas urinary TGF-β1 levels did not segregate animals having high vs low GAI that represents glomerular inflammation, elevated urinary TGF-β1 levels clearly segregated animals having high CIs and TIIs vs animals with low indices (*, p < 0.05; data not shown). Taken together, these data show that tissue TGF-β levels correlate with chronic fibrotic lesions but not with inflammatory lesions.
TGF-β1 inhibits production of lupus-like autoantibodies
A previous study using Tgfb1−/− mice on a mixed genetic background showed induction of lupus-like autoantibodies in the absence of TGF-β1 (8). However, admixture of genes from different backgrounds in these animals may pose difficulty in data interpretation. To test whether reduction in TGF-β1 accelerates autoantibody production in a lupus-prone background, we began to introgress the TGF-β1-null mutation onto the genetically lupus-prone MRL/Mp-lpr/lpr mice. Tgfb1+/− MRL/Mp-lpr/lpr mice, however, failed to breed beyond the N5 backcross due to their early mortality (D. Albuquerque and R. R. Singh; data not shown). Next, we backcrossed the stock Tgfb1+/− mixed strain onto the BALB/c background for eight generations. We found a significant increase in IgG ANA in Tgfb1−/− BALB/c mice as compared with wild-type littermates (Fig. 4A). All Tgfb1−/− BALB/c mice died within 3 wk of birth (our unpublished data). We then studied the effect of TGF-β1 on autoantibody production in vitro. Addition of TGF-β1 to spleen cells from lupus mice (BWF1 or pristane-injected BALB/c mice) reduced levels of IgG ANA or anti-dsDNA Ab. Such inhibitory effects were abrogated in the presence of an anti-TGF-β mAb (Fig. 4, B–D). We have previously shown that vaccination with a plasmid DNA vector that encodes a self peptide induces T cells that produce TGF-β1 (30). As shown in Fig. 4E, a T cell line raised from the vaccinated BWF1 mice reduced levels of IgG ANA in cultures, which was abrogated by the addition of an anti-TGF-β mAb, but not by the addition of a control IgG. Taken together, we provide several lines of evidence that TGF-β inhibits the production of lupus-like autoantibodies.
FIGURE 4.
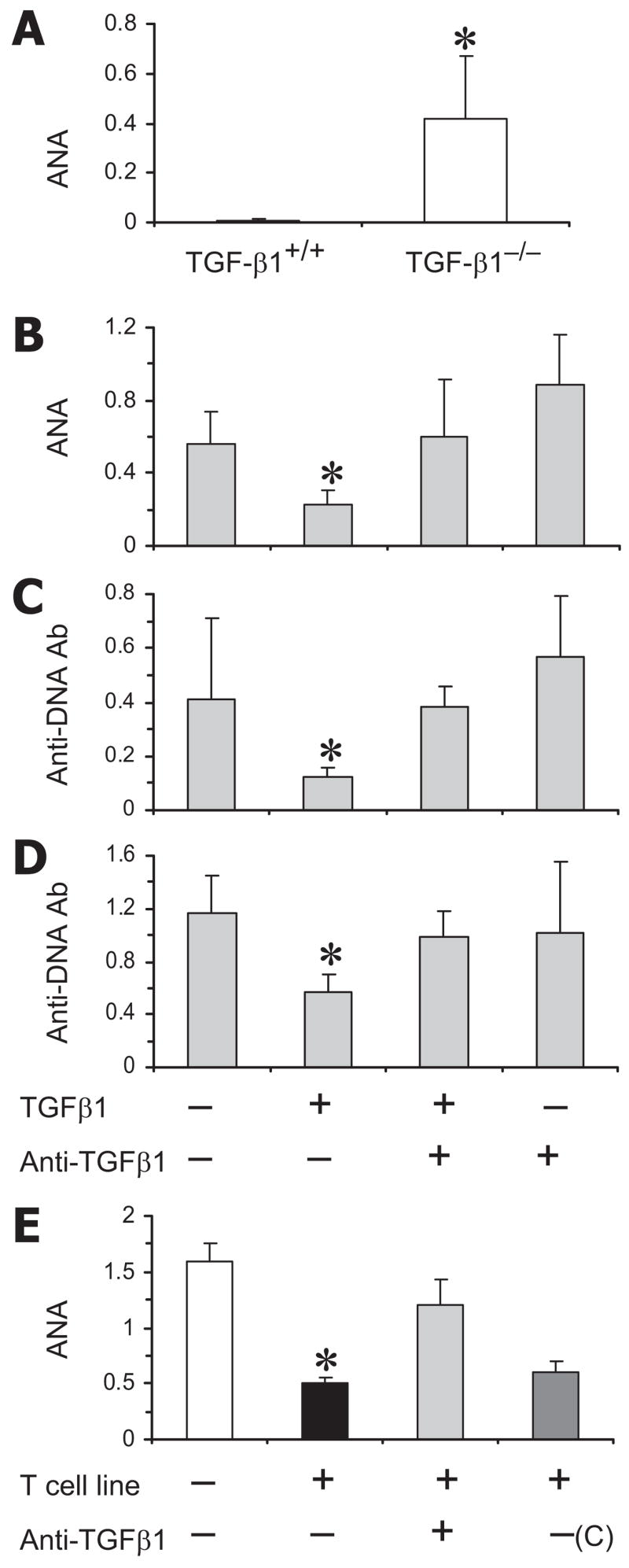
Regulatory role of TGF-β on autoantibody production. A, Sera from BALB/c Tgfb1−/− and their wild-type littermates were tested for IgG ANA by ELISA (n = 4 mice per group). Results are expressed as the mean ± SD OD values at 1/100 sera dilutions. B–D, Spleen cells from 22-wk-old pristane-injected BALB/c (B and C) or BWF1 (D) mice were cultured without or with TGF-β1 (0.01–50 ng/ml) and anti-TGF-β1 mAb (1 μg/ml) for 6 days. Culture supernatants were assayed for IgG ANA or anti-dsDNA Ab. Results are expressed as the mean ± SD OD values at 1/2 supernatant dilutions (n = 6–9 mice per group) in the presence of 20 ng/ml TGF-β1. E, BWF1 spleen cells were cultured with a TGF-β1-producing CD8+ inhibitory T cell line derived from a young BWF1 mice vaccinated with a minigene that encodes a CTL epitope (30). An anti-TGF-β1 mAb or a control IgG (C) was added at 1 μg/ml to some cultures. Supernatants collected on day 6 were assayed for IgG ANA that are shown as the mean ± SD of triplicate OD.*, p < 0.05–0.01. Results are representative of at least three separate experiments.
Effect of Ab blockade of TGF-β on lupus
To further understand the role of TGF-β in the pathogenesis of lupus, we treated BWF1 mice at 17–19 wk of age with anti-TGF-β mAb (1D11) at a dose of 500 μg per mouse twice a week for 6 wk in one experiment and for 10 wk in another experiment. Control animals were treated with control IgG at the same dosage regimen. Because TGF-β1 deficiency exacerbates autoantibody production and inflammation (5–8, 13, 34), animals were also monitored for general health, skin lesions, lymphadenopathy, and serum autoantibody levels. Surprisingly, we did not find evidence of increased autoimmune disease in the anti-TGF-β mAb-treated animals. None of the treated and 18% of control animals died during the monitoring, and anti-TGF-β mAb-treated animals did not exhibit skin lesions, weight loss, lymphoid organ enlargement, or increased cellularity when compared with controls (data not shown). Serum levels of IgG anti-cardiolipin Ab and RF, but not of IgG anti-dsDNA Ab, were slightly increased in anti-TGF-β Ab-treated animals, but the differences were not statistically significant (Fig. 5A). Importantly, organs (liver, lung, and intestine) harvested from anti-TGF-β Ab-treated animals did not show any increase in inflammation (data not shown).
FIGURE 5.

Effect of anti-TGF-β Ab treatment on autoantibody and cytokine production. BWF1 mice were treated with 1D11 or control IgG, as described in Materials and Methods. Sera, spleen cell culture supernatants, and kidney protein or RNA extracts harvested from these mice were tested for autoantibodies and cytokines by ELISA, Western blot, or RT-PCR. Results from six to nine mice in each group are expressed as the mean ± SE levels. (A) IgG anti-dsDNA, anti-cardiolipin Ab, rheumatoid factor, and total IgG levels were measured by ELISA in serum samples. No significant differences were observed between the treated and control animals. B, Serum TGF-β1 levels as detected by ELISA (*, p < 0.001). C, Renal TGF-β1 protein. A representative Western blot shows TGF-β1 in kidney extracts from five mice per group, and the band densities are expressed as the mean ± SE units (*, p < 0.001). An equal loading for protein in various lanes was seen in the Coomassie blue-stained gels (not shown in the figure). D, Renal Tgfb1 mRNA levels by RT-PCR. A representative RT-PCR and the mean ± SE DNA band density of Tgfb1 compared with GAPDH are shown. E, Spleen cell culture supernatants were assayed for cytokines IFN-γ, IL-2, IL-4, and TGF-β1 by ELISA (*, p < 0.05). Spleen cell production of TGF-β1 was very low to undetectable in both treated and control BWF1 mice (data not shown). Results represent two similar experiments.
The lack of exacerbation of autoimmunity and inflammation in anti-TGF-β mAb-treated mice in our experiments suggested that the mAb treatment might cause only partial in vivo neutralization of TGF-β, which may not be sufficient to induce the full TGF-β1 deficiency syndrome. To address this, we first determined TGF-β1 levels in serum by ELISA and protein and mRNA levels in the kidneys by Western blot and RT-PCR, respectively. Serum TGF-β1 was decreased to undetectable levels in the treated animals as compared with the control IgG-injected animals (Fig. 5B). TGF-β1 protein levels were also significantly decreased in the kidney lysates (Fig. 5C). The RT-PCR analysis for Tgfb1 mRNA levels, however, did not show any difference between the treated and control animals (Fig. 5D). TGF-β2 and TGF-β3 protein and mRNA levels in the kidneys were quite low in both treated and control animals (data not shown). Next, we measured key type 1 and 2 cytokines because TGF-β1 deficiency increases the production of type 1 cytokine IFN-γ (7). We found, however, that spleen cell production of IFN-γ and IL-2 was not significantly different between the treated and control animals (Fig. 5E). This suggests that the mAb neutralization of TGF-β1 may not be sufficient to cause enhanced proinflammatory cytokine production. Alternatively, the mAb-treated animals may have developed other mechanisms that might inhibit the production of proinflammatory cytokines. Indeed, IL-4 production was increased in spleen cell cultures from the anti-TGF-β mAb-treated mice as compared with control IgG-treated mice (Fig. 5E). Interestingly, Il4 mRNA levels were also modestly increased in the kidney tissues from 1D11-treated mice compared with control IgG-treated mice (data not shown). Such an increase in type 2 cytokines in spleen and kidneys might be responsible for the lack of induction of inflammatory disease induced by reduced levels of TGF-β after mAb treatment.
Next, to determine the role of TGF-β in the development of lupus nephritis, we monitored anti-TGF-β mAb-treated BWF1 mice for renal disease (Fig. 6). The cumulative frequency of severe proteinuria was significantly lower in the mAb-treated mice than in control IgG-injected animals (Fig. 6A). The treated and control mice were sacrificed at 30–32 wk of age or at an earlier time point when the animals were moribund, and their stained kidney sections analyzed for histological lesions. Control IgG-treated mice developed severe glomerulosclerosis, fibrous crescents, tubular disease, and interstitial fibrosis, whereas the anti-TGF-β mAb-treated animals had minimal renal fibrosis, though they continued to have glomerular infiltration (Fig. 6B). The composite kidney biopsy index was significantly lower in anti-TGF-β mAb-treated mice than in control animals (Fig. 6C). The treated mice had particularly reduced extent of chronic renal lesions (CI and TII), whereas there was no significant effect of treatment on GAI that mostly represents glomerular inflammation (Fig. 6C) or on renal IgG deposition (data not shown). Among chronic renal lesions, the tubulointerstitial and glomerulosclerosis indices were particularly reduced in the treated animals as compared with controls (Fig. 6C). To further assess the effect of TGF-β blockade on renal fibrosis, RNA isolated from the kidneys of treated and control mice was analyzed for pro-αI collagen I and IV RNA expression by RT-PCR. Pro-αI collagen mRNA levels were reduced, but the differences were not statistically significant (data not shown). The mean band density of the collagen IV as compared with the GAPDH house-keeping gene was lower in anti-TGF-β mAb-treated mice than in control mice (Fig. 6D).
FIGURE 6.
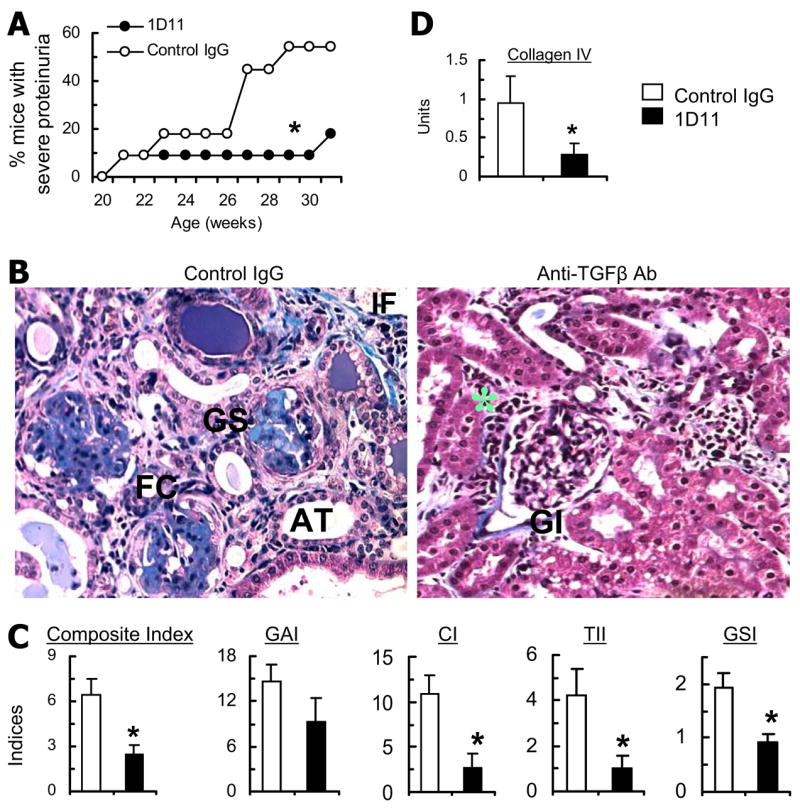
Effect of anti-TGF-β Ab treatment on autoantibody production and lupus nephritis. BWF1 mice treated with an anti-TGF-β mAb (1D11) or control mouse-IgG were bled at different time points, monitored for proteinuria, and sacrificed at 30–32 wk of age to harvest kidneys (n = 11 per group). A, Cumulative frequency of severe proteinuria (*, p < 0.05, log rank test). B, Representative Masson’s trichrome-stained kidney sections. The control animals show the presence of sclerotic lesions (GS), dilated atrophic tubules (AT), peri-glomerular (fibrous crescent, FC), and interstitial fibrosis (IF). Kidneys from the treated mice do show glomerular (GI) and periglomerular (*) infiltration. Magnification ×10. C, Renal sections stained with PAS, H&E, and Masson’s trichrome were scored and the individual scores converted into indices, including a composite kidney biopsy index, GAI, CI, TII, and GSI, which are shown as the mean ± SE of the mean individual indices (*, p < 0.05). D, Collagen IV mRNA levels. Results of DNA band densitometry are expressed as the ratio of band density for collagen to GAPDH in the mean ± SE units. Similar results were obtained in two independent experiments. GSI, glomerular sclerosis index.
Discussion
We report that TGF-β1 expression is reduced in the spleen of lupus-prone mice. TGF-β1 or TGF-β1-producing T cells down-regulate autoantibody production. In contrast, expression of TGF-β1 and its signaling molecules, TβRII, SMAD3, and phospho-SMAD3, is markedly increased in the target organ, i.e., kidneys, of these animals. In fact, TGF-β1 levels in the kidneys or urine correlate with the extent of organ damage in kidneys, particularly with the chronic fibrotic lesions but not with inflammatory lesions. Consistently, in vivo blockade of TGF-β by mAb treatment selectively inhibits the development of chronic fibrotic lesions but does not appear to affect local inflammation and autoantibody production. These data indicate dual, apparently paradoxical, roles of TGF-β in the development of early autoimmune vs late organ damage phases of systemic immune-mediated inflammatory diseases, such as lupus.
TGF-β1 prevents activation of self-reactive T cells (7, 9). TGF-β1-deficient mice develop multiorgan inflammation and production of lupus-like autoantibodies ((5, 6, 8); Fig. 4A). We have shown that nonautoimmune mice, upon immunization that breaks tolerance in autoreactive T and B cells, develop autoantibodies and mild renal disease (25). The course of such induced autoimmunity, however, is self-limited, and the recovery from disease temporally coincides with the appearance of T cells that secrete TGF-β and limit autoantibody production (25). In contrast, lupus-prone mice have impaired activation of such TGF-β-producing regulatory T cells (25). Such TGF-β1-producing T cells are also deficient in humans with SLE (13, 35). Here we show that TGF-β1 expression and production by spleen cells is reduced in lupus-prone BWF1 mice as compared with BALB/c and CWF1 mice (Fig. 1, A–D). This reduction starts before the onset of autoantibody production in BWF1 mice and is also seen in another lupus-prone strain, NZM.2410, as compared with MHC-matched control NZW mice (data not shown). Among spleen cells, surface TGF-β1 expression was particularly reduced on a subset of CD8+ T cells from BWF1 mice as compared with BALB/c mice (our unpublished data). Furthermore, addition of TGF-β1 or TGF-β1-producing T cells to spleen cells from lupus mice inhibits the production of antinuclear and anti-DNA Ab (Fig. 4, B–E) that are hallmarks of SLE. The adoptive transfer of such TGF-β1-producing T cells into young BWF1 mice suppresses autoantibody production and lupus disease (36). Genetic vaccination of BWF1 mice with peptide-encoding minigenes that activate TGF-β1-producing T cells also inhibits lupus disease (30). Taken together, TGF-β1 deficiency appears to predispose for the development of lupus-like autoantibodies and multiorgan inflammation.
The above observations have led many investigators to propose that re-implantation of ex vivo TGF-β-primed T cells might have therapeutic efficacy against systemic inflammatory diseases (35). However, we found that TGF-β1 protein and mRNA expression increases in the target organs, i.e., kidneys, of BWF1 mice as they age and develop lupus nephritis (Fig. 1, E–H). Molecules involved in TGF-β signaling, TβRII and SMAD3, are also increased in lupus kidneys (Fig. 2). Nuclear localization for SMAD3 and phospho-SMAD3 in the kidneys of lupus mice suggests that the TGF-β system is activated in these organs (Fig. 2, L–N). Furthermore, tissue concentration of TGF-β1 in kidneys correlates with the extent of chronic kidney disease (Fig. 3A). This relationship appears more robust when TGF-β1 was measured in the urine of lupus mice. Urinary TGF-β levels strongly correlate with chronic kidney damage, particularly with tubulointerstitial disease and glomerulosclerosis (Fig. 3D). Thus, our animal model study may have important implications for identifying chronic fibrotic kidney disease, as the rising levels of urinary TGF-β1 may serve as a “non-invasive” marker for active fibrogenesis, suggesting impending progression to, or the presence of, the more advanced and irreversible phase of kidney disease. This is important since studies in lupus nephritis patients suggest that the prognosis for renal survival correlates with CI that represents glomerulosclerosis and tubulointerstitial disease, and that CI is the best predictor of chronic renal failure (16–18). The mechanisms driving increased TGF-β production in inflamed renal tissues remain to be determined. We hypothesize that local parenchymal and infiltrating cells increase the production of TGF-β and other anti-inflammatory cytokines to counter inflammation and initiate local tissue repair.
The increase in target organ TGF-β expression described above is relevant for the progression of renal disease because anti-TGF-β mAb treatment of BWF1 mice with early lupus nephritis reduces the development of chronic renal lesions that are fibrotic in nature (Fig. 6). Anti-TGF-β mAb can also prevent the progression of fibrosis in primary renal diseases that are not characterized by immune system abnormalities of SLE (19, 37–39). Thus, common mechanisms appear to mediate end-organ damage after a variety of tissue injuries.
We were concerned that TGF-β blockade might enhance autoimmunity in SLE-prone mice, since TGF-β-producing T cells are known to inhibit autoantibody production (25, 34, 35). Surprisingly, anti-TGF-β Ab treatment, when given to BWF1 mice with lupus nephritis, did not worsen humoral autoimmunity (Fig. 5A) or inflammation in tissues, such as gut, lung, liver, and skin (data not shown), and did not enhance the production of inflammatory cytokines, such as IFN-γ (Fig. 5E). This was unexpected, as TGF-β1-deficient animals exhibit lymphocyte activation, enhanced proinflammatory cytokine release, and multiorgan inflammation and lupus-like autoantibodies (5–8); Fig. 4A). One possibility is that partial TGF-β deficiency rendered by Ab blockade may not be sufficient to trigger autoimmunity. Treatment with a higher dose, i.e., 3 mg per wk (instead of 1 mg per wk in our study) of anti-TGF-β Ab did exacerbate autoimmune diabetes in another study (40). Nevertheless, serum TGF-β1 was undetectable (Fig. 5B), and TGF-β1 protein in the renal tissue (Fig. 5C) and spleen cells (data not shown) was markedly reduced in anti-TGF-β Ab-treated mice. Our data suggest another, previously unappreciated, mechanism that in vivo TGF-β blockade induces the production of immuno-regulatory cytokines, such as IL-4, that may suppress tissue inflammation (41). In lupus-prone NZM.2410 mice, IL-4 blockade inhibits glomerulosclerosis while increasing the number of inflammatory cells in glomeruli and the level of anti-DNA Abs in serum (31), suggesting that IL-4, like TGF-β, might have both anti-inflammatory as well as profibrotic roles in lupus. Here, we found that IL-4 levels were indeed increased in the spleen of anti-TGF-β Ab-treated mice (Fig. 5E). This observation is consistent with a previous report that TGF-β can inhibit differentiation to Th2 cells by inhibiting IL-4 induction (42).
It is believed that infiltration with T cells or deposition of autoantibody-containing immune complexes in target organs, such as kidney, drives the early inflammatory phase of autoimmune diseases, such as lupus. Our data suggest that a lack of the immuno-regulatory influence of TGF-β on autoreactive immune cells could be a mechanism driving this phase of autoimmune diseases (Fig. 7I). The early immune-mediated injury is believed to trigger a series of events, including complement activation, chemokine production, further inflammatory cell infiltration and inflammatory cytokine release, and activation of local cells in target organs (14, 15). Our data lead to the hypothesis that the activated intrinsic renal or infiltrating cells secrete anti-inflammatory cytokines, such as TGF-β, as a local counter-regulatory measure (Fig. 7II). Such increase in TGF-β in injured tissues may cause the production of extracellular matrix for local tissue repair. An expansion of extra-cellular matrix caused by exuberant local TGF-β expression might result in dysregulated tissue repair with organ fibrosis (15, 20, 23) and eventuate end-organ failure (Fig. 7III). In summary, we envision that in immunoinflammatory diseases, immune dysregulation, induction of autoimmunity, and the initiation and progression of organ damage occur across a continuum of time and tissue, emphasizing that the role of multifunctional cytokines such as TGF-β in complex systemic diseases is highly dependent on the context in which the molecule is found. Thus, while TGF-β1 may protect against autoimmunity and inflammation, its continued up-regulation might worsen the outcome of inflammatory disease. Finally, our data in the animal model suggest that TGF-β blockade should be explored as a safe and effective means of inhibiting end-stage organ damage in patients with immunoinflammatory diseases. It is important because there is no effective treatment for patients whose autoimmune disease has progressed to end-organ fibrosis.
FIGURE 7.
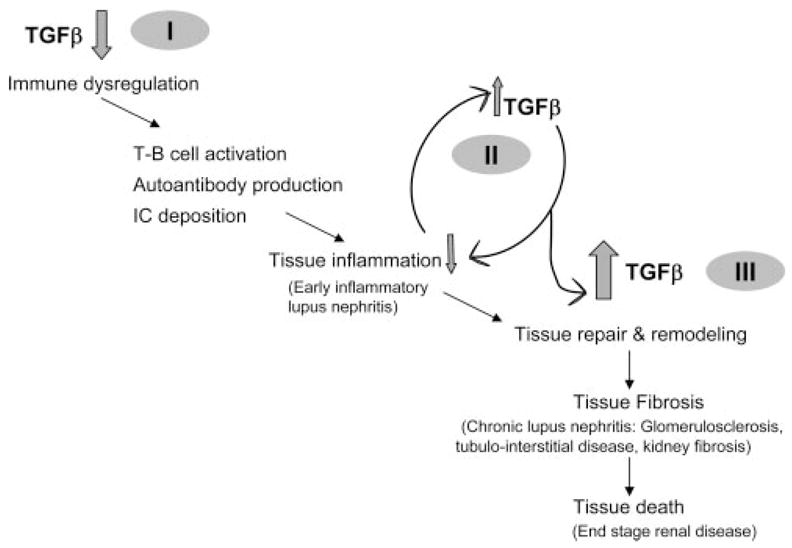
Dual roles of TGF-β in the pathogenesis of systemic immunoinflammatory diseases. According to our working model, lupus-prone subjects have reduced lymphoid tissue production of TGF-β1 (I), which predisposes to enhanced T cell activation and autoantibody production. The presence of immune complexes (IC) and inflammatory cell infiltrates in the early stage of nephritis stimulates the local counterproduction of immuno-regulatory or anti-inflammatory cytokines, such as TGF-β (II). Persistent and/or excessive production of these cytokines, however, initially results in tissue repair and remodeling but eventually culminates in tissue fibrosis (III) and end-stage organ failure.
Acknowledgments
We thank David Adams, Deijanira Albuquerque, Monica Chiaramonte, Fred Finkelman, Robert Luke, and David Witte for insightful suggestions, reagents, or technical assistance, Jeffery Kopp for providing TGF-β1 trans-genic mouse kidney, Mark Shlomchik for RF reagents, and Harold Moses for TGF-β riboprobes.
Footnotes
This work was supported in part by National Institutes of Health Grants AR47322, AR050797, and DK69282.
Abbreviations used in this paper: SLE, systemic lupus erythematosus; AP, alkaline phosphatase; BWF1, (New Zealand Black and White (NZB x NZW))F1; CI, chronicity index; CWF1, (BALB/c x NZW)F1; ANA, antinuclear Ab; GAI, glomerular activity index; PAS, periodic acid Schiff; RF, rheumatoid factor; TβRII, TGF-β receptor type II; TβRI, TGF-β receptor type I; TII, tubulointerstitial index.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345:340–350. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- 2.Singh RR. SLE: translating lessons from model systems to human disease. Trends Immunol. 2005;26:572–579. doi: 10.1016/j.it.2005.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niculescu F, Nguyen P, Niculescu T, Rus H, Rus V, Via CS. Pathogenic T cells in murine lupus exhibit spontaneous signaling activity through phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways. Arthritis Rheum. 2003;48:1071–1079. doi: 10.1002/art.10900. [DOI] [PubMed] [Google Scholar]
- 4.Bouzahzah F, Jung S, Craft J. CD4+ T cells from lupus-prone mice avoid antigen-specific tolerance induction in vivo. J Immunol. 2003;170:741–748. doi: 10.4049/jimmunol.170.2.741. [DOI] [PubMed] [Google Scholar]
- 5.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor β 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bommireddy R, Saxena V, Ormsby I, Yin M, Boivin GP, Babcock GF, Singh RR, Doetschman T. TGF-β 1 regulates lymphocyte homeostasis by preventing activation and subsequent apoptosis of peripheral lymphocytes. J Immunol. 2003;170:4612–4622. doi: 10.4049/jimmunol.170.9.4612. [DOI] [PubMed] [Google Scholar]
- 8.Dang H, Geiser AG, Letterio JJ, Nakabayashi T, Kong L, Fernandes G, Talal N. SLE-like autoantibodies and Sjogren’s syndrome-like lymphoproliferation in TGF-β knockout mice. J Immunol. 1995;155:3205–3212. [PubMed] [Google Scholar]
- 9.Bommireddy R, Pathak LJ, Martin J, Ormsby I, Engle SJ, Boivin GP, Babcock GF, Eriksson AU, Singh RR, Doetschman T. Self-antigen recognition by TGF-β1-deficient T cells causes their activation and systemic inflammation. Lab Invest. 2006;86:1008–1019. doi: 10.1038/labinvest.3700460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams DE, Yang JQ, Singh RR. Conditional TGF-β receptor II deletion in murine thymocytes results in spontaneous T cell activation and multi-organ inflammation. FASEB J. 2004;18:A1136. [Google Scholar]
- 11.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-β receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 13.Ohtsuka K, Gray JD, Stimmler MM, Toro B, Horwitz DA. Decreased production of TGF-β by lymphocytes from patients with systemic lupus erythematosus. J Immunol. 1998;160:2539–2545. [PubMed] [Google Scholar]
- 14.Grande JP. Mechanisms of progression of renal damage in lupus nephritis: pathogenesis of renal scarring. Lupus. 1998;7:604–610. doi: 10.1191/096120398678920721. [DOI] [PubMed] [Google Scholar]
- 15.Strutz F, Neilson EG. New insights into mechanisms of fibrosis in immune renal injury. Springer Semin Immunopathol. 2003;24:459–476. doi: 10.1007/s00281-003-0123-5. [DOI] [PubMed] [Google Scholar]
- 16.Hill GS, Delahousse M, Nochy D, Thervet E, Vrtovsnik F, Remy P, Glotz D, Bariety J. Outcome of relapse in lupus nephritis: roles of reversal of renal fibrosis and response of inflammation to therapy. Kidney Int. 2002;61:2176–2186. doi: 10.1046/j.1523-1755.2002.00357.x. [DOI] [PubMed] [Google Scholar]
- 17.Ravinal RC, Costa RS, Coimbra TM, Pastorello MT, Coelho EB, Dantas M, dos RM. Classes, activity and chronicity indices, and α-smooth muscle actin expression as prognostic parameters in lupus nephritis outcome. Lupus. 2002;11:82–87. doi: 10.1191/0961203302lu153oa. [DOI] [PubMed] [Google Scholar]
- 18.Daniel L, Sichez H, Giorgi R, Dussol B, Figarella-Branger D, Pellissier JF, Berland Y. Tubular lesions and tubular cell adhesion molecules for the prognosis of lupus nephritis. Kidney Int. 2001;60:2215–2221. doi: 10.1046/j.1523-1755.2001.00055.x. [DOI] [PubMed] [Google Scholar]
- 19.Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor β 1. Nature. 1990;346:371–374. doi: 10.1038/346371a0. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto T, Noble NA, Miller DE, Border WA. Sustained expression of TGF-β 1 underlies development of progressive kidney fibrosis. Kidney Int. 1994;45:916–927. doi: 10.1038/ki.1994.122. [DOI] [PubMed] [Google Scholar]
- 21.Yoshioka K, Takemura T, Murakami K, Okada M, Hino S, Miyamoto H, Maki S. Transforming growth factor-β protein and mRNA in glomeruli in normal and diseased human kidneys. Lab Invest. 1993;68:154–163. [PubMed] [Google Scholar]
- 22.Braley-Mullen H, Chen K, Wei Y, Yu S. Role of TGF-β in development of spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2001;167:7111–7118. doi: 10.4049/jimmunol.167.12.7111. [DOI] [PubMed] [Google Scholar]
- 23.Kopp JB, V, Factor M, Mozes M, Nagy P, Sanderson N, Bottinger EP, Klotman PE, Thorgeirsson SS. Transgenic mice with increased plasma levels of TGF-β 1 develop progressive renal disease. Lab Invest. 1996;74:991–1003. [PubMed] [Google Scholar]
- 24.Hahn BH, Singh RR. Animal models of systemic lupus erythematosus. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus. 7. Lippincott Williams and Wilkins; Philadelphia: 2007. pp. 299–355. [Google Scholar]
- 25.Singh RR, Ebling FM, Albuquerque DA, Saxena V, Kumar V, Giannini EH, Marion TN, Finkelman FD, Hahn BH. Induction of autoantibody production is limited in nonautoimmune mice. J Immunol. 2002;169:587–594. doi: 10.4049/jimmunol.169.1.587. [DOI] [PubMed] [Google Scholar]
- 26.Singh RR, Hahn BH, Sercarz EE. Neonatal peptide exposure can prime T cells and, upon subsequent immunization, induce their immune deviation: implications for antibody vs. T cell-mediated autoimmunity. J Exp Med. 1996;183:1613–1621. doi: 10.1084/jem.183.4.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Albuquerque DA, Saxena V, Adams DE, Boivin GP, Brunner HI, Witte DP, Singh RR. An ACE inhibitor reduces Th2 cytokines and TGF-β1 and TGF-β2 isoforms in murine lupus nephritis. Kidney Int. 2004;65:846–859. doi: 10.1111/j.1523-1755.2004.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, McCormick LL, Desai SR, Wu C, Gilliam AC. Murine sclerodermatous graft-versus-host disease, a model for human scleroderma: cutaneous cytokines, chemokines, and immune cell activation. J Immunol. 2002;168:3088–3098. doi: 10.4049/jimmunol.168.6.3088. [DOI] [PubMed] [Google Scholar]
- 29.Ling H, Li X, Jha S, Wang W, Karetskaya L, Pratt B, Ledbetter S. Therapeutic role of TGF-β-neutralizing antibody in mouse cyclosporin A nephropathy: morphologic improvement associated with functional preservation. J Am Soc Nephrol. 2003;14:377–388. doi: 10.1097/01.asn.0000042168.43665.9b. [DOI] [PubMed] [Google Scholar]
- 30.Fan GC, Singh RR. Vaccination with minigenes encoding V(H)-derived major histocompatibility complex class I-binding epitopes activates cytotoxic T cells that ablate autoantibody-producing B cells and inhibit lupus. J Exp Med. 2002;196:731–741. doi: 10.1084/jem.20020223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JQ, Saxena V, Xu H, Van Kaer L, Wang CR, Singh RR. Repeated α-galactosylceramide administration results in expansion of NK T cells and alleviates inflammatory dermatitis in MRL-lpr/lpr mice. J Immunol. 2003;171:4439–4446. doi: 10.4049/jimmunol.171.8.4439. [DOI] [PubMed] [Google Scholar]
- 32.Hill GS, Delahousse M, Nochy D, Tomkiewicz E, Remy P, Mignon F, Mery JP. A new morphologic index for the evaluation of renal biopsies in lupus nephritis. Kidney Int. 2000;58:1160–1173. doi: 10.1046/j.1523-1755.2000.00272.x. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 34.Ohtsuka K, Gray JD, Quismorio FP, Jr, Lee W, Horwitz DA. Cytokine-mediated down-regulation of B cell activity in SLE: effects of inter-leukin-2 and transforming growth factor-β. Lupus. 1999;8:95–102. doi: 10.1191/096120399678847498. [DOI] [PubMed] [Google Scholar]
- 35.Horwitz DA, Gray JD, Zheng SG. The potential of human regulatory T cells generated ex vivo as a treatment for lupus and other chronic inflammatory diseases. Arthritis Res. 2002;4:241–246. doi: 10.1186/ar414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karpouzas GA, La Cava A, Ebling FM, Singh RR, Hahn BH. Differences between CD8+ T cells in lupus-prone (NZB x NZW) F1 mice and healthy (BALB/c x NZW) F1 mice may influence autoimmunity in the lupus model. Eur J Immunol. 2004;34:2489–2499. doi: 10.1002/eji.200424978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc Natl Acad Sci USA. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benigni A, Zoja C, Corna D, Zatelli C, Conti S, Campana M, Gagliardini E, Rottoli D, Zanchi C, Abbate M, et al. Add-on anti-TGF-β antibody to ACE inhibitor arrests progressive diabetic nephropathy in the rat. J Am Soc Nephrol. 2003;14:1816–1824. doi: 10.1097/01.asn.0000074238.61967.b7. [DOI] [PubMed] [Google Scholar]
- 39.Islam M, Burke JF, Jr, McGowan TA, Zhu Y, Dunn SR, McCue P, Kanalas J, Sharma K. Effect of anti-transforming growth factor-β antibodies in cyclosporine-induced renal dysfunction. Kidney Int. 2001;59:498–506. doi: 10.1046/j.1523-1755.2001.059002498.x. [DOI] [PubMed] [Google Scholar]
- 40.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-β-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 41.Huaux F, Liu T, McGarry B, Ullenbruch M, Phan SH. Dual roles of IL-4 in lung injury and fibrosis. J Immunol. 2003;170:2083–2092. doi: 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- 42.Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-β inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000;165:4773–4777. doi: 10.4049/jimmunol.165.9.4773. [DOI] [PubMed] [Google Scholar]